

Abstract
Protein glycosylation is a heterogeneous post-translational modification (PTM) that plays an essential role in biological regulation. However, the diversity found in glycoproteins has undermined efforts to describe the intact glycoproteome via mass spectrometry (MS). We present IsoTaG, a mass-independent chemical glycoproteomics platform for characterization of intact, metabolically labeled glycopeptides at the whole-proteome scale. In IsoTaG, metabolic labeling of the glycoproteome is combined with (i) chemical enrichment and isotopic recoding of glycopeptides to select peptides for targeted glycoproteomics using directed MS and (ii) mass-independent assignment of intact glycopeptides. We structurally assigned 32 N-glycopeptides and over 500 intact and fully elaborated O-glycopeptides from 250 proteins across three human cancer cell lines and also discovered unexpected peptide sequence polymorphisms (pSPs). The IsoTaG platform is broadly applicable to the discovery of PTM sites that are amenable to chemical labeling, as well as previously unknown protein isoforms including pSPs.
Glycosylation of proteins generates greater proteomic diversity than any other PTM. In eukaryotes, most glycans are attached to proteins at asparagine (N-glycans) or serine or threonine (O-glycans) side chains. These forms of glycosylation regulate a range of biological processes, including protein folding and trafficking1, signaling2 and cell-cell communication3. Dynamic changes in protein glycosylation have been found to accompany disease states such as Alzheimer’s4 and cancer progression5, suggesting that specific protein glycoforms6, and the enzymes that generate them7, may serve as disease biomarkers and therapeutic targets.
Methods to profile the intact glycoproteome would enable correlation of glycosylation variants with disease state, yet structural characterization of glycoproteins poses a great challenge in biology8,9. MS is often the method of choice for profiling the proteome, but expansion of MS methods for profiling the glycoproteome is uniquely problematic. In contrast to proteomic profiling of uniform PTMs (such as phosphorylation10,11 and acetylation12), intact-glycopeptide analysis faces challenges on two fronts: detection by MS and computational identification via database searching. Glycan heterogeneity produces a number of substoichiometric modifications to the glycopeptide that effectively reduce the quantity of any particular glycoform. Because of this, as well as the lower ionization efficiency of glycoconjugates13, the detection of intact glycopeptides often requires increased sample quantities and enrichment. Moreover, the heterogeneity and nontemplated nature of glycopeptides stymie computational analysis. Crucially, most high-throughput proteomic platforms rely on database searches for peptide identification14. Nontemplated PTMs (for example, glycosylation) present significant computational challenges on the proteome level. Whereas a consensus sequence exists for N-glycosylation (N-X-S/T, where X is not proline), there are no defined consensus motifs for the various types of O-glycosylation. Indeed, the sheer number of possible glycoforms associated with the entire proteome yields an unmanageable sum of combinations for database search algorithms to match.
Without a universal method to identify glycoforms by database searches, enrichment of specific glycoprotein subtypes and reduction of glycan heterogeneity are presently required for glycoproteomic profiling. Enrichment of specific glycoprotein subtypes has been achieved via metabolic labeling15–18, chemical tagging19,20, enzymatic labeling21 and lectin chromatography22–24. Nonetheless, identification of glycopeptides from whole proteomes requires reduction of the glycans to a uniform and predictable mass. Reduction of the glycan has been achieved for N-glycoproteins by enzymatic digestion (by PNGase F and EndoH)25 and for O-glycans by genetic engineering (SimpleCell)26,27. However, truncation of the glycan destroys information correlating glycan structural heterogeneity to the protein attachment site and undermines further correlation with biological activity. Methods for broadly profiling intact glycopeptides on the whole-proteome scale have until now remained elusive.
Here we present a mass-independent chemical glycoproteomics platform that enabled us to identify hundreds of intact glycopeptides from whole cell glycoproteomes. The platform, termed isotope-targeted glycoproteomics (IsoTaG), comprises four central components: (i) metabolic labeling with a chemically functionalized glycan, (ii) chemical tagging and enrichment using an isotopic recoding affinity probe, (iii) directed tandem MS and (iv) targeted glycopeptide assignment. In earlier work, we developed a computational algorithm, termed isotopic signature transfer and mass pattern prediction (IsoStamp)28, for the detection of recoded species in full-scan mass spectra. Here, we show that an IsoTaG approach improved glycopeptide targeting and led to the identification of 32 intact N-glycopeptides and over 500 intact O-glycopeptides from 250 glycoproteins across three cancer cell lines. Approximately 50% of the identified proteins were not previously known to be glycosylated. Furthermore, the specific peptide sequences for 60% of the glycopeptides were assigned as glycosylated for the first time.
RESULTS
Design and implementation of the IsoTaG platform
Critical to the performance of IsoTaG is the design and synthesis of dibrominated biotin tag 1 that allows for click chemistry–based affinity enrichment, recovery of the glycopeptide, and isotopic recoding of the intact glycopeptide (Fig. 1, Supplementary Fig. 1 and Supplementary Methods). Several cleavable linkers have been developed for proteomics applications;29 we selected the silane scaffold30 because of our observation that glycosidic linkages tolerate its mild acid cleavage conditions (2% formic acid in H2O). The natural abundance of the stable isotopes, 79Br and 81Br (1:1), provided a ready source for isotope recoding. In previous work we found that the dibromide motif, which convolutes the mass envelope of a peptide with a triplet signature (1:2:1 (M, M + 2, M + 4)), could be detected computationally using a pattern-recognition algorithm that we developed in-house28.
Figure 1.
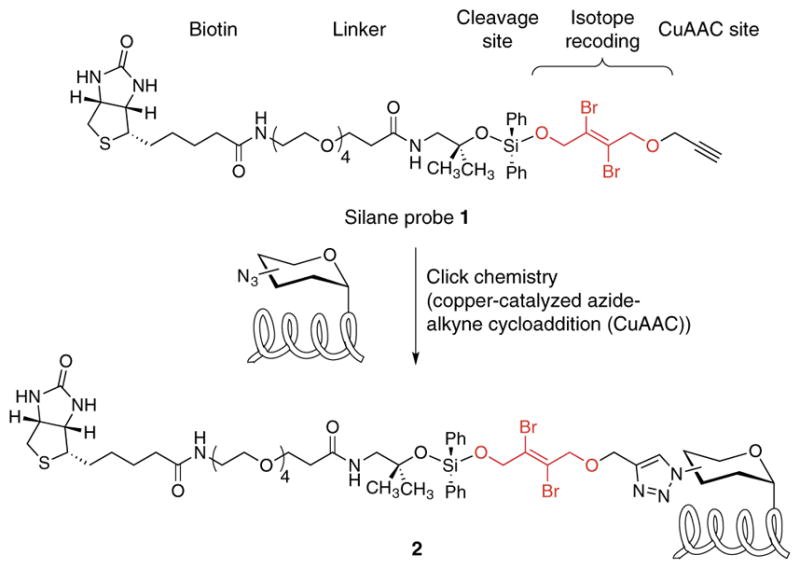
Structure of cleavable probe 1 for mass-independent glycoproteomics. Probe 1 tags glycoproteins via click chemistry, resulting in tagged glycoproteins 2.
We tested probe 1 in a glycoproteomics analysis of Jurkat cell lysates using the IsoTaG workflow (Fig. 2a). To demonstrate the flexibility of the platform for various glycoprotein subtypes, we labeled Jurkat cells separately with tetraacetylated N-azidoacetylgalactosamine (Ac4GalNAz), tetraacetylated N-azidoacetylmannosamine (Ac4ManNAz) or dimethylsulfoxide (DMSO) vehicle for 48 h. Ac4ManNAz is metabolized to the corresponding N-azidoacetyl sialic acid (SiaNAz) to yield labeled sialoglycoproteins31. Ac4GalNAz is processed by the cellular GalNAc salvage pathway to form UDP-GalNAz, which is inter-converted by the UDP-galactose 4-epimerase (GALE) with its C4 epimer, UDP-GlcNAz15. Thus, metabolic labeling with Ac4GalNAz produces GalNAz-labeled mucin-type O-glycoproteins, GlcNAz-labeled N-glycoproteins and O-GlcNAzylated proteins. We harvested labeled Jurkat cells by centrifugation and collected the media by centrifugal filtration for analysis of secreted glycoproteins. In addition, the cell pellet was homogenized and separated into soluble and insoluble fractions. Three milligrams of protein per fraction were used as a starting point for glycoproteomics enrichment.
Figure 2.

IsoTaG enrichment and mass spectrometry workflow. (a) Enrichment strategy using dibrominated silane probe 1, shown in cartoon form. Metabolically labeled glycoproteins are enriched using streptavidin-agarose affinity chromatography. On-bead digestion removes nonglycosylated peptides, and isotopically recoded glycopeptides are cleaved and eluted from the resin. (b) Isotopically recoded glycopeptides are analyzed by reversed-phase liquid chromatography coupled to a mass spectrometer. (c) IsoTaG uses the pattern-searching algorithm to direct tandem MS (i.e., MS2 and MS3) analysis to isotopically recoded species. Targeted searching of selected species results in high-confidence glycopeptide identification. The tagged glycan is denoted with “Br2.”
Fractionated Jurkat cell lysates were conjugated to probe 1 via copper-catalyzed azide-alkyne [3+2] cycloaddition (CuAAC; Fig. 2a). Excess probe was removed by methanol precipitation, and proteins were resuspended in 1% RapiGest surfactant in PBS. Glycoproteins were enriched by streptavidin-agarose affinity chromatography and digested on-bead with trypsin to release all nonconjugated peptides for glycoprotein identification. Treatment with 2% formic acid recovered the glycopeptides from the beads. The efficiency of glycoprotein capture and release was assessed by western blot (Supplementary Figs. 2–4). The released glycopeptides were analyzed by reversed-phase nanoflow liquid chromatography coupled to a Thermo LTQ–Orbitrap XL mass spectrometer.
In traditional proteomics approaches, tandem MS is performed on the most abundant species in the full-scan mass spectra to the exclusion of lower-abundance species (Fig. 2b). In contrast, IsoTaG enables mass-independent, targeted glycoproteomics (Fig. 2c). Glycopeptides displaying the isotopic signature are computationally detected by the pattern-searching algorithm28, which produces an inclusion list of m/z values and retention times (±2-min window) for ions bearing isotopically recoded envelopes. We then performed an inclusion list–directed tandem MS run, where we typically observed a <0.5-min shift in retention time. Femtomole quantities of tagged proteins at a signal-to-noise ratio of 2.5:1 were detectable with the pattern-recognition algorithm28. Owing to the versatility of the algorithm28, species up to 12 kDa that have incorporated multiple tags are also directed to the inclusion list and have an equal chance of selection for tandem MS.
IsoTaG improves selection of low-abundance glycopeptides
To assess the advantage of IsoTaG over traditional data-dependent acquisition, we performed a series of back-to-back runs with and without the inclusion list and evaluated the total number of isotopically recoded species selected for tandem MS. A fourfold improvement in tagged glycopeptides selected for tandem MS was observed across all fractions and glycan types (Supplementary Fig. 5). In total, over 1,500 isotopically recoded species were selected for tandem MS from Ac4GalNAz-labeled and Ac4ManNAz-labeled Jurkat cell lysates using the inclusion list, as compared to 385 without. This selection factor indicates that IsoTaG effectively targets low-abundance species and mitigates the need for extensive fractionation, mass spectrometer analysis time and computation time. To assess the rate of off-target labeling, we quantified the number of isotopically recoded species selected for tandem MS from the DMSO control samples. Using the same procedure, we found 52 isotopically recoded off-target species in the DMSO control (off-target rate = 3%).
We then repeated the enrichment procedure with PC-3 and MCF-7 cell lines. Because of our interest in identifying low-abundance glycopeptides, tandem MS was performed using collision-induced dissociation (CID) to achieve the highest sensitivity. Figure 2c illustrates the identification of an N-glycoprotein from Ac4GalNAz-labeled Jurkat lysates. Inclusion list–triggered tandem MS of an isotopically recoded precursor ion generated a tandem mass spectrum (MS2) for assignment of glycan structure from charged and neutral losses. Subsequent data-dependent CID of the three most intense ions in the MS2 spectrum generated MS3 spectra for glycopeptide amino acid sequence identification by database searching. In a typical MS duty cycle, we performed inclusion list–driven MS2 on the top three precursor ions, and the three most intense fragment ions in each MS2 spectrum were selected for MS3 analysis, for a total of nine events following MS1.
By virtue of their isotopically recoded mass envelopes, glycopeptides could be identified in a targeted, mass-independent manner (Fig. 3). MS2 and MS3 spectra of isotopically recoded precursor ions were searched against the Swiss-Prot human proteome using the Sequest HT algorithm within Proteome Discoverer software. Unassigned spectra were then searched in an iterative fashion using the UniProtKB human proteome and Byonic, a program specialized for variable-modification searches. Glycans were assigned manually from the MS2 spectra, and glycan structures were inferred from the observed neutral or charged losses (Supplementary Fig. 6). Ambiguous glycan types (for example, GlcNAz vs. GalNAz) were inferred from the subcellular localization of the underlying peptide or the most likely glycan structure (for example, O10 and O11; Fig. 3b). All assignments were validated for isotopic distribution, exact mass (mass error, Δmass < 5 p.p.m.), charged or neutral loss of the glycan, and peptide fragment ions. None of the assigned glycopeptides was detected in the DMSO control samples. To assess reproducibility, we performed biological replicates of the enrichment procedure with the Jurkat cell line and found that approximately half of the glycopeptides were identified in all three replicates, and 78% were found in two of three replicates (Fig. 4a). In aggregate, 32 N- and over 500 O-glycopeptides from 250 glycoproteins were identified, of which 220 peptides and 120 proteins were not previously known to be glycosylated.
Figure 3.

IsoTaG identified a diversity of intact glycopeptides. (a) N-glycan structures (N1–N16) identified. (b) O-glycan structures identified include O-GlcNAc (O1), Tn (O2), STn (O5), core 1 (O3), core 2 (O11), core 3 (O4), core 4 (O10) and sialylated variants (O6–O9). A purple peptide (bar) indicates a glycan and peptide sequence were identified for at least one glycopeptide. These species generated interpretable peptide fragments. An orange peptide indicates that only the glycan was assigned. These species gave unassignable peptide fragments. (c,d) Frequencies of observed peptides bearing N-glycans (c) and O-glycans (d).
Figure 4.

Distribution of identified glycopeptides. (a,b) Glycopeptides identified with three biological replicates (R1–R3) of Jurkat cell lysate enrichment (a) and from GalNAz- or ManNAz-labeled lysates (b). (c,d) Glycoproteins identified via IsoTaG are distributed throughout the cell (c) and are highly cell specific (d).
Mass spectrometry data analysis
The identified glycopeptides represent a range of proteins and glycan types. Sixteen N-glycan types were identified (N1–N16; Fig. 3a), in which immature and high-mannose structures were more predominant than we expected (Fig. 3c). In addition to the 32 fully characterized N-glycopeptides, we partially characterized 156 N-glycopeptide spectra (for example, the glycan was sequenced but not the underlying peptide). The lower number of fully identifiable N-glycopeptides mainly reflects difficult elucidation by MS (up to four SiaNAz glycans were observed in a single species, but the entire glycoform was unresolved) but may also be caused by hindered access of probe 1 or reduced ionization efficiency. Alternatively, metabolically labeled immature N-glycans may be more abundant than elaborated N-glycans in the samples evaluated, a hypothesis supported by previous observations using lectin chromatography24. To fully characterize the peptides carrying N-glycans, we performed the enrichment using conditioned medium from PC-3 cells metabolically labeled with Ac4GalNAz, Ac4ManNAz or DMSO. After trypsin digestion, we used PNGase F in 18O-water to release only peptides with N-glycans and found 54 formerly N-glycosylated peptides at 1% false discovery rate. These samples represent one-ninth of the total fractions evaluated herein, indicating that N-glycoproteins are labeled and captured by our enrichment procedure. Unfortunately, we could no longer characterize the structures that occupied the glycosite using this established method23.
Identified O-glycan types corresponded to O-GlcNAc (O1), Tn (O2), STn (O5), Cores 1–4 (O3, O11, O4 and O10, respectively) and sialylated glycans (O6, O7, O8 and O9) (Fig. 3b). The preponderance of O-GlcNAcylated peptides (Fig. 3d) reflects, at least in part, the efficiency of the GALE epimerase to biosynthesize UDP-GlcNAz from Ac4GalNAz15. Additionally, terminal glycans (for example, GlcNAz) may be easier to tag with probe 1. Nonetheless, O-GlcNAcylated proteins are highly abundant on the basis of other analyses32.
Fragmentation of the glycan occurs before fragmentation of the peptide backbone with CID, enabling the assignment of glycan structure—but not the underlying peptide—for several of the elaborated glycopeptides. Whereas electron-transfer dissociation (ETD) is an orthogonal fragmentation method for peptides bearing CID-labile PTMs33, we found that halogenated glycopeptides have low ETD fragmentation efficiency. The mucin-type O-glycans identified were predominantly various sialylation states of core 1 O-glycans from PC-3 and MCF-7 cell lines. Jurkat cells displayed highly heterogeneous O-glycopeptides owing to the knockdown of core 1 O-glycan elaboration via truncation of the COSMC chaperone34. Notably, core 1 O-glycans from Jurkat cells were still identified, as truncated COSMC maintains 2–5% activity of the full-length protein. The presence of core 1 glycoproteins in the conditioned medium from Jurkat cells was confirmed by lectin staining (Supplementary Fig. 7).
Interestingly, unique subpopulations of glycopeptides were enriched with either GalNAz or ManNAz labeling (Fig. 4b). The predicted subcellular localization of the identified glycoproteins reflected the prevalence of nuclear and cytoplasmic O-GlcNAzylated peptides identified (Fig. 4c). Nonetheless, the identified glycoproteins were highly cell specific. Of the characterized glycoproteins, 60% were found exclusively in one of the three cell types, and 30% in two of the three cell types (Fig. 4d).
We identified intact glycopeptides with multiple glycoforms (Supplementary Table 1). Elaborated glycopeptides possessed a high degree of glycoform variation, including peptides derived from the same protein (O94907) or from different proteins (for example, O94907 and Q14766). Up to five glycoforms were observed for a single peptide (P14314), and several peptides possessed four detected glycoforms (P02786, Q02818, Q12841, Q14242 and Q92854).
Identification of peptide sequence polymorphisms
Approximately 60% of isotopically recoded species were assigned by database searching methods described above. The remaining 40% of tandem mass spectra of isotopically recoded species were unassigned from database searching alone. In some cases, spectral nonassignment was related to low signal-to-noise ratios of fragment ions or the presence of unidentified chemical modifications (for example, unexpected fragments and CuAAC-related off-target reactivity). In other cases, however, we suspected that failure to identify glyco-peptides from database searching was due to the absence of a matching amino acid sequence in the protein database. To investigate this possibility, we collected an additional data set on Ac4ManNAz-labeled PC-3 cell lysates, with full-scan mass spectra and MS2 spectra acquired in the Orbitrap analyzer to obtain high-resolution measurement of both precursor and fragment ions. Targeted analysis of this data set revealed 21 glycopeptides carrying pSPs that were not present in the protein database (UniProtKB) and one pSP previously observed by genetic sequencing35. For example, a total of 15 isoforms were identified, representing four glycan types and six pSPs derived from prostate-associated microseminoprotein36 (Fig. 5 and Supplementary Fig. 8). Polymorphic peptides additionally carry glycoform variants (for example, P14314 and Q1L6U9; Supplementary Table 1); this is potentially related to variable levels of abundance or indicative of differential biosynthetic processing of the underlying peptide.
Figure 5.

Targeted tandem MS analysis of isotopically recoded glycopeptides identifies both glycan and peptide isoforms. The novel O-glycopeptide from prostate-associated microseminoprotein was found as four glycoforms by MS2 and six pSPs by MS3. The tagged glycan is denoted with “Br2.” Amino acid variants are in boldface red font. Fragmentation spectra for three peptides are shown (b ions in red, y ions in blue).
Identification of protein polymorphisms in cancer is an area of intense study on the genomic37 and proteomic38,39 levels. Interestingly, all pSPs identified from PC-3 cells, a metastatic prostate cancer cell line, were mutations of alanine to serine or of proline to threonine. The selectivity of mutations observed in our data may be related to the glycan-dependent enrichment strategy. Alternatively, as aberrant glycosylation is a hallmark of cancer, particularly in metastasis40, an increase in O-glycosylation sites may play a significant role and is a subject for future investigation.
DISCUSSION
Unlike previous methods, which require cleavage or truncation of glycans for glycosite mapping, IsoTaG identifies fully elaborated glycopeptides, revealing the inherent structural details of the glycoproteome. Use of the IsoStamp algorithm increased selection of glycopeptide species for tandem MS by fourfold and facilitated mass-independent targeted database searching for high-confidence assignment. As described, the enrichment and recovery of metabolically labeled glycopeptides with probe 1, MS analysis and initial data analysis were all performed in 3 d. In our preliminary study we found that IsoTaG was highly reproducible using biological replicates from Jurkat lysates. With Jurkat, PC-3 and MCF-7 cell lines, 50% of identified glycoproteins and 60% of identified glycopeptides were novel, suggesting that many more glycoproteins, and particularly O-glycoproteins, remain to be discovered. Indeed, the assignment of a large number of novel O-glycopeptides further highlights the ability of IsoTaG to survey new classes of glycoproteins. A 3% off-target CuAAC rate was found with the Jurkat cell line, as quantified by the number of isotopically recoded species selected for tandem MS in labeled versus control samples. Protein glycosylation was also highly cell specific, as 60% of the identified glycoproteins were found exclusively in one of the three cell types (30% in two of three cell types). As less than 2% of proteins are expressed in a cell-specific manner41, analysis of glycoproteins may be an important source of tissue-specific diversity and disease biomarkers.
Notably, any covalent labeling strategy may be integrated into the IsoTaG platform to achieve enrichment and targeted proteomics of low-abundance species. We have illustrated the generality of the platform by using two metabolically labeled glycans: Ac4GalNAz and Ac4ManNAz. Use of highly glycan-specific metabolic17,18 or enzymatic42 labels may enable focused analysis of particular glycan types. Incorporation of the isotopic pattern via metabolic heavy-atom labeling is also a viable alternative43. An Orbitrap mass spectrometer was used in the present study; however, other types of mass spectrometers that can achieve high resolution (for example, quadrupole time-of-flight analyzers) can also be employed28. Recent advances in nested fragmentation techniques (such as higher-energy collisional dissociation with accurate mass-product–dependent ETD) show promise toward a targeted glycoproteomics approach and may be incorporated into an IsoTaG strategy44,45.
IsoTaG represents the first proteome-scale platform in which the glycan structure and peptide can be identified for both intact N- and O-glycopeptides. Recognition of tagged glycopeptides by full-scan MS improved rates of selection and characterization, such that 60% of isotopically recoded species were characterized (glycan structure and peptide), including the identification of novel pSPs. These results point to a wealth of information in the proteome and glycome that are not covered in current databases. Given that on average 20% of spectra in a data set are assigned by database searching46, we suggest that a targeted isotopic recoding approach may be broadly useful for the future discovery of novel protein isoforms, including nontemplated PTMs and pSPs.
ONLINE METHODS
Materials
Commercial solvents and reagents were used as received with the following exceptions. Dichloromethane and N,N-dimethylformamide were purified according to the method of Pangborn and coworkers47. Triethylamine was distilled from calcium hydride under an atmosphere of nitrogen immediately before use. RapiGest was prepared according to the method of Lee and Compton48. 3-[4-({Bis[(1-tert-butyl-1H-1,2,3-triazol-4-yl)methyl]amino}methyl)-1H-1,2,3-triazol-1-yl]propanol (BTTP) was prepared according to the method of Wu and coworkers49. Tetraacetylated N-azidoacetyl galactosamine was prepared according to the method of Bertozzi and coworkers50. Tetraacetylated N-azidoacetyl mannosamine was prepared according to the method of Bertozzi and coworkers51. EDTA-free protease inhibitor cocktail was obtained from Roche Diagnostics (Version 11). Streptavidin-agarose beads were obtained from Thermo Scientific and washed with PBS before use. High-sensitivity streptavidin-HRP was obtained from Pierce (catalog number 21130). PNGase F was purchased from NEB (catalog number P0704S).
Cell culture
All cell lines were obtained from the American Type Culture Collection (ATCC) and maintained at 37 °C and 5% CO2 in a water-saturated incubator. Jurkat and PC-3 cells were maintained in RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin. MCF-7 cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Cell lines were metabolically labeled between passages 4–7 (MCF-7), 6–10 (Jurkat), or 17–22 (PC-3) and visually inspected daily for the presence of mycoplasma.
Cell culture flasks were seeded with 100 μM of Ac4GalNAz, Ac4ManNAz, or vehicle control containing DMSO (3.0 μL). A suspension of cells (2 × 105 cells/mL) was added to the flask, and the flask was incubated for 48 h at 37 °C. Flasks containing adherent cells were aspirated, and the cells were washed with PBS. The contents of flasks containing suspension cells were transferred to centrifuge tubes, and the cells were pelleted (300g, 3 min). The medium was aspirated, and cell pellets were washed with PBS. Cells were resuspended in complete medium containing 100 μM glycan metabolite without FBS additive, and the cells were incubated an additional 48 h at 37 °C.
Medium (100 mL) was harvested and cleared by centrifugation (300g, 3 min). Clarified medium was spin concentrated (Amicon, 15-mL 10-kDa spin filter) to 1 mL. The concentrated residue was washed with PBS (three washes of 15 mL each: 3 × 15 mL) and transferred to an Eppendorf microcentrifuge tube as the “conditioned medium fraction.” Adherent cells were washed with PBS (1 × 10 mL) and trypsinized for 5 min at 37 °C. Cells were harvested, centrifuged (300g, 3 min), and washed with PBS. Suspension cells were pelleted by centrifugation (300g, 3 min) and washed with PBS. Cell pellets were resuspended in lysis buffer (2 mL: 10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5% Triton X-100, 1× protease inhibitors, 1 μM thiamet G), swelled for 5 min on ice, and broken by Dounce homogenization using a tight glass hand pestle (Wheaton, 30 strokes). Insoluble material was pelleted by centrifugation (3,700g, 10 min, 4 °C). The supernatant was collected as the “soluble fraction” and the pellet kept as the “insoluble fraction.” The insoluble fraction was resuspended in 1% RapiGest/PBS and briefly probe sonicated. The conditioned medium and soluble fractions were adjusted to 1% RapiGest/PBS. Protein concentration from the three fractions was measured by bicinchonic acid assay (Pierce) and normalized to 4.5 mg/mL.
CuAAC-assisted enrichment
GalNAz-labeled, ManNAz-labeled, or DMSO vehicle–treated cell fractions were divided into aliquots containing 3.0 mg protein. Click-chemistry reagents (40.0 μL, 200 μM 1, 300 μM CuSO4, 600 μM BTTP, 1.50 mM sodium ascorbate, mixed immediately before addition to lysates) were added, and the reaction was incubated for 3.5 h at 24 °C. Proteins were precipitated in methanol (1 mL) for 1 h at −80 °C. Precipitated proteins were pelleted by centrifugation, and the supernatant was discarded. Protein pellets were resuspended in 400 μL 1% RapiGest/PBS and solubilized by probe sonication. Streptavidin-agarose resin was added, and the resulting mixture was incubated for 12 h at 24 °C with rotation. The beads were pelleted by centrifugation, and the supernatant containing uncaptured proteins was separated. The beads were washed with 1% RapiGest/PBS (1 mL), 6 M urea (2 × 1 mL), and PBS (5 × 1 mL).
The washed beads were reduced (5 mM DTT) and alkylated (10 mM iodoacetamide). On-bead trypsin (1.5 μg) digestion proceeded for 12 h at 37 °C. Beads were pelleted by centrifugation, and the supernatant digest was collected. The beads were washed with PBS (1 × 200 μL) and H2O (2 × 200 μL). Washes were combined with the supernatant digest to form the trypsin digest. The silane tag 1 was cleaved with two treatments of 2% formic acid/H2O (200 μL) for 30 min at 24 °C with rotation, and the eluent was collected. The beads were washed with 50% acetonitrile-water + 1% formic acid (2 × 200 μL), and the washes were combined with the eluent to form the cleavage fraction. The trypsin digest and cleavage fraction were concentrated using a vacuum centrifuge to 50–100 μL. Samples were desalted by ZipTip P10 and stored at −20 °C until analysis.
Mass spectrometry
Recovered glycopeptides were analyzed using a Thermo Dionex UltiMate3000 RSLCnano liquid chromatograph that was connected in-line with an LTQ Orbitrap XL mass spectrometer equipped with a nanoelectrospray ionization (nanoESI) source. Full-scan mass spectra were acquired in the positive-ion mode over the range m/z = 400–1,800 using the Orbitrap mass analyzer, in profile format, with a mass resolution setting of 60,000. Inclusion lists were generated using the IsoStamp algorithm (http://www.cchem.berkeley.edu/crbgrp/IsoStamp/). In the data-dependent mode, the three most intense ions exceeding an intensity threshold of 50,000 counts were selected from each full-scan mass spectrum for tandem mass spectrometry (MS/MS, i.e., MS2) analysis using collision-induced dissociation (CID). MS2 spectra were acquired using the linear ion trap or the Orbitrap analyzer (in the latter case, with a resolution setting of 7,500 at m/z = 400, FWHM), in centroid format, with the following parameters: isolation width, 4 m/z units; normalized collision energy, 28%; default charge state, 3+; activation Q, 0.25; and activation time, 30 ms. The three most intense fragment ions in each MS2 spectrum exceeding an intensity threshold of 1,000 counts were selected for MS3 analysis using CID. MS3 spectra were acquired using the linear ion trap, in centroid format, with the same parameters as those used for MS2. Global parent mass lists (i.e., inclusion lists) were enabled to specify the m/z values and retention times of glycopeptide precursor ions detected in full-scan mass spectra by the IsoStamp isotope pattern-searching algorithm. Data acquisition was controlled using Xcalibur software (version 2.0.7, Thermo). The raw data are available upon request.
Data analysis
The raw data were processed using Proteome Discoverer 1.4 software and searched against the human-specific SwissProt-reviewed database downloaded 18 July 2014. Tandem MS data were screened for glycopeptide signifiers including isotopically recoded precursor ion in full-scan mass (i.e., MS1) spectra, and neutral or charged glycan losses in MS2 spectra. Selected MS2/MS3 spectra were documented and saved separately. Saved spectra were manually annotated for glycoform and peptide mass. Saved spectra were then searched iteratively using the Byonic search algorithm v2.0 as a node in Proteome Discoverer 1.4. Computational assignments of all spectra were validated by manual inspection for glycan and peptide fragments. High probability assignments were inspected for validity, and unassigned spectra were archived for further analysis. Unassigned spectra from the initial searches were sorted on the basis of glycan type derived from fragment ions observed in the MS2 spectra (for example, HexNAzBr2OH, elaborated O-glycan, or elaborated N-glycan) and searched with variable modification using a focused glycan database. Finally, spectra that remained low-confidence assignments were manually inspected for similarities to assigned spectra (i.e., characteristic peptide fragment ions) or searched against the UniProtKB database (downloaded 30 September 2014) with variable modification on the specific glycan structure. Mass spectrometry spectra are provided as Supplementary Data 1–4 and annotated as losses from the MS(n − 1) precursor mass as follows: glycan(number of glycan units) in reverse order of the observed losses. Glycans at separate sites are separated by a comma. The glycan annotations used are HexNAzBr2OH = C15H20Br2N4O7 (+527.9678, abbreviated HexNAz*), HexNAz = C8H12N4O5 (+244.0808), HexNAcNH2 = C8H14N2O5 (+218.0903), HexNAc = C8H13NO5 (+203.0794), NeuAzBr2OH = C18H24Br2N4O10 (+615.9839, abbreviated NeuAz*), NeuAz = C11H16N4O8 (+332.0968), NeuAcNH2 = C11H18N2O8 (+306.1063), NeuAc = C11H17NO8 (+291.0954), Hex = C6H10O5 (+162.0528), +CO = CO (+27.9949, occurs on the O-terminus of the tag).
Western blotting procedures
PNGase F release of formerly N-glycosylated peptides
GalNAz-labeled, ManNAz-labeled, or DMSO vehicle–treated medium was enriched and trypsin digested as described above. The beads were washed with PBS (1 × 200 μL) and H2O (1 × 200 μL). The beads were washed with 40 mM ammonium carbonate in 18O-water (1 × 40 μL). N-glycosylated peptides were cleaved by treatment with PNGase F (5 U) in ammonium carbonate in 18O-water (40 mM, 100 μL) in a 10-kDa spin filter (Corning) for 6 h at 37 °C. The eluent from the PNGase F treatment was collected by centrifugation (7,000 r.c.m., 5 min). The beads were washed with 50% acetonitrile-water (2 × 400 μL), and the washes were combined with the eluent to form the PNGase F fraction. The PNGase F fraction was concentrated using a vacuum centrifuge (i.e., a SpeedVac, 40 °C) to 50–100 μL. Samples were desalted with a ZipTip P10 and stored at −20 °C until analysis.
Anti-biotin immunoblotting
Aliquots collected during the enrichment procedure (10.0 μL) were reduced and separated by standard SDS-PAGE (Bio-Rad, Criterion system), electroblotted onto nitrocellulose, blocked in 5% bovine serum albumin (Sigma) in PBS with 0.1% Tween-20, and analyzed by standard enhanced chemiluminescence immunoblotting methods (Pierce). The staining agent was streptavidin-HRP (Pierce, 1:100,000).
Lectin staining
GalNAz-labeled or DMSO-vehicle Jurkat conditioned medium (100 μg) in buffer (25.0 μL, 50 mM NaOAc, pH 5.5, 4 mM CaCl2) was divided into two aliquots. One aliquot from each condition was treated with neuraminidase (4.0 μL, Vibrio cholerae, Roche). Aliquots were mixed and incubated at 37 °C for 12 h. Aliquots (10.0 μL) were reduced and separated by standard SDS-PAGE (Bio-Rad, Criterion system), electro-blotted onto nitrocellulose, blocked in PBS with 0.5% Tween-20, and analyzed by standard fluorescent imaging (Typhoon 9410, GE Healthcare). The staining agent was peanut agglutinin–FITC (Vector Laboratories, 1:100).
Supplementary Material
Acknowledgments
We thank P. Robinson and N. Rumachik for critical reading. Financial support from the US National Institutes of Health (CA200423, C.R.B.), Jane Coffin Childs Memorial Fund (C.M.W.), US National Science Foundation (Graduate Research Fellowship, D.R.S.) and Howard Hughes Medical Institute (C.R.B.) are gratefully acknowledged.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
C.R.B. conceived of and directed the project. C.M.W. designed and synthesized the probe. C.M.W. designed and performed cell culture and enrichment studies. A.T.I. and C.M.W. collected MS data. D.R.S. and C.M.W. optimized the IsoStamp algorithm. C.M.W. analyzed the data and composed the manuscript. K.K.P. performed preliminary studies. All authors revised the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Gouyer V, et al. Inhibition of the glycosylation and alteration in the intracellular trafficking of mucins and other glycoproteins by GalNAcα-O-bn in mucosal cell lines: an effect mediated through the intracellular synthesis of complex GalNAcα-O-bn oligosaccharides. Front Biosci. 2001;6:D1235–D1244. doi: 10.2741/gouyer. [DOI] [PubMed] [Google Scholar]
- 2.Zachara NE, Hart GW. Cell signaling, the essential role of O-GlcNAc! Biochim Biophys Acta. 2006;1761:599–617. doi: 10.1016/j.bbalip.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Hudak JE, Canham SM, Bertozzi CR. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat Chem Biol. 2014;10:69–75. doi: 10.1038/nchembio.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuzwa SA, et al. Increasing O-Glcnac slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol. 2012;8:393–399. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- 5.Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer. 2005;5:526–542. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- 6.Büll C, Stoel MA, den Brok MH, Adema GJ. Sialic acids sweeten a tumor’s life. Cancer Res. 2014;74:3199–3204. doi: 10.1158/0008-5472.CAN-14-0728. [DOI] [PubMed] [Google Scholar]
- 7.Radhakrishnan P, et al. Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc Natl Acad Sci USA. 2014;111:E4066–E4075. doi: 10.1073/pnas.1406619111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mariño K, Bones J, Kattla JJ, Rudd PM. A systematic approach to protein glycosylation analysis: a path through the maze. Nat Chem Biol. 2010;6:713–723. doi: 10.1038/nchembio.437. [DOI] [PubMed] [Google Scholar]
- 9.Pan S, Chen R, Aebersold R, Brentnall TA. Mass spectrometry based glycoproteomics–from a proteomics perspective. Mol Cell Proteomics. 2011;10:R110 003251. doi: 10.1074/mcp.R110.003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou H, Watts JD, Aebersold R. A systematic approach to the analysis of protein phosphorylation. Nat Biotechnol. 2001;19:375–378. doi: 10.1038/86777. [DOI] [PubMed] [Google Scholar]
- 11.Olsen JV, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 12.Choudhary C, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 13.Pouria S, et al. Glycoform composition profiling of O-glycopeptides derived from human serum IgA1 by matrix-assisted laser desorption ionization-time of flight-mass spectrometry. Anal Biochem. 2004;330:257–263. doi: 10.1016/j.ab.2004.03.053. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Fonslow BR, Shan B, Baek MC, Yates JR., III Protein analysis by shotgun/bottom-up proteomics. Chem Rev. 2013;113:2343–2394. doi: 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyce M, et al. Metabolic cross-talk allows labeling of O-linked β-N-acetylglucosamine-modified proteins via the N-acetylgalactosamine salvage pathway. Proc Natl Acad Sci USA. 2011;108:3141–3146. doi: 10.1073/pnas.1010045108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hubbard SC, Boyce M, McVaugh CT, Peehl DM, Bertozzi CR. Cell surface glycoproteomic analysis of prostate cancer-derived PC-3 cells. Bioorg Med Chem Lett. 2011;21:4945–4950. doi: 10.1016/j.bmcl.2011.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chuh KN, Zaro BW, Piller F, Piller V, Pratt MR. Changes in metabolic chemical reporter structure yield a selective probe of O-GlcNAc modification. J Am Chem Soc. 2014;136:12283–12295. doi: 10.1021/ja504063c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaro BW, Yang YY, Hang HC, Pratt MR. Chemical reporters for fluorescent detection and identification of O-GlcNAc-modified proteins reveal glycosylation of the ubiquitin ligase NEDD4-1. Proc Natl Acad Sci USA. 2011;108:8146–8151. doi: 10.1073/pnas.1102458108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 20.Nilsson J, et al. Enrichment of glycopeptides for glycan structure and attachment site identification. Nat Methods. 2009;6:809–811. doi: 10.1038/nmeth.1392. [DOI] [PubMed] [Google Scholar]
- 21.Khidekel N, et al. Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nat Chem Biol. 2007;3:339–348. doi: 10.1038/nchembio881. [DOI] [PubMed] [Google Scholar]
- 22.Vosseller K, et al. O-linked N-acetylglucosamine proteomics of postsynaptic density preparations using lectin weak affinity chromatography and mass spectrometry. Mol Cell Proteomics. 2006;5:923–934. doi: 10.1074/mcp.T500040-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Zielinska DF, Gnad F, Wiśniewski JR, Mann M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 24.Trinidad JC, Schoepfer R, Burlingame AL, Medzihradszky KF. N- and O-Glycosylation in the murine synaptosome. Mol Cell Proteomics. 2013;12:3474–3488. doi: 10.1074/mcp.M113.030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hägglund P, Bunkenborg J, Elortza F, Jensen ON, Roepstorff P. A new strategy for identification of N-glycosylated proteins and unambiguous assignment of their glycosylation sites using HILIC enrichment and partial deglycosylation. J Proteome Res. 2004;3:556–566. doi: 10.1021/pr034112b. [DOI] [PubMed] [Google Scholar]
- 26.Steentoft C, et al. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat Methods. 2011;8:977–982. doi: 10.1038/nmeth.1731. [DOI] [PubMed] [Google Scholar]
- 27.Steentoft C, et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013;32:1478–1488. doi: 10.1038/emboj.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palaniappan KK, et al. Isotopic signature transfer and mass pattern prediction (IsoStamp): an enabling technique for chemically-directed proteomics. ACS Chem Biol. 2011;6:829–836. doi: 10.1021/cb100338x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bielski R, Witczak Z. Strategies for coupling molecular units if subsequent decoupling is required. Chem Rev. 2013;113:2205–2243. doi: 10.1021/cr200338q. [DOI] [PubMed] [Google Scholar]
- 30.Szychowski J, et al. Cleavable biotin probes for labeling of biomolecules via azide-alkyne cycloaddition. J Am Chem Soc. 2010;132:18351–18360. doi: 10.1021/ja1083909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prescher JA, Dube DH, Bertozzi CR. Chemical remodelling of cell surfaces in living animals. Nature. 2004;430:873–877. doi: 10.1038/nature02791. [DOI] [PubMed] [Google Scholar]
- 32.Hart GW, Akimoto Y. In: Essentials of Glycobiology. 2. Varki A, et al., editors. Ch 18. Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 33.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ju T, et al. Human tumor antigens Tn and sialyl Tn arise from mutations in Cosmc. Cancer Res. 2008;68:1636–1646. doi: 10.1158/0008-5472.CAN-07-2345. [DOI] [PubMed] [Google Scholar]
- 35.Gerhard DS, et al. The status, quality, and expansion of the NIH full-length cDNA project: the Mammalian Gene Collection (MGC) Genome Res. 2004;14:2121–2127. doi: 10.1101/gr.2596504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valtonen-André C, et al. A highly conserved protein secreted by the prostate cancer cell line PC-3 is expressed in benign and malignant prostate tissue. Biol Chem. 2007;388:289–295. doi: 10.1515/BC.2007.032. [DOI] [PubMed] [Google Scholar]
- 37.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tran JC, et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480:254–258. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang B, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paszek MJ, et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature. 2014;511:319–325. doi: 10.1038/nature13535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pontén F, et al. A global view of protein expression in human cells, tissues, and organs. Mol Syst Biol. 2009;5:337. doi: 10.1038/msb.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clark PM, et al. Direct in-gel fluorescence detection and cellular imaging of O-GlcNAc-modified proteins. J Am Chem Soc. 2008;130:11576–11577. doi: 10.1021/ja8030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Breidenbach MA, Palaniappan KK, Pitcher AA, Bertozzi CR. Mapping yeast N-glycosites with isotopically recorded glycans. Mol Cell Proteomics. 2012;11:M111.015339. doi: 10.1074/mcp.M111.015339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yin X, et al. Glycoproteomic analysis of the secretome of human endothelial cells. Mol Cell Proteomics. 2013;12:956–978. doi: 10.1074/mcp.M112.024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu SW, Pu TH, Viner R, Khoo KH. Novel LC-MS2 product dependent parallel data acquisition function and data analysis workflow for sequencing and identification of intact glycopeptides. Anal Chem. 2014;86:5478–5486. doi: 10.1021/ac500945m. [DOI] [PubMed] [Google Scholar]
- 46.Elias JE, Haas W, Faherty BK, Gygi SP. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods. 2005;2:667–675. doi: 10.1038/nmeth785. [DOI] [PubMed] [Google Scholar]
- 47.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Safe and convenient procedure for solvent purification. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 48.Lee PJJ, Compton BJ. Destructible surfactants and uses thereof. 7,229,539. US patent. 2007
- 49.Wang W, et al. Sulfated ligands for the copper(I)-catalyzed azide-alkyne cycloaddition. Chem Asian J. 2011;6:2796–2802. doi: 10.1002/asia.201100385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hang HC, Yu C, Kato DL, Bertozzi CR. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc Natl Acad Sci USA. 2003;100:14846–14851. doi: 10.1073/pnas.2335201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prescher JA, Dube DH, Bertozzi CR. Chemical remodelling of cell surfaces in living animals. Nature. 2004;430:873–877. doi: 10.1038/nature02791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.