

Abstract
Medulloblastoma is the most common malignant brain tumor diagnosed in children. Over the last few decades advances in radiation and chemotherapy have significantly improved the odds of survival. Nevertheless, one third of all patients still succumb to their disease, and many long-term survivors are afflicted with neurocognitive sequelae. Large scale multi-institutional efforts have provided insight into the transcriptional and genetic landscape of medulloblastoma. Four distinct subgroups of medulloblastoma have been identified, defined by distinct transcriptomes, genetics, demographics and outcomes. Integrated genomic profiling of each of these subgroups has revealed distinct genetic alterations, driving pathways and in some instances cells of origin. In this review we highlight, in a subgroup specific manner, our current knowledge of the genetic and molecular alterations in medulloblastoma and underscore the possible avenues for future therapeutic intervention.
Keywords: Medulloblastoma, molecular genetics, therapy, subgroups
INTRODUCTION
Medulloblastoma is the most common malignant paediatric brain cancer, having an incidence of approximately 0.74/100000 person-year [1, 2]. They are located in the cerebellum and 30% of cases present with metastatic dissemination over the cranial and spinal leptomeninges [3]. Initial treatment for medulloblastoma is maximal safe surgical resection followed by adjuvant craniospinal irradiation and/or high dose cytotoxic platinum based chemotherapy. Radiation as per current protocols in North America and Western Europe is risk adapted, in that, metastatic patients receive 36 Gy and non-metastatic patients receive 23.4 Gy of craniospinal irradiation with a boost to the tumour bed. With the current standard of care, overall patient survival has reached 70%, however, metastatic patients and infants are both high risk groups with poor survival [3–7]. Despite successful completion of treatment, patients frequently present with neurocognitive sequelae — long term neurological deficits in cognition [6, 7]. As such, there is an urgent need for more specific targeted therapies which minimize the impact on the developing brain.
Recent integrated genomic studies have now shown that medulloblastoma is not one single morphological entity, and is in fact at the molecular level, comprised of several different diseases. Large scale efforts focused on studying the transcriptional landscape have revealed 4 distinct subgroups (WNT, SHH, Group 3, Group 4), each with their own unique survival, age demographics, and genetic aberrations [8, 9]. These subgroups are stable at recurrence and across tumour compartments [4, 10] and are likely reminiscent of the cell of origin. The next generation of clinical trials is already taking subgroup into account to rationally stratify patients and tailor therapy. Accurate, robust, and inexpensive subgroup prediction methods are essential; molecular subgroups can be reliably assigned by either expression profiling or through the use of genome wide methylation arrays [13, 14]. Further investigation into the molecular genetics of medulloblastoma will hopefully pave the way for new targeted therapeutic strategies to cure this devastating childhood disease.
Familial Predisposition Syndromes
Initial insights into the pathways driving medulloblastoma were inferred from familial predispositions associated with medulloblastoma. The most common being Li-Framumeni syndrome with germline mutations in TP53 [11]. These mutations can drive a variety of other cancers, but in medulloblastoma both somatic and germline TP53 mutations are frequently present in childhood SHH patients and are known to facilitate catastrophic large scale rearrangements via chromothripsis [16–18]. Somatic TP53 mutations can also occur in the WNT subgroup. Less frequent is Gorlin syndrome which is an autosomal dominate disease characterised by mutations of the transmembrane receptor Patched1 (PTCH1). The majority of these patients will acquire basal cell carcinoma, while about 5–20% will get medulloblastoma [13, 14]. Deletion of the PTCH1 locus results in higher Smoothened (SMO) activity and upregulation of the Sonic Hedgehog (Shh) signalling pathway, a marker of the SHH subgroup. Less common predispositions are: i) Turcot Syndrome adenomatous polyposis coli (APC) germline mutations which are associated with a multitude of other central nervous system tumours and colorectal cancer [15, 16], and ii) autosomal dominant mutations in CREB binding protein (CREBBP) causing Rubinstein-Taybi syndrome [23]. These familial predispositions are not all encompassing and only account for a subset of patients. There are many other genetic factors which can lead to the development of medulloblastoma which will be covered in the sections that follow.
WNT SUBGROUP
Clinical Attributes
Of all the subgroups, the WNT subgroup has the most favourable prognosis with over 95% of patients surviving the disease (Table 1). WNT tumors exhibit classic histology, are rarely metastatic and have an even gender predisposition. WNT is the least common subtype, with a rate of 10% among medulloblastoma patients. The hallmark alteration in WNT tumors is somatic activating mutations in exon 3 of β-catenin (CTNNB1). Monosomy 6 is the main recurrent structural alteration and is usually found in an otherwise balanced genome [17–19].
Table 1.
| WNT | SHH | GROUP 3 | GROUP 4 | |
|---|---|---|---|---|
| AGE DISTRIBUTION | 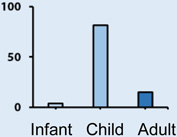 |
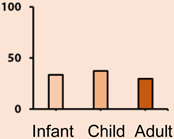 |
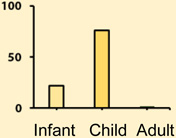 |
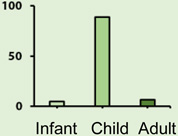 |
| GENDER (F|M) | ||||
| HISTOLOGY | Classic, Rarely LCA | Desmoplastic, Classic, LCA | Classic, LCA | Classic, LCA |
| METASTATIC RATE | Low | Low | High | High |
| PROGNOSIS | Excellent | Intermediate | Poor | Intermediate |
| SCNA | - | MYCN (12%) GLI2 (8%) |
MYC (17%) PVT1 (12%) OTX2 (8%) |
SNCAIP (10%) MYCN (6%) CDK6 (5%) |
| SNVS | CTNNB1 (91%) DDX3X (50%) SMARCA4 (26%) MLL2 (13%) TP53 (13%) |
TERT (60%) PTCH1 (46%) SUFU (24%) MLL2 (16%) SMO (14%) TP53 (13%) |
SMARCA4 (11%) MLL2 (4%) |
KDM6A (13%) MLL3 (5%) |
| BROAD EVENTS | 6 Loss | 3q Gain 9q, 10q, 14q Loss |
1q, 7, 17q, 18q Gain 8, 10q, 11, 16p, 17p Loss |
7, 17q, 18q Gain 8, 11p, X Loss |
| EXPRESSION | WNT Signaling | SHH Signaling | MYC/Retinal Signature | Neuronal Signature |
| RECURRENCE | - | Local | Metastatic | Metastatic |
Molecular Biology
The Wnt signalling pathway plays an essential role in embryonic development, controlling cell fate specification, cell proliferation, cell migration and body axis patterning. In the developing brain, the Wnt pathway has broad regulatory effects on neuronal maturation and synapse formation. This pathway is activated through binding of WNT ligands to the Frizzled receptors, which relay signals into the nucleus through the release of CTNNB1 (Fig. 1 Left). Important negative regulators of this pathway are APC and SUFU which normally prevent the accumulation of CTNNB1 in the nucleus [20, 21]. Nearly all (90%) of WNT patients have somatic missense mutations in CTNNB1 which promote protein stabilization. The next most common mutation is in DDX3X, with mutations clustering in its two helicase domains hypothesized to alter its RNA binding capacity rather than abolish it. In vivo and in vitro functional studies of DDX3X suggest that it enhances cellular and/or maintains proliferation of the WNT progenitor cells. It is also possible that these mutations help enhance transformation by β-catenin activation [22–24]. Also commonly found in WNT are missense mutations in TP53. Despite being a marker of high risk in the SHH subgroup and other cancers, TP53 mutations confer no difference in survival for patients diagnosed with WNT subgroup medulloblastomas [25].
Figure 1.

Dysregulated pathways in WNT and SHH medulloblastoma. (a) WNT patients normally have activating alteration in β-cat which promote its stabilization and allow it to upregulate target genes. (b) Alterations in the SHH subgroup usually fall within the Shh signalling as well as cooperating PI3K/ mTOR pathways and converge on the upregulation of GLI. The most common are inactivating alterations in PTCH or SMO or activating mutations in SMO. High risk patients typically have co-amplifications of MYCN, GLI2 and mutations in P53 which results in genomic instability and/or chromothripsis [26, 29–31, 81]. Activating mutations (green star); inactivating mutations (red star); amplifications (red arrow); DNA damage (yellow star); amplification (up arrow).
Models
The progenitors of the lower rhombic lip are the likely cell of origin for WNT tumours. CTNNB1 stabilization and nuclear localization is the most characteristic feature of the WNT subgroup and in mouse models its action is not sufficient to transform external granule cells, which are the SHH cells of origin. Furthermore, WNT tumours in humans are found adjacent to the brainstem unlike SHH which arise from within the cerebellum. During development, postmitotic mossy-fibre neuron precursors in the dorsal brainstem migrate into the central brainstem. Targeted expression of activated beta-catenin in mouse postmitotic mossy-fibre neuron precursors using a brain lipid-binding protein (Blbp) promoter, coupled with a knockout of TP53 leads to the formation of a WNT tumour with high latency and low penetrance [26]. Subsequent work established that through the addition of a phosphoinositide 3-kinase (PI3K) catalytic-α polypeptide mutant allele (Pik3caE545K) identified in WNT medulloblastomas, the penetrance in the mouse model was increased to 100% with highly representative WNT tumours forming within 3 months [23, 27]
SHH SUBGROUP
Clinical attributes
The SHH subgroup accounts for a third of all medulloblastoma patients and has an intermediate prognosis with a five year survival ranging between 60–80%. The age distribution is bimodal with the majority of infant and adult medulloblastomas being SHH. The histological classification can be any of the 5 described variants from the WHO classification system; however the desmoplastic variant is more common in children and adults. Large cell and/or anaplastic histology is common in children harbouring germline or somatic mutations in TP53. SHH patients commonly have focal amplifications of GLI2, and MYCN, as well as loss of 17p (Table 1) [11, 25, 34, 35].
Molecular Biology
During early cerebellar development, Purkinje cells release Shh glycoproteins and stimulate the proliferation and subsequent migration of granule cells into the internal granule cell layer. Excessive activation of the Shh pathway overdrives the expression of GLI2 transcription factor targets which induce uncontrolled proliferation of the granule cells and the formation of a tumour [28, 36]. Alterations in this subgroup most often fall within the Shh signalling pathway and less frequently in cooperating pathways such as PI3K and mTOR (Fig. 1 Right). The most common are somatic or germline inactivating alterations or loss of PTCH1 and SUFU, or somatic missense mutations activating SMO [26, 29–31]. A subset of high-risk patients present with co-amplification of MYCN and GLI2 accompanied by inactivation of TP53. Within the SHH subgroup there is also a difference in the molecular biology and risk factors for different age groups. SUFU mutations are found predominantly in infants, while the high risk GLI2 amplifications are found in older children and teenagers [17, 37]. In adults, the most common are somatic mutations in SMO and C228T or C250T of the TERT promoter [21], which creates an E-twenty-six binding motif [22, 23].
Models
There are a large variety of mouse models that recapitulate SHH subgroup, and these function mainly through dysregulation of the hedgehog signalling pathway. The first medulloblastoma mouse model used a single allele knockout of the PTCH1 gene, a negative inhibitor of the SMO pathway which drives tumorigenesis in granule cells [41]. Since then there have been other models that cross Ptch1+/− with other aberrations which confer a more aggressive phenotype, such as deletions of cyclin-dependant kinases Ink4c and Kip1 [42, 43], or the master regulator TP53 [44]. NeuroD2 dependant overexpression of mutant SMO in granule cells is also able to drive highly penetrant tumours with leptomeningeal metastasis [45, 46]. In addition, even though SHH medulloblastoma are traditionally thought to arise from granule cells, there have been mouse models that demonstrate that aberrant Shh signalling in cochlear nuclei and neural stem cells are capable of forming a tumour [47, 48].
A model that has shown great utility in screening for novel driver genes and cooperating events has been the medulloblastoma Sleeping Beauty (SB) mouse model [49] which utilizes random transposon integration to drive tumorigenesis. The transposons contain elements which are capable of overexpressing or truncating genes depending on the insertion location and orientation. Insertion events are mediated by a transposase, which is limited to granule cell precursors through the use of the MATH1 promoter to drive expression of the transposase. Nearly all the mice develop tumours with a high rate of leptomeningeal metastasis by 3 months. The SB model has identified a large number of primary tumour drivers such as MyoD [50] and Nfia [51] and has also revealed the large degree of divergence between primary and metastatic tumours (discussed below).
GROUP 3
Clinical Attributes
Group 3 medulloblastoma comprise about 20% of all medulloblastomas (Table 1). These patients have the worst survival and the highest rate of metastatic dissemination. Group 3 patients recur almost exclusively with metastatic dissemination with a clean tumour bed [4]. Patients diagnosed with this subgroup are commonly infants and younger children with a male to female discordance of 2:1. The histology of this tumour is commonly classic or large cell anaplastic (LCA) and the genome of these tumours is very unbalanced with a large number of broad alterations such as gain of chromosome 7 and isochromosome 17q; many of these alterations are also shared with Group 4.
Molecular Biology
There are several recurrent somatic copy-number alterations in Group 3, but unusually few recurrent single nucleotide variants and indels. The Group 3 transcriptome is dominated by photoreceptor and GABAergic expression signatures [10]. The most common event is amplification of MYC in 10–20% of patients, which in many cases occurs with a fusion between the promoter of PVT1 and the second exon of MYC [27]. In many cancers the MYC locus is often co-amplified with the non-coding RNA PVT1, which is able to stabilize the MYC protein [28]. In medulloblastoma, it is likely that these fusions create a positive feedback loop since the PVT1 promoter contains canonical E-boxes which are activated by the MYC protein [29]. Amplification of the transcription factor OTX2 is another common copy number alteration occurring in 10% of patients, and mutually exclusive of MYC amplification. OTX2 is known to play an important role in controlling cell fate and differentiation of various progenitors in the developing brain and is able to repress the myogenic differentiation of medulloblastoma cells. It also plays a role in the TGF-B signalling pathway which contains numerous other less recurrent copy-number alterations indicating that deregulation of this pathway may be a driver event [54–56].
Models
Two orthotopic transplantation models of Group 3 have been created which couple the overexpression of MYC with the inactivation of TP53 [57, 58]. MYC expression leads to a higher rate of proliferation as well as a higher rate of TP53 mediated apoptosis which necessitates the need to inactivate the TP53 locus. In the first model, Pei et al isolated CD133-positive and glial lineage marker-negative neural stem cells from the postnatal cerebellum and infected these cells with MYC and dominate negative TP53 (DNp53) [59]. These cells were shown to be unresponsive to Shh stimulation and capable of differentiating into neurons, astrocytes and oligodendrocytes. Infection of these cells with a stabilized MYC (MycT58A) followed by transplantation into a mouse led to the formation of a number of transient hyperplastic lesions with a high rate of apoptosis. By introducing DNp53, the apoptotic effects were abolished and tumours were formed with LCA histology, prominent necrosis, and nuclear molding. The second model was produced by isolating granule progenitor cells from Atoh1-GFP mice that express GFP in external granule layer progenitor cells, from postnatal TP53−/− mice. Atoh1-GFP enriched cells transduced with MYC were able to form aggressive tumours with LCA histology even after multiple passages in mice. In Group 3, focal events in TP53 are exceedingly rare but there is frequent loss of 17p (where TP53 resides). The resistance to Shh pathway inhibition and the similarity in Group 3 signature genes suggest that these two models are highly representative of the human disease.
GROUP 4
Clinical Attributes
Group 4 is the most common of the medulloblastoma subgroups and has an intermediate overall survival of 75% (Table 1). The histology is most commonly classic. It has a high rate of metastasis and a 2:1 male to female discordance. The Group 4 genome is commonly tetraploid, and the most common structural alteration is isochromosome 17q, which is found in 80% of patients.
Molecular Biology
Group 4 has a neuronal and glutaminergic expression signature and, like Group 3, few recurrent single nucleotide variants and indels. The most frequently mutated somatic gene in Group 4 medulloblastoma is KDM6A, a histone H3 Lys27 (H3K27) demethylase; nonsense mutation of which are found in 13% of patients [60–62]. KDM6A belongs to the Jumonji C family of histone demethylases along with KDM6B, which is also found to be mutated in medulloblastoma. The proto-oncogenes MYCN and cyclin-dependant kinase CDK6 are recurrently amplified in Group 4. More common are focal amplifications / tandem duplications of the alpha-synuclein interacting protein (SNCAIP) gene on chromosome 5q23 [26], which encodes a protein previously implicated in Parkinson’s disease [63]. It is still unknown if SNCAIP is the driver for these patients, more research needs to be done to uncover its specific role in Group 4 medulloblastoma.
Models
Due to the low number of focal events and many broad rearrangements the search for a model of Group 4 has proven elusive. MYCN is commonly upregulated in medulloblastoma and is the site of one of the most recurrent focal amplifications in Group 4. Swartling et al created a mouse model of MYCN driven medulloblastoma by targeting its expression with Glt1, a brain specific promoter highly expressed in the cerebellum throughout development until adulthood. The tumours had a low latency and metastatic rate and were either a classic or LCA histology type. MYCN was required for both the initiation and maintenance of the tumour but was likely cooperating with other events since the genome was unbalanced and had a number of recurrent copy number alterations. This model is showing great promise as a Group 4 model, but additional studies need to be performed in order to characterize the expression signatures and identify the cell of origin [33].
EPIGENETICS
There have been a number of recurrent somatic single nucleotide and copy number variants within chromatin modifiers identified across all of the subgroups. Most common are truncating mutations in myeloid/lymphoid or mixed-lineage leukemia protein 2 (MLL2) and MLL3 suggesting a role as a universal oncogenic driver. In Group 3 and Group 4 there are a large variety of recurrent somatic mutations in SMARCA4 (exclusive to Group 3), and KDM6A (exclusive to Group 4), and less commonly in CHD7, ARID1B, KDM4C, and ZMYM3 [26, 29–31]. There is also over-expression of EZH2, a H3K27 methyltransferase that is part of the polycomb repressive complex essential for regulating development and differentiation. These events are largely mutually exclusive of each other and the amplifications of MYC and MYCN. The mechanism of their pathogenesis is still a subject of intense investigation, but it is possible that these events preserve Group 3 and Group 4 tumours in a stem cell-like state by maintaining high levels of the repressive H3K27me3 mark (EZH2 upregulation or KDM6A inactivation) and/or disruption of H3K4me3 associated transcription (ZMYM3 and CHD7 inactivation) [55, 64, 65].
Enhancer-promoter interactions play an essential role in tissue specific regulation of genes and development [66]. The three-dimensional localization of active enhancers ultimately decides which genes can be activated by the enhancer and any disruption can lead to the aberrant regulation of genes. In Group 3 and Group 4 medulloblastoma it has recently been demonstrated that structural rearrangements are able to alter the intended targets of enhancers to drive tumorigenesis in medulloblastoma [67]. In particular, a series of seemingly unrelated deletion, duplication and translocation events were able to activate expression of the transcription factors GFI1 or GFI1B through the repositioning of distal enhancers. These somatic events were highly recurrent, constituting a third of Group 3 and 10% of Group 4 tumours. When these genes were co-expressed with MYC in murine neural stem cells, they were also able to drive the formation of an aggressive tumour in recipient mice with a high rate of metastasis.
METASTASES
In medulloblastoma patients, metastasis is a sign of dismal prognosis. It is most common in Group 3 and Group 4 patients, both of which almost exclusively recur with metastatic dissemination [4]. Little is known about the genes driving dissemination and the context in which they operate since matching patient primary and metastatic samples are exceedingly rare. Studies with the SB mouse model have shown that there is a large genetic divergence between the primary and metastatic compartments with almost no overlap in common insertion sites [50] suggesting that the primary tumour is a poor indicator of the therapeutic targets present in the metastatic lesions. These PTCH1-driven SB models have revealed a number of possible drivers of metastasis such as Eras, Lhx1, Ccrk, and Gdi2 which potentially drive dissemination in SHH patients [70, 71]. Tumour-stromal interactions also appear to play an essential role in medulloblastoma tumorigenesis and metastatic dissemination. Tumour cell-induced expression of the placental growth factor (PIGF) in the stroma was shown to activate pro-survival pathways through the Nrp1 receptor [68] and promote tumour growth and metastasis.
THERAPIES
The WNT subgroup has the best prognosis out of all the subgroups; nearly all patients surviving after surgery, radiation and chemotherapy. For this subgroup there have been international efforts to de-escalate therapy to help reduce the long term cognitive deficits common in children after receiving radiotherapy.
There are a number of proposed therapies for SHH patients which all aim to lower the aberrant activity of the Shh pathway. One of the most promising class of drugs are SMO inhibitors, which are already in phase II clinical trials for a number of cancers including medulloblastoma [69–71]. Unfortunately, acquired resistance is inevitable in both animals and humans; a recurrent mutation in a conserved aspartic acid residue within the G protein–coupled receptor domain of SMO has been shown to disrupt the functionality of inhibitors, leaving Shh signalling intact [71, 72]. Furthermore, the drug is only effective for patients with alterations within or upstream of SMO, and therefore high-risk children with amplifications of MYCN and GLI2 would not be able to benefit from this treatment [17]. Another class of drugs called bromodomain inhibitors may be able to circumvent this problem by inhibiting the Shh at the level of GLI2. BRD4 is a bromodomain protein which binds to ε-N-lysine acetylation motifs on open chromatin and is known to facilitate transcription at promoter regions of key transcription factors such as GlI2 and MYC. Treatment of SHH with BRD4 inhibitors has shown great promise in pre-clinical models even in the presence of SMO drug resistance mutations [72, 73].
The search for specific therapies for Group 3 and 4 patients has proven elusive. In Group 3, TGF-beta signaling is commonly dysregulated and pathway antagonists, to target this, are already being explored for a multitude of cancers, including glioblastoma; varying success has been observed [74]. MYC inhibition is another attractive but challenging therapeutic strategy. There are no known direct inhibitors of MYC — studies have focused on inhibiting the expression of MYC RNA [75] or inhibiting its heterodimer MAX [76, 77]. So far the most promising approach is inhibition of BRD4 using bromodomain inhibitor JQ1 to reduce the transcription of MYC [78]. There is also some evidence that JQ1 may be effective for treatment of MYCN driven neuroblastoma in pre-clinical models; suggesting that it could be effective for Group 4 patients [79]. In both Group 3 and Group 4, epigenetic alterations are a characteristic feature and there are a number of approved drugs in clinical trials for several adult and pediatric brain tumours. In particular, there are several specific inhibitors of the polycomb repressive complex 2 as well as EZH2 which act to antagonize the levels of H3K27me3 [80].
CONCLUSION
Large scale genomic and transcriptional studies have completely revolutionized our understanding of medulloblastoma pathogenesis. They have made sense of a diverse set of dysregulated pathways, and genetic alterations, and have presented a more rational way to stratify patients for targeted therapy. With these new paradigms in mind, clinical trials are already underway which address the tumour biology, particularly for the SHH and WNT subgroups. With so few recurrent genetic events, the focus is shifting towards the relationship between epi- and molecular genetics. This is an exciting time, and research into the molecular genetics of medulloblastoma will no doubt help maximize survival while minimizing long term developmental defects.
ACKNOWLEDGMENTS
FUNDING INFORMATION
MDT is funded by the Canadian Institutes of Health Research, National Institutes of Health (R01CA159859 and R01CA148699), Pediatric Brain Tumor Foundation, he is the Garron Family Chair in Childhood Cancer Research at The Hospital for Sick Children and The University of Toronto. VR is supported by a Canadian Institutes of Health Research fellowship, AIHS Clinical Fellowship and ALSF Young Investigator award. PS is funded through Natural Sciences and Engineering Research Council of Canada (NSERC), University of Toronto, and the Research Training Competition (RESTRACOMP) at The Hospital for Sick Children.
REFERENCES
- 1.Pui C-H, Gajjar AJ, Kane JR, Qaddoumi IA, Pappo AS. Challenging issues in pediatric oncology. Nat Rev Clin Oncol. 2011;8:540–549. doi: 10.1038/nrclinonc.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 3.Rutkowski S, Von Hoff K, Emser A, Zwiener I, Pietsch T, Figarella-Branger D, Giangaspero F, Ellison DW, Garre ML, Biassoni V, et al. Survival and prognostic factors of early childhood medulloblastoma: An international meta-analysis. J Clin Oncol. 2010;28:4961–4968. doi: 10.1200/JCO.2010.30.2299. [DOI] [PubMed] [Google Scholar]
- 4.Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho Y-J, Shih DJ, Luu B, Dubuc AM, Northcott PA, et al. Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol. 2013;14:1200–1207. doi: 10.1016/S1470-2045(13)70449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, Woo S, Wheeler G, Ahern V, Krasin MJ, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813–820. doi: 10.1016/S1470-2045(06)70867-1. [DOI] [PubMed] [Google Scholar]
- 6.Shih DJH, Northcott Pa, Remke M, Korshunov A, Ramaswamy V, Kool M, Luu B, Yao Y, Wang X, Dubuc AM, et al. Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol. 2014;32:886–896. doi: 10.1200/JCO.2013.50.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Packer RJ, Gajjar A, Vezina G, Rorke-Adams L, Burger PC, Robertson PL, Bayer L, LaFond D, Donahue BR, Marymont MH, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202–4208. doi: 10.1200/JCO.2006.06.4980. [DOI] [PubMed] [Google Scholar]
- 8.Mabbott DJ, Spiegler BJ, Greenberg ML, Rutka JT, Hyder DJ, Bouffet E. Serial evaluation of academic and behavioral outcome after treatment with cranial radiation in childhood. J Clin Oncol. 2005;23:2256–2263. doi: 10.1200/JCO.2005.01.158. [DOI] [PubMed] [Google Scholar]
- 9.Spiegler BJ, Bouffet E, Greenberg ML, Rutka JT, Mabbott DJ. Change in neurocognitive functioning after treatment with cranial radiation in childhood. J Clin Oncol. 2004;22:706–713. doi: 10.1200/JCO.2004.05.186. [DOI] [PubMed] [Google Scholar]
- 10.Taylor MD, Northcott Pa, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012;123:465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, Berhoukim R, Amani V, Goumnerova L, Eberhart CG, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol. 2011;29:1424–1430. doi: 10.1200/JCO.2010.28.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Dubuc AM, Ramaswamy V, Mack S, Gendoo DMa, Remke M, Wu X, Garzia L, Luu B, Cavalli F, et al. Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol. 2015 doi: 10.1007/s00401-015-1389-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pietsch T, Schmidt R, Remke M, Korshunov A, Hovestadt V, Jones DTW, Felsberg J, Kaulich K, Goschzik T, Kool M, et al. Prognostic significance of clinical, histopathological, and molecular characteristics of medulloblastomas in the prospective HIT2000 multicenter clinical trial cohort. Acta Neuropathol. 2014;128:137–149. doi: 10.1007/s00401-014-1276-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Northcott Pa, Shih DJH, Remke M, Cho Y-J, Kool M, Hawkins C, Eberhart CG, Dubuc A, Guettouche T, Cardentey Y, et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol. 2012;123:615–626. doi: 10.1007/s00401-011-0899-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varley JM, Evans DGR, Birch JM Gene THETP. Li-Fraumeni syndrome - a molecular and clinical review. 1997;76:1–14. doi: 10.1038/bjc.1997.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rausch T, Jones DT, Zapatka M, Stütz AM, Zichner T, Weischenfeldt J, Jäger N, Remke M, Shih D, Northcott PA, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kool M, Jones DT, Jäger N, Northcott Pa, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M, et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJH, Martin DC, Castelo-Branco P, Baskin B, Ray PN, Bouffet E, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013;31:2927–2935. doi: 10.1200/JCO.2012.48.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans DG, Farndon PA, Burnell LD, Gattamaneni HR, Birch JM. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer. 1991;64:959–961. doi: 10.1038/bjc.1991.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farndon PA, Del Mastro RG, Evans DG, Kilpatrick MW. Location of gene for Gorlin syndrome. Lancet. 1992;339:581–582. doi: 10.1016/0140-6736(92)90868-4. [DOI] [PubMed] [Google Scholar]
- 21.Huang H, Mahler-Araujo BM, Sankila A, Chimelli L, Yonekawa Y, Kleihues P, Ohgaki H. APC mutations in sporadic medulloblastomas. Am J Pathol. 2000;156:433–437. doi: 10.1016/S0002-9440(10)64747-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995;332:839–847. doi: 10.1056/NEJM199503303321302. [DOI] [PubMed] [Google Scholar]
- 23.Kushner BH, LaQuaglia MP, Wollner N, Meyers PA, Lindsley KL, Ghavimi F, Merchant TE, Boulad F, Cheung NK, Bonilla MA, et al. Rubinstein–Taybi Syndrome Predisposing to Non-WNT, Non-SHH, Group 3 Medulloblastoma. J Clin Oncol. 1996;14:1526–1531. doi: 10.1200/JCO.1996.14.5.1526. [DOI] [PubMed] [Google Scholar]
- 24.Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE, Pearson AD, Clifford SC. β-catenin status predicts a favorable outcome in childhood medulloblastoma: The United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005;23:7951–7957. doi: 10.1200/JCO.2005.01.5479. [DOI] [PubMed] [Google Scholar]
- 25.Kool M, Korshunov A, Remke M, Jones DTW, Schlanstein M, Northcott PA, Cho YJ, Koster J, Schouten-Van Meeteren A, Van Vuurden D, et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123:473–484. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Northcott PA, Shih DJH, Peacock J, Garzia L, Morrissy AS, Zichner T, Stütz AM, Korshunov A, Reimand J, Schumacher SE, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patapoutian A, Reichardt LF. Roles of Wnt proteins in neural development and maintenance. Curr Opin Neurobiol. 2000;10:392–399. doi: 10.1016/s0959-4388(00)00100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marino S. Medulloblastoma: Developmental mechanisms out of control. Trends Mol Med. 2005;11:17–22. doi: 10.1016/j.molmed.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 29.Jones DTW, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, Cho Y-J, Pugh TJ, Hovestadt V, Stütz AM, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson G, Parker M, Kranenburg Ta, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–48. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–1099. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pöschl J, Stark S, Neumann P, Gröbner S, Kawauchi D, Jones DTW, Northcott PA, Lichter P, Pfister SM, Kool M, et al. Genomic and transcriptomic analyses match medulloblastoma mouse models to their human counterparts. Acta Neuropathol. 2014;128:123–136. doi: 10.1007/s00401-014-1297-8. [DOI] [PubMed] [Google Scholar]
- 34.Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramaswamy V, Remke M, Adamski J, Bartels U, Tabori U, Wang X, Huang a, Hawkins C, Mabbott D, Laperriere N, et al. Medulloblastoma subgroup-specific outcomes in irradiated children: who are the true high-risk patients? Neuro Oncol. 2015:1–7. doi: 10.1093/neuonc/nou357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hatten ME, Roussel MF. Development and cancer of the cerebellum. Trends Neurosci. 2011;34:134–142. doi: 10.1016/j.tins.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Northcott PA, Hielscher T, Dubuc A, MacK S, Shih D, Remke M, Al-Halabi H, Albrecht S, Jabado N, Eberhart CG, et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011;122:231–240. doi: 10.1007/s00401-011-0846-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Remke M, Ramaswamy V, Peacock J, Shih DJH, Koelsche C, Northcott PA, Hill N, Cavalli FMG, Kool M, Wang X, et al. TERT promoter mutations are highly recurrent in SHH subgroup medulloblastoma. Acta Neuropathol. 2013;126:917–929. doi: 10.1007/s00401-013-1198-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. 2013:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 40.Lamb ME, Sternberg K, Wan C, Lin G, Jin Q, Leichtman MD, White SH, Meidinger C, Rapoport B, Kolm SC, et al. Highly Recurrent TERT Promoter Mutations in Human Melanoma. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 42.Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, Rehg JE, Calabrese C, Solecki D, Eberhart CG, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev. 2005;19:2656–2567. doi: 10.1101/gad.1368605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ayrault O, Zindy F, Rehg J, Sherr CJ, Roussel MF. Two tumor suppressors, p27Kip1 and patched-1, collaborate to prevent medulloblastoma. Mol Cancer Res. 2009;7:33–40. doi: 10.1158/1541-7786.MCR-08-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001;61:513–516. [PubMed] [Google Scholar]
- 45.Hatton Ba, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B, Hansen S, Knoblaugh SE, Lee D, Eberhart CG, et al. The Smo/Smo model: Hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008;68:1768–1776. doi: 10.1158/0008-5472.CAN-07-5092. [DOI] [PubMed] [Google Scholar]
- 46.Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, Russell TL, Ellenbogen RG, Bernstein ID, Beachy PA, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of Sonic Hedgehog-induced medulloblastomas. Cancer Res. 2004;64:7794–7800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 47.Grammel D, Warmuth-Metz M, Von Bueren AO, Kool M, Pietsch T, Kretzschmar HA, Rowitch DH, Rutkowski S, Pfister SM, Schüller U. Sonic hedgehog-associated medullobla stoma arising from the cochlear nuclei of the brainstem. Acta Neuropathol. 2012;123:601–614. doi: 10.1007/s00401-012-0961-0. [DOI] [PubMed] [Google Scholar]
- 48.Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schüller U, Machold R, Fishell G, Rowitch DH, et al. Medulloblastoma Can Be Initiated by Deletion of Patched in Lineage-Restricted Progenitors or Stem Cells. Cancer Cell. 2008;14:135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJH, Witt H, Croul S, Bouffet E, Fults DW, Eberhart CG, et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature. 2012;482:529–533. doi: 10.1038/nature10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dey J, Dubuc AM, Pedro KD, Thirstrup D, Mecham B, Northcott PA, Wu X, Shih D, Tapscott SJ, LeBlanc M, et al. MyoD is a tumor suppressor gene in medulloblastoma. Cancer Res. 2013;73:6828–6837. doi: 10.1158/0008-5472.CAN-13-0730-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Genovesi LA, Ng CG, Davis MJ, Remke M, Taylor MD, Adams DJ, Rust AG, Ward JM, Ban KH, Jenkins Na, et al. Sleeping Beauty mutagenesis in a mouse medulloblastoma model defines networks that discriminate between human molecular subgroups. Proc Natl Acad Sci U S A. 2013;110:E4325–E4334. doi: 10.1073/pnas.1318639110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tseng Y-Y, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, Ronning P, Reuland B, Guenther K, Beadnell TC, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014 doi: 10.1038/nature13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carramusa L, Contino F, Ferro A, Minafra L, Perconti G, Giallongo A, Feo S. The PVT-1 Oncogene Is a Myc Protein Target That Is Overexpressed in Transformed Cells. J Cell Physiol. 2007;213:440–444. doi: 10.1002/jcp.21133. [DOI] [PubMed] [Google Scholar]
- 54.Bai RY, Staedtke V, Lidov HG, Eberhart CG, Riggins GJ. OTX2 represses myogenic and neuronal differentiation in medulloblastoma cells. Cancer Res. 2012;72:5988–6001. doi: 10.1158/0008-5472.CAN-12-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bunt J, Hasselt NA, Zwijnenburg DA, Koster J, Versteeg R, Kool M. OTX2 sustains a bivalent-like state of OTX2-bound promoters in medulloblastoma by maintaining their H3K27me3 levels. Acta Neuropathol. 2013;125:385–394. doi: 10.1007/s00401-012-1069-2. [DOI] [PubMed] [Google Scholar]
- 56.Young MD, Willson TA, Wakefield MJ, Trounson E, Hilton DJ, Blewitt ME, Oshlack A, Majewski IJ. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 2011;39:7415–7427. doi: 10.1093/nar/gkr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, Witt H, Korshunov A, Read TA, Sun JL, et al. An Animal Model of MYC-Driven Medulloblastoma. Cancer Cell. 2012;21:155–167. doi: 10.1016/j.ccr.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, Finkelstein D, Qu C, Pounds S, Ellison DW, et al. A Mouse Model of the Most Aggressive Subgroup of Human Medulloblastoma. Cancer Cell. 2012;21:168–180. doi: 10.1016/j.ccr.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee A, Kessler JD, Read T-A, Kaiser C, Corbeil D, Huttner WB, Johnson JE, Wechsler-Reya RJ. Isolation of neural stem cells from the postnatal cerebellum. Nat Neurosci. 2005;8:723–729. doi: 10.1038/nn1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sengoku T, Yokoyama S. Structural basis for histone H3 lys 27 demethylation by UTX/KDM6A. Genes Dev. 2011;25:2266–2277. doi: 10.1101/gad.172296.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim E, Song JJ. Diverse ways to be specific: A novel zn-binding domain confers substrate specificity to UTX/KDM6a histone H3 lys 27 demethylase. Genes Dev. 2011;25:223–2226. doi: 10.1101/gad.179473.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13:297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- 63.Wan OW, Chung KKK. The role of alpha-synuclein oligomerization and aggregation in cellular and animal models of Parkinson’s disease. PLoS One. 2012;7:1–14. doi: 10.1371/journal.pone.0038545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dubuc AM, Remke M, Korshunov A, Northcott PA, Zhan SH, Mendez-Lago M, Kool M, Jones DTW, Unterberger A, Morrissy AS, et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2013;125:373–384. doi: 10.1007/s00401-012-1070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Northcott Pa, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009;41:465–472. doi: 10.1038/ng.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ong C-T, Corces VG. Enhancers: emerging roles in cell fate specification. EMBO Rep. 2012;13:423–430. doi: 10.1038/embor.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D, Shih DJH, Hovestadt V, Zapatka M, Sturm D, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014 doi: 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Snuderl M, Batista A, Kirkpatrick ND, Ruiz de Almodovar C, Riedemann L, Walsh EC, Anolik R, Huang Y, Martin JD, Kamoun W, et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell. 2013;152:1065–1076. doi: 10.1016/j.cell.2013.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–1178. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Chang I, Darbonne WC, et al. Phase I Trial of Hedgehog Pathway Inhibitor Vismodegib (GDC-0449) in Patients with Refractory, Locally Advanced or Metastatic Solid Tumors. Clin Cancer Res. 2011;17:2502–2511. doi: 10.1158/1078-0432.CCR-10-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rodon J, Tawbi HA, Thomas AL, Stoller RG, Turtschi CP, Baselga J, Sarantopoulos J, Mahalingam D, Shou Y, Moles MA, et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin Cancer Res. 2014;20:1900–1909. doi: 10.1158/1078-0432.CCR-13-1710. [DOI] [PubMed] [Google Scholar]
- 72.Long J, Li B, Rodriguez-Blanco J, Pastori C, Volmar C-H, Wahlestedt C, Capobianco A, Bai F, Pei X-H, Ayad NG, et al. The BET Bromodomain Inhibitor I-BET151 Acts Downstream of Smoothened Protein to Abrogate the Growth of Hedgehog Protein-driven Cancers. J Biol Chem. 2014;289:35494–35502. doi: 10.1074/jbc.M114.595348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tang Y, Gholamin S, Schubert S, Willardson MI, Lee A, Bandopadhayay P, Bergthold G, Masoud S, Nguyen B, Vue N, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 2014;20:732–740. doi: 10.1038/nm.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brooks TA, Hurley LH. Targeting MYC Expression through G-Quadruplexes. Genes Cancer. 2010;1:641–649. doi: 10.1177/1947601910377493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22:6151–6159. doi: 10.1038/sj.onc.1206641. [DOI] [PubMed] [Google Scholar]
- 77.Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Metallo SJ, Prochownik EV. Improved low molecular weight Myc-Max inhibitors. Mol Cancer Ther. 2007;6:2399–2408. doi: 10.1158/1535-7163.MCT-07-0005. [DOI] [PubMed] [Google Scholar]
- 78.Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res. 2014;20:912–925. doi: 10.1158/1078-0432.CCR-13-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3:309–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 81.Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, Pomeroy SL, Korshunov A, Lichter P, Taylor MD, et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer. 2012;12:818–834. doi: 10.1038/nrc3410. [DOI] [PMC free article] [PubMed] [Google Scholar]