

Abstract
von Willebrand factor (VWF) is a large multimeric glycoprotein that mediates the attachment of platelets to damaged endothelium and also serves as the carrier protein for coagulation factor VIII (FVIII), protecting it from proteolytic degradation. Quantitative or qualitative defects in VWF result in von Willebrand disease (VWD), a common inherited bleeding disorder. VWF is synthesized with a very large propeptide (VWFpp) that is critical for intracellular processing of VWF. VWFpp actively participates in the process of VWF multimerization and is essential for trafficking of VWF to the regulated storage pathway. Mutations identified within VWFpp in VWD patients are associated with altered VWF structure and function. The assay of plasma VWFpp has clinical utility in assessing acute and chronic vascular perturbation associated with diseases such as thrombotic thrombocytopenic purpura, sepsis, and diabetes among others. VWFpp assay also has clear utility in the diagnosis of VWD subtypes, particularly in discriminating true type 3 subjects from type 1C (reduced plasma survival of VWF), which is clinically important and has implications for therapeutic treatment.
Historical perspective: identification of the von Willebrand factor propeptide (VWFpp)
Historically, VWF has also been known as “factor VIII-related antigen” (or VIIIag).1 VWFpp was initially identified by Montgomery and Zimmerman in 1978 as a second von Willebrand disease (VWD) antigen (vW AgII) that was deficient in platelets and plasma of patients with severe VWD.2 VWFpp (vW AgII) was released from platelets during blood clotting, demonstrating its presence in platelets as well as in plasma. VWF and VWFpp were shown to be immunologically distinct, leading to the (premature) conclusion that the 2 proteins did not share a precursor-product relationship. Subsequent studies showed VWFpp to be released from endothelial cells and platelets, together with VWF.3,4 Cloning of the full-length complementary DNA for human VWF revealed the presence of a “prosequence” that was postulated to be identical to VWFpp.5,6 Sequencing of VWFpp and a circulating 100 kDa plasma glycoprotein were demonstrated to be identical to amino acid sequences predicted from the complementary DNA clone encoding the 5′ end of VWF.7 This study confirms that VWFpp is a very large propeptide that is cleaved from pro-VWF during intracellular processing and subsequently released into plasma. The delineation of VWF full-length sequence and identification of the VWFpp led to intensive biological and clinical studies.
Biology of VWFpp
Domain structure and functional binding sites of VWF
VWF is synthesized exclusively by endothelial cells and megakaryocytes,8,9 as pre-pro–VWF with a 22- amino acid (aa) signal peptide, 741 aa propeptide, and 2050 aa mature VWF protein (Figure 1A).10 VWF is comprised of several homologous domains (A-D). The propeptide contains 2 D domains (D1 and D2), whereas the mature VWF protein begins at the D′ domain and continues through the CT/cysteine knot domain (Figure 1B). The domain structure of VWF was recently re-annotated (Figure 1C) by Zhou et al to assign VWF sequence to specific modules, and these modules were related to structure using electron microscopy.11 Several VWF domains contain functional binding sites or alternatively, cleavage sites (Figure 1B).10 A furin cleavage site is located between the end of VWFpp and the D′ domain. The D′/D3 domains contain a binding site for FVIII. The A1 domain is important for binding of VWF to GPIbα on platelets. VWF binds to types IV and VI collagen via the A1 domain, and to types I and III collagen via the A1 and A3 domains. The VWF-cleaving protease, a disintegrin and metalloprotease with thrombospondin type 1 repeats, cleaves VWF between tyrosine 1605 and methionine 1606 within the A2 domain. An Arg-Gly-Asp sequence for binding to α2β3 on platelets is located within the C4 domain.
Figure 1.
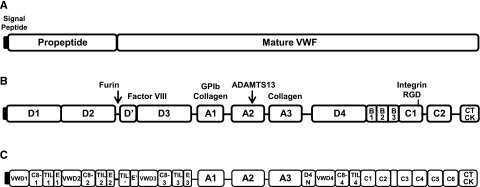
VWF structure and binding/cleavage sites. (A) VWF contains a signal peptide, propeptide, and mature VWF portion. (B) VWF is comprised of homologous domains (A-D), where specific binding or cleavage sites have been mapped. (C) Refined domain structure of VWF.
Life cycle of VWF and VWFpp
VWF is subjected to extensive intracellular processing.12 VWF is translocated to the endoplasmic reticulum (ER) where signal peptide is removed, protein is folded, and disulfide bonds are formed involving the majority of VWF’s 234 cysteine residues (Figure 2). VWF is posttranslationally glycosylated at its 17 N-linked glycosylation sites (4 in VWFpp and 13 in mature VWF).13 Before leaving the ER, VWF forms a carboxyl-terminal (C-terminal) dimer. In the Golgi, 10 O-linked glycans are added, carbohydrates are further processed, and sulfation occurs. C-terminal dimers form amino-terminal (N-terminal) multimers with variable molecular weight, ranging from 500 to 20 000 kDa. VWFpp is cleaved from mature VWF by furin, although the 2 proteins remain noncovalently associated.14,15 VWF and VWFpp are packaged into regulatory secretory granules, Weibel-Palade bodies (WPBs) in endothelial cells, and α-granules in megakaryocytes/platelets.16,17
Figure 2.

VWF intracellular processing. VWF is transcribed as pre-pro–VWF (nucleus, blue) and is translocated to the ER (green), where it undergoes signal peptide removal, glycosylation, and dimer formation. In the Golgi (yellow) O-linked glycans are added, carbohydrates are processed, VWF is sulfated and multimerized, and propeptide is cleaved from mature VWF but remains noncovalently associated. VWF is trafficked to the regulated secretory pathway (peach) and stored in WPBs or α-granules before release into plasma (red), where VWFpp and VWF dissociate and circulate independently of one another.
Although early reports on VWF secretion refer to both constitutive and regulated VWF secretion, recent data suggests that regulated secretion from endothelial cell WPBs may be the primary pathway for release of VWF and VWFpp into plasma.18 In plasma, VWF and VWFpp dissociate and circulate independently of one another. VWF binds FVIII in plasma, protecting it from degradation. VWF can bind to exposed subendothelial collagen, bind to platelets at the sites of injury, promote platelet-platelet interaction, self-associate, and be degraded by a disintegrin and metalloprotease with thrombospondin type 1 repeats.1,19,20 The mechanisms regulating the clearance of VWF and VWFpp are not clearly defined and are active areas of investigation.21,22
Role of VWFpp in multimerization of VWF
VWFpp is indispensible for proper intracellular processing of VWF. VWFpp is a required participant in the VWF multimerization process. VWF multimer assembly begins in the ER, where pro-VWF monomers form C-terminal dimers (Figure 2). In the Golgi, pro-VWF dimers are assembled into large multimers through N-terminal interchain disulfide bonds (Figure 2). Deletion of VWFpp does not prevent the formation of C-terminal dimers,23 but does prevent N-terminal multimerization.23,24 Multimerization of VWFpp-deleted VWF can be re-established by co-expression with VWFpp (in trans using 2 separate plasmids).23 Both D domains of VWFpp are required for multimerization and deletion of either D1 or D2, and results in complete loss of VWF multimerization with formation of only dimeric VWF.25 VWF multimer assembly has been demonstrated to proceed in a cell-free system optimally at pH 5.8, to a lesser extent at pH 6.2, and not at pH 7.4, conditions typical of the WPB, Golgi, and ER, respectively.26,27 The acidic pH in the Golgi, together with the lack of chaperones and oxidoreductases, make for an unlikely environment for the disulfide bond formation required for multimerization. VWF appears to overcome these limitations by using its own propeptide in a manner similar to that employed by disulfide isomerases. Both the D1 and D2 domains contain vicinal cysteine sequences in a CGLC motif similar to the active site of disulfide isomerases. Insertion of a glycine into either of these vicinal cysteine sequences within VWF inhibits VWF multimerization, but does not inhibit secretion of dimeric VWF.28 Purvis et al demonstrated the formation of a transient intracellular disulfide bond between VWFpp and the VWF D3 domain.29 They identified cysteines 1099 and 1142 in the D3 domain of VWF to be oxidized when multimerization proceeds and reduced when multimerization does not occur.30 When either cysteine is mutated to alanine, multimerization does not proceed. More recent evidence suggests that protonation of one or more histidine residues in VWFpp mediates the pH dependence of multimerization.31 By using histidine to alanine mutagenesis, Dang et al demonstrated that replacement of histidine 395 or 460 in VWFpp caused a profound defect in inter-subunit disulfide formation, suggesting that these 2 residues in VWFpp contribute to the pH-dependent regulation of VWF multimer assembly.31 VWFpp clearly has a critical role in the multimerization of VWF.
VWFpp is essential for regulated storage of VWF
VWF is stored for regulated release together with VWFpp in endothelial cell WPBs and platelet α-granules.16 In the rod-like WPB body, VWF multimers form a tubular conformation that correlates with WPB elongation.32 VWF expression is necessary for the biogenesis of the WPB, and VWF-deficient animals do not have WPB within their endothelial cells.33,34 In VWF-deficient aortic endothelial cells from type 3 VWD dogs, WPB formation can be re-established by expression of wild-type (WT) human or canine VWF.34 In addition, heterologous VWF expression can induce the formation of striated, rod-shaped WPB-like granules in other, non-endothelial cell lines such as AtT-20, CV-1, RIN5f, and HEK293.15,35-37 However, in other cells such as COS-1 and hepatocytes, WPB are not formed after VWF expression, suggesting that these cell types may lack the necessary chaperone or adaptor proteins. Collectively, these data demonstrate that VWF induces the formation of its own storage granule.
Wagner et al first demonstrated the requirement for VWFpp in the formation of the VWF storage granule.35 Expression of VWFpp-deleted VWF abolished the formation of WPB-like storage granules, a finding that has been confirmed by others.15,36 Co-expression of VWFpp with VWFpp-deleted VWF restores the formation of VWF-containing storage granules, in addition to restoring multimerization.15,36 Although VWFpp is required for both VWF multimerization and regulated storage, these 2 events are independent of one another; VWF multimerization is not required for its regulated storage.15,35
Several studies have demonstrated that the entire mature VWF is not required for the formation of WPB-like granules.32,38 A VWF protein truncated before the cysteine knot domain formed WPB-like granules.35 Michaux et al showed that a VWF protein truncated after the D3 domain (D1-D3 domains) formed mostly round, but sometimes elongated, WPB.32 Inclusion of the A1 domain (D1-A1 domains) resulted in elongated WPBs with VWF tubules similar to that found after expression of full-length VWF. Formation of WPB-like tubules has also been recreated in vitro using purified VWFpp and disulfide-linked dimers of D1-D3 domains, and this process required low pH and calcium ions.38 These studies indicate that VWF storage granule formation requires VWFpp, and at a minimum, the D′-D3 domains of mature VWF.
Still other studies have focused more specifically on the role of VWFpp in trafficking of VWF to the regulated storage pathway. Disruption of either of the vicinal cysteine motifs in the D1 domain of VWFpp resulted in defective multimerization, but had no effect on regulated storage of VWF.28 Deletion of either the D1 or D2 domain results in loss of granular sorting,25 although this may be secondary to a loss of a sustained VWFpp/VWF noncovalent interaction. VWFpp can independently sort to preexisting adrenocorticotropin (ACTH)-containing storage granules in AtT-20 cells.15 VWFpp appears to contain the necessary signal or conformation for navigating the regulated secretory pathway and cotraffics mature VWF to storage by virtue of a noncovalent association involving arginine 416 in VWFpp and threonine 869 in mature VWF.15,39 These studies also confirmed that VWF multimerization is not a prerequisite for regulated storage as many multimerization-defective variants efficiently formed storage granules. The “storage signal” within VWFpp has been further localized to the D2 domain. A series of C-terminal or N-terminal truncations of VWFpp (Figure 3A) were expressed in AtT-20 cells and intracellular localization assessed by confocal microscopy (S.L.H., unpublished data). Although some variants appeared to be localized in granules similar to WT VWF, other variants demonstrated a diffuse staining pattern reminiscent of ER-localization (Figure 3B). Further analysis showed colocalization of several VWFpp truncations in granules with endogenous ACTH (Figure 3C). Figure 3A summarizes the variants stored in granules (red) and those that appeared ER-localized (black). Some of the ER-localized variants were not efficiently secreted from the cell, and may be conformationally challenged rather than lacking a storage signal. The minimal area of sequence overlap between stored truncations begins at the D2 domain (aa 387) and extends to aa 567 (Figure 3A, blue box), indicating the location of a VWFpp “storage signal.” However, although VWFpp expression is necessary for granule biogenesis, VWFpp expression alone is not sufficient to support the formation of WPB-like granules in VWF-deficient endothelial cells or HEK293 cells.32,34
Figure 3.

Location of VWF “storage signal.” (A) Schematic representation of N-terminal and C-terminal truncations of VWFpp. Each construct contains the VWF signal peptide followed directly by the VWFpp sequence depicted. WT VWFpp is located in the middle. VWFpp truncations that were observed to traffic to storage granules are shown in red, whereas those truncations that appeared to be ER-localized are shown in black. The minimum area of sequence overlap between truncations that sorted to storage granules is highlighted in the blue box. (B) AtT-20 cells were transiently transfected with VWFpp truncation constructs to determine the region of VWFpp containing the VWF “storage signal.” After 72 hours, cells were fixed, permeabilized, stained with monoclonal antibodies to VWFpp, and detected with Alexa Fluor-488–conjugated goat anti-mouse immunoglobulin (Ig)G (green) by confocal microscopy. Expression of VWFpp truncations E, F, and I (panels 3, 4, and 6) resulted in granular storage of VWFpp similar to WT (panel 1), whereas truncations D and G appeared to be ER-localized (panels 2 and 5). (C) AtT-20 cells were transiently transfected with VWFpp truncations to address co-localization with endogenous ACTH-containing granules. After 72 hours, cells were fixed, permeabilized, dual stained with monoclonal antibodies to VWFpp and a polyclonal antibody to ACTH, and detected with Alexa Fluor-488–conjugated goat anti-mouse IgG (VWFpp/green) and Alexa Fluor-594–conjugated goat anti-rabbit IgG (ACTH/red) by confocal microscopy. Colocalization of VWFpp and ACTH is shown in yellow. Truncations E and F (panels 2 and 3) co-localized with endogenous ACTH-containing granules similar to WT VWFpp (panel 1). Total magnification, ×1500. Bar, 10 μm. Collectively, these results demonstrate that VWFpp contains a “storage signal” that appears to be localized in the D2 domain of VWFpp.
The existence of a signal within VWFpp for trafficking to the regulated secretory pathway has been exploited in order to traffic unrelated proteins to storage granules in endothelial cells and platelets.40,41 Full-length VWFpp or a C-terminal truncation of VWFpp (aa 201-763) were used to traffic complement C3 to storage granules in AtT-20 cells and bovine endothelial cells.40 The D2 domain fused to coagulation factor VIII was demonstrated to traffic to platelet α-granules in dogs receiving gene therapy for hemophilia A.41
VWFpp may also have a role in the exocytosis of VWF from WPB as proposed by Michaux et al.32 They suggest that a pH-dependent interaction of VWFpp with VWF within WPBs is necessary to constrain VWF into a spring-like structure, and that upon exocytosis, VWFpp dissociation is what allows filament unfurling. VWFpp then rapidly dissociates from the cell into circulation. VWFpp clearly is indispensable in the biogenesis of VWF-containing storage granules and can potentially be useful in trafficking other proteins or therapeutics of interest into the regulated secretory pathway.
Mutations in VWFpp: impact on VWF structure and function
A number of candidate mutations identified in VWD patients are located in the D1 and D2 domains of VWFpp. Many of these sequence variations are listed in the International Society on Thrombosis and Haemostasis VWD database and have been associated with types 1, 3, and 2A VWD.32,34,37,42-55 Because the role of VWFpp in VWF multimerization has been clearly established, it is not surprising that VWFpp mutations may be associated with type 2A VWD, which is characterized by the lack of high molecular weight multimers. Expression studies in various cell lines have demonstrated a negative impact of some VWFpp mutations on VWF multimerization, often accompanied by retention of VWF in the ER resulting in reduced secretion.37,46,48-51,56 Expression of VWF variants Y87S, D141Y, C275S, del(437-442), and C570S demonstrated a profound impact on VWF multimerization with secretion of only dimeric VWF, indicating that mutations in either VWFpp D domain may impact VWF multimerization.46,48,49,51
Fewer studies have examined the impact of mutations in VWFpp on the regulated storage of VWF.32,34,37,47-50 D1 domain variant Y87S is targeted to storage granules in several cell types. However, Y87S-containing storage granules are not elongated and lack tubular packaging.32,34,48 The D1 domain variant R273W is retained in the ER and also targeted to round storage granules with disordered tubular packaging.37 Three VWF D2 domain mutations (del[437-442], N528S, and C584F) have been reported with impaired regulated storage of VWF.47,49,50 VWFpp mutations are found in patients with types 1, 3, and 2A VWD, and these mutations may cause altered intracellular processing.
Extracellular function of VWFpp
The 741 aa VWFpp is considerably larger than many similar protein propeptides that function as intracellular chaperones.57-59 In addition, the fact that VWFpp is stored and then secreted with a reasonably long half-life (2 to 3 hours) alludes to a specific function in plasma for VWFpp. Early work suggested that VWFpp may inhibit the interaction of platelets with collagen.60 Other studies utilizing bovine-derived VWFpp demonstrated a domain within bovine VWFpp that bound bovine type 1 collagen.61 Bovine VWFpp was also found to serve as a substrate for factor XIIIa and could be crosslinked to laminin.62 In addition, bovine VWFpp was found to be a ligand for very late antigen-4 integrin.63 Many of these studies examined only VWFpp derived from bovine platelets and these proposed functions have not been confirmed using human-derived components.
In another recent study, Madabhushi et al demonstrated that a fraction of VWFpp actually circulates bound to VWF and can regulate the hemostatic potential of VWF by reducing binding of VWF to platelets.64 VWFpp was shown to bind to VWF exclusively through the D′D3 domains. Low pH was found to enhance the affinity of VWFpp binding to other VWFpp units, as well as to D′D3. This VWFpp/VWF-D′D3 interaction attenuated VWF-mediated platelet activation and adhesion. In contrast to previous studies utilizing bovine VWFpp, binding of human VWFpp to equine collagen was not detected in either microfluidic or enzyme-linked immunosorbent assays. Although VWFpp regulation of VWF-A1 function may represent one extracellular function for VWFpp, it seems very likely that VWFpp may have additional extracellular function(s), given its large size and complexity of intracellular processing. These other potential extracellular VWFpp functions remain to be elucidated.
Assay of plasma VWFpp: clinical utility
Vascular disorders and endothelial cell perturbation
One extracellular function for VWFpp is as a biomarker. VWFpp and mature VWF are released into plasma and circulate largely independently of one another with disparate half-lives (∼2 and 12 hours, respectively).65,66 Vischer et al demonstrated that the VWFpp level could be used to assess acute VWF secretion from endothelium. They later described the utility of using parallel determination of VWF and VWFpp as a marker of endothelial activation in diabetes.67 van Mourik et al established that the assay of both VWFpp and VWF can be used to distinguish between acute and chronic endothelial cell perturbation by examining patients with TTP, sepsis, and diabetes, and a control group consisting of healthy individuals subjected to desmopressin (DDAVP), low dose endotoxin, or exercise.68 In the healthy controls, the concomitant rise of VWFpp and VWF levels, presumably released from endothelial cell storage, was followed by a rapid decline in the VWFpp level, whereas VWF levels returned to baseline slowly. During the acute phase of vascular perturbation, both VWF and VWFpp were elevated. At later time points, only VWF levels continued to be elevated. Thus, elevated VWFpp, together with elevated VWF, indicates acute but transient endothelial cell activation. In patients with symptoms of acute vascular disease, such as TTP, hemolytic uremic syndrome, and sepsis, both VWFpp and VWF levels were elevated.68,69 In contrast, VWFpp levels are only slightly elevated, even with elevated VWF levels, in patients with extended periods of vascular disease such as diabetes, ischemic heart disease, or peripheral vascular disease, indicating that marginally increased VWFpp levels in combination with elevated VWF levels reflect chronic stimulation of the endothelium. Evaluation of VWF and the VWFpp level can also be useful in assessing drug-induced vascular injury caused by vasoactive agents such DDAVP, lipopolysaccharide/endotoxin, fenoldopam, dopamine, or potassium channel opener, which may result in endothelial cell activation, perturbation, or injury.70 Although the increased level of VWFpp in these clinical situations is assumed to be due to release primarily from endothelial cells, release from activated platelets cannot be ruled out. Additionally, acute release of VWF and VWFpp from activated platelets during phlebotomy or sample processing are pre-analytical variables affecting sample quality.
The parallel assay of VWFpp and VWF has been demonstrated in several studies to be a marker of acute endothelial cell activation in cerebral and noncerebral severe malaria.71-74 These findings implicate acute, regulated release of endothelial cell WPBs in the pathogenesis of Plasmodium falciparum malaria. VWFpp and VWF levels have also been used to identify endothelial cell activation in diabetes.67,69,75-77 A significant increase in VWF and VWFpp were identified in insulin-dependent diabetic patients with microalbunimuria or overt diabetic nephropathy.67,75 The study by Verrotti et al demonstrated that endothelial dysfunction is present at least 2 years before the development of persistent microalbuminuria.75 Endothelial activation, as indicated by an elevated VWFpp level has been implicated in chronic renal failure, aortic stenosis, ischemic stroke, sepsis, meningococcal disease, dengue, systemic sclerosis, sickle cell disease, HELLP syndrome, and asthma.78-86 In addition to utility in identifying endothelial activation, VWFpp and VWF may also be used in a similar manner to identify platelet activation.87 In some clinical circumstances, an elevated VWFpp level may indicate activation of both platelets and endothelial cells.
VWD
The assay of VWFpp can also be useful in the diagnosis of VWD, particularly in defining the VWD subtype. The VWFpp level is used as an indicator of VWF synthesis/secretion. The parallel assay of VWF and VWFpp can identify a reduced survival of VWF as is found in acquired, type 1C, type 2B, and some type 2A VWD patients.88-98 Type 1C VWD patients are characterized by markedly reduced plasma survival of VWF, with half-life of VWF as short as 1.0 hour, in contrast to the 8 to 12 hour half-life in healthy individuals and other type 1 patients (Figure 4A).89,99 By definition, there is 1 unit each of VWF and VWFpp in 1 mL of plasma in healthy individuals with a VWFpp/VWF:Ag ratio at steady-state of ∼1.0. Blood group O individuals have lower VWF levels and slightly elevated VWFpp/VWF:Ag than non-O due to slightly faster VWF clearance (Figure 4B).89,100 Although the VWF is quickly cleared from plasma in type 1C patients, a normal VWFpp half-life and elevated VWFpp/VWF:Ag are expected. In type 1C VWD patients with a W1144G or S2179F mutation and reduced VWF half-life, VWFpp/VWF:Ag levels were found to be elevated, whereas unaffected family members had ratios within the normal range (Figure 4C).89 One caveat in interpreting the VWFpp/VWF:Ag ratio is the assumption that VWFpp half-life is relatively fixed. Although VWF half-life is fairly routinely determined, VWFpp clearance is rarely followed. It is conceivable that VWFpp survival may be prolonged or reduced in some individuals, which would lead to an increase or decrease in VWFpp/VWF:Ag, respectively.
Figure 4.

Reduced VWF survival in type 1C VWD is predicted by increased baseline plasma VWFpp/VWF:Ag ratio. (A) Time course of VWF:RCo after administration of DDAVP in type 1 subjects without a reduced VWF survival phenotype (▲ and ♦), and in a patient with type 1C VWD and VWF S2179F sequence variation with reduced VWF survival (●). Although VWF is released after DDAVP administration in the type 1C VWD subject, released VWF is rapidly cleared from circulation. (B) VWFpp/VWF:Ag ratio in healthy individuals with blood groups: O (▪), A (▲), B (▾), and AB (♦). The horizontal line indicates the median VWFpp/VWF:Ag ratio. VWFpp/VWF:Ag is slightly increased in blood group O individuals. (C) Ratio of VWFpp to VWF:Ag in family members with a type 1C VWF mutation (S2179F or W1144G, circles) or without a mutation (triangles). The horizontal line represents the median value of the ratio. An elevated VWFpp/VWF:Ag ratio is identified in family members with a type 1C VWF mutation and reduced VWF survival. Adapted with permission from Haberichter et al.89
In a large study of types 1, 2, and 3 VWD patients, Sanders et al reported the use of VWFpp/VWF:Ag and FVIII:C/VWF:Ag ratios to help define the pathological mechanism (increased VWF clearance, reduced VWF secretion, or combination of both mechanisms) underlying a patient’s VWD phenotype.94 Perhaps the most striking use for the VWFpp assay is in identifying true type 3 VWD patients. In some patients with very low VWF levels (<5 IU/dL), detectable or even normal levels of VWFpp may be measured.94,101 These moderate or normal VWFpp levels in patients with severely reduced VWF levels indicate reduced VWF survival, as found in type 1C VWD. In type 3 VWD, VWF is not stored in endothelial cell WPB or platelet α-granules, whereas type 1C patients are expected to have normal WPB and α-granule stores of VWF and may have a milder bleeding phenotype.94 The identification of patients with reduced VWF survival impacts treatment options. DDAVP is the most common treatment of type 1 VWD, releasing VWF from WPBs to increase plasma VWF levels. Many type 1C VWD patients release VWF after DDAVP, but the released VWF is quickly cleared. However, this released VWF may be sufficient to achieve hemostasis in minor bleeding situations, but is likely inadequate for major bleeding or surgery. Type 3 VWD patients routinely require VWF replacement therapy rather than DDAVP administration. The routine assay of VWFpp in suspected type 3 VWD patients is likely to lead to re-classification of some as type 1C VWD, which may expand the treatment options. Similarly, the assay of VWFpp may help to identify patients with acquired VWD associated with clearance of high molecular weight multimers such as found in monoclonal gammopathy.102 These subjects often have a markedly increased VWFpp/VWF:Ag ratio.
The VWFpp assay is not routinely included in the diagnostic workup for VWD; however, extensive published data suggests this assay would be a valuable addition to the diagnostic panel, particularly in patients with low VWF:Ag levels. Although this assay may provide limited additional information in type 2 VWD, the ability to identify true type 3, type 1C, and other type 1 VWD patients are clinically important and have implications for therapeutic treatment.
VWFpp clearly plays an active role in the intracellular processing of VWF, and is indispensible for proper multimerization and regulated storage of VWF. The assay of plasma VWFpp has well-defined clinical utility in assessing acute or chronic endothelial activation, as well as in discriminating between subtypes of VWD, which may have implications for treatment strategies. An extracellular role for VWF has been demonstrated in attenuating platelet activation and adhesion. However, given the large size of VWFpp, and the complexities involved in granular storage and secretion of this large propeptide, it seems likely that other extracellular roles for VWFpp remain to be discovered.
Acknowledgments
The author thanks Robert R. Montgomery and Veronica H. Flood for valuable advice and reviewing the manuscript.
This work was supported in part by a grant from the National Institutes of Health, National Heart, Lung, and Blood Institute (HL-081588).
Authorship
Contribution: S.L.H. wrote the manuscript.
Conflict-of-interest disclosure: The author declares no competing financial interests.
Correspondence: Sandra L. Haberichter, Diagnostic Labs and Blood Research Institute, BloodCenter of Wisconsin, 638 N 18th St, PO Box 2178, Milwaukee, WI 53201; e-mail: sandra.haberichter@bcw.edu.
References
- 1.Sadler JE, Budde U, Eikenboom JC, et al. Working Party on von Willebrand Disease Classification. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost. 2006;4(10):2103–2114. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 2.Montgomery RR, Zimmerman TS. von Willebrand’s disease antigen II. A new plasma and platelet antigen deficient in severe von Willebrand’s disease. J Clin Invest. 1978;61(6):1498–1507. doi: 10.1172/JCI109070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott JP, Montgomery RR. Platelet von Willebrand’s antigen II: active release by aggregating agents and a marker of platelet release reaction in vivo. Blood. 1981;58(6):1075–1080. [PubMed] [Google Scholar]
- 4.McCarroll DR, Ruggeri ZM, Montgomery RR. The effect of DDAVP on plasma levels of von Willebrand antigen II in normal individuals and patients with von Willebrand’s disease. Blood. 1984;63(3):532–535. [PubMed] [Google Scholar]
- 5.Ginsburg D, Handin RI, Bonthron DT, et al. Human von Willebrand factor (vWF): isolation of complementary DNA (cDNA) clones and chromosomal localization. Science. 1985;228(4706):1401–1406. doi: 10.1126/science.3874428. [DOI] [PubMed] [Google Scholar]
- 6.Verweij CL, Diergaarde PJ, Hart M, Pannekoek H. Full-length von Willebrand factor (vWF) cDNA encodes a highly repetitive protein considerably larger than the mature vWF subunit [published correction appears in EMBO J. 1986;5(11):3074]. EMBO J. 1986;5(8):1839–1847. doi: 10.1002/j.1460-2075.1986.tb04435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fay PJ, Kawai Y, Wagner DD, et al. Propolypeptide of von Willebrand factor circulates in blood and is identical to von Willebrand antigen II. Science. 1986;232(4753):995–998. doi: 10.1126/science.3486471. [DOI] [PubMed] [Google Scholar]
- 8.Jaffe EA, Hoyer LW, Nachman RL. Synthesis of antihemophilic factor antigen by cultured human endothelial cells. J Clin Invest. 1973;52(11):2757–2764. doi: 10.1172/JCI107471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nachman R, Levine R, Jaffe EA. Synthesis of factor VIII antigen by cultured guinea pig megakaryocytes. J Clin Invest. 1977;60(4):914–921. doi: 10.1172/JCI108846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sadler JE, Mancuso DJ, Randi AM, Tuley EA, Westfield LA. Molecular biology of von Willebrand factor. Ann N Y Acad Sci. 1991;614:114–124. doi: 10.1111/j.1749-6632.1991.tb43697.x. [DOI] [PubMed] [Google Scholar]
- 11.Zhou YF, Eng ET, Zhu J, Lu C, Walz T, Springer TA. Sequence and structure relationships within von Willebrand factor. Blood. 2012;120(2):449–458. doi: 10.1182/blood-2012-01-405134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denis CV. Molecular and cellular biology of von Willebrand factor. Int J Hematol. 2002;75(1):3–8. doi: 10.1007/BF02981972. [DOI] [PubMed] [Google Scholar]
- 13.Canis K, McKinnon TA, Nowak A, et al. Mapping the N-glycome of human von Willebrand factor. Biochem J. 2012;447(2):217–228. doi: 10.1042/BJ20120810. [DOI] [PubMed] [Google Scholar]
- 14.Vischer UM, Wagner DD. von Willebrand factor proteolytic processing and multimerization precede the formation of Weibel-Palade bodies. Blood. 1994;83(12):3536–3544. [PubMed] [Google Scholar]
- 15.Haberichter SL, Fahs SA, Montgomery RR. von Willebrand factor storage and multimerization: 2 independent intracellular processes. Blood. 2000;96(5):1808–1815. [PubMed] [Google Scholar]
- 16.Wagner DD, Olmsted JB, Marder VJ. Immunolocalization of von Willebrand protein in Weibel-Palade bodies of human endothelial cells. J Cell Biol. 1982;95(1):355–360. doi: 10.1083/jcb.95.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cramer EM, Meyer D, le Menn R, Breton-Gorius J. Eccentric localization of von Willebrand factor in an internal structure of platelet alpha-granule resembling that of Weibel-Palade bodies. Blood. 1985;66(3):710–713. [PubMed] [Google Scholar]
- 18.Giblin JP, Hewlett LJ, Hannah MJ. Basal secretion of von Willebrand factor from human endothelial cells. Blood. 2008;112(4):957–964. doi: 10.1182/blood-2007-12-130740. [DOI] [PubMed] [Google Scholar]
- 19.Savage B, Sixma JJ, Ruggeri ZM. Functional self-association of von Willebrand factor during platelet adhesion under flow. Proc Natl Acad Sci USA. 2002;99(1):425–430. doi: 10.1073/pnas.012459599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276(44):41059–41063. doi: 10.1074/jbc.C100515200. [DOI] [PubMed] [Google Scholar]
- 21.Casari C, Lenting PJ, Wohner N, Christophe OD, Denis CV. Clearance of von Willebrand factor. J Thromb Haemost. 2013;11(suppl 1):202–211. doi: 10.1111/jth.12226. [DOI] [PubMed] [Google Scholar]
- 22.Rawley O, O’Sullivan JM, Chion A, et al. von Willebrand factor arginine 1205 substitution results in accelerated macrophage-dependent clearance in vivo. J Thromb Haemost. 2015;13(5):821–826. doi: 10.1111/jth.12875. [DOI] [PubMed] [Google Scholar]
- 23.Wise RJ, Pittman DD, Handin RI, Kaufman RJ, Orkin SH. The propeptide of von Willebrand factor independently mediates the assembly of von Willebrand multimers. Cell. 1988;52(2):229–236. doi: 10.1016/0092-8674(88)90511-9. [DOI] [PubMed] [Google Scholar]
- 24.Voorberg J, Fontijn R, van Mourik JA, Pannekoek H. Domains involved in multimer assembly of von willebrand factor (vWF): multimerization is independent of dimerization. EMBO J. 1990;9(3):797–803. doi: 10.1002/j.1460-2075.1990.tb08176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Journet AM, Saffaripour S, Wagner DD. Requirement for both D domains of the propolypeptide in von Willebrand factor multimerization and storage. Thromb Haemost. 1993;70(6):1053–1057. [PubMed] [Google Scholar]
- 26.Mayadas TN, Wagner DD. In vitro multimerization of von Willebrand factor is triggered by low pH. Importance of the propolypeptide and free sulfhydryls. J Biol Chem. 1989;264(23):13497–13503. [PubMed] [Google Scholar]
- 27.Erent M, Meli A, Moisoi N, et al. Rate, extent and concentration dependence of histamine-evoked Weibel-Palade body exocytosis determined from individual fusion events in human endothelial cells. J Physiol. 2007;583(pt 1):195–212. doi: 10.1113/jphysiol.2007.132993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayadas TN, Wagner DD. Vicinal cysteines in the prosequence play a role in von Willebrand factor multimer assembly. Proc Natl Acad Sci USA. 1992;89(8):3531–3535. doi: 10.1073/pnas.89.8.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Purvis AR, Sadler JE. A covalent oxidoreductase intermediate in propeptide-dependent von Willebrand factor multimerization. J Biol Chem. 2004;279(48):49982–49988. doi: 10.1074/jbc.M408727200. [DOI] [PubMed] [Google Scholar]
- 30.Purvis AR, Gross J, Dang LT, et al. Two Cys residues essential for von Willebrand factor multimer assembly in the Golgi. Proc Natl Acad Sci USA. 2007;104(40):15647–15652. doi: 10.1073/pnas.0705175104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dang LT, Purvis AR, Huang RH, Westfield LA, Sadler JE. Phylogenetic and functional analysis of histidine residues essential for pH-dependent multimerization of von Willebrand factor. J Biol Chem. 2011;286(29):25763–25769. doi: 10.1074/jbc.M111.249151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michaux G, Abbitt KB, Collinson LM, Haberichter SL, Norman KE, Cutler DF. The physiological function of von Willebrand’s factor depends on its tubular storage in endothelial Weibel-Palade bodies. Dev Cell. 2006;10(2):223–232. doi: 10.1016/j.devcel.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 33.Denis CV, André P, Saffaripour S, Wagner DD. Defect in regulated secretion of P-selectin affects leukocyte recruitment in von Willebrand factor-deficient mice. Proc Natl Acad Sci USA. 2001;98(7):4072–4077. doi: 10.1073/pnas.061307098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haberichter SL, Merricks EP, Fahs SA, Christopherson PA, Nichols TC, Montgomery RR. Re-establishment of VWF-dependent Weibel-Palade bodies in VWD endothelial cells. Blood. 2005;105(1):145–152. doi: 10.1182/blood-2004-02-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner DD, Saffaripour S, Bonfanti R, et al. Induction of specific storage organelles by von Willebrand factor propolypeptide. Cell. 1991;64(2):403–413. doi: 10.1016/0092-8674(91)90648-i. [DOI] [PubMed] [Google Scholar]
- 36.Voorberg J, Fontijn R, Calafat J, Janssen H, van Mourik JA, Pannekoek H. Biogenesis of von Willebrand factor-containing organelles in heterologous transfected CV-1 cells. EMBO J. 1993;12(2):749–758. doi: 10.1002/j.1460-2075.1993.tb05709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michaux G, Hewlett LJ, Messenger SL, et al. Analysis of intracellular storage and regulated secretion of 3 von Willebrand disease-causing variants of von Willebrand factor. Blood. 2003;102(7):2452–2458. doi: 10.1182/blood-2003-02-0599. [DOI] [PubMed] [Google Scholar]
- 38.Huang RH, Wang Y, Roth R, et al. Assembly of Weibel-Palade body-like tubules from N-terminal domains of von Willebrand factor. Proc Natl Acad Sci USA. 2008;105(2):482–487. doi: 10.1073/pnas.0710079105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haberichter SL, Jacobi P, Montgomery RR. Critical independent regions in the VWF propeptide and mature VWF that enable normal VWF storage. Blood. 2003;101(4):1384–1391. doi: 10.1182/blood-2002-07-2281. [DOI] [PubMed] [Google Scholar]
- 40.Haberichter SL, Jozwiak MA, Rosenberg JB, Christopherson PA, Montgomery RR. The von Willebrand factor propeptide (VWFpp) traffics an unrelated protein to storage. Arterioscler Thromb Vasc Biol. 2002;22(6):921–926. doi: 10.1161/01.atv.0000017063.36768.87. [DOI] [PubMed] [Google Scholar]
- 41.Du LM, Nurden P, Nurden AT, et al. Platelet-targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A. Nat Commun. 2013;4:2773. doi: 10.1038/ncomms3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodeve A, Eikenboom J, Castaman G, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM-1VWD). Blood. 2007;109(1):112–121. doi: 10.1182/blood-2006-05-020784. [DOI] [PubMed] [Google Scholar]
- 43.Casonato A, Sartorello F, Cattini MG, et al. An Arg760Cys mutation in the consensus sequence of the von Willebrand factor propeptide cleavage site is responsible for a new von Willebrand disease variant. Blood. 2003;101(1):151–156. doi: 10.1182/blood-2002-04-1046. [DOI] [PubMed] [Google Scholar]
- 44.Gaucher C, Diéval J, Mazurier C. Characterization of von Willebrand factor gene defects in two unrelated patients with type IIC von Willebrand disease. Blood. 1994;84(4):1024–1030. [PubMed] [Google Scholar]
- 45.Hampshire DJ, Goodeve AC. The international society on thrombosis and haematosis von Willebrand disease database: an update. Semin Thromb Hemost. 2011;37(5):470–479. doi: 10.1055/s-0031-1281031. [DOI] [PubMed] [Google Scholar]
- 46.Lanke E, Kristoffersson AC, Philips M, Holmberg L, Lethagen S. Characterization of a novel mutation in the von Willebrand factor propeptide in a distinct subtype of recessive von Willebrand disease. Thromb Haemost. 2008;100(2):211–216. [PubMed] [Google Scholar]
- 47.Daidone V, Barbon G, Pontara E, et al. Loss of cysteine 584 impairs the storage and release, but not the synthesis of von Willebrand factor. Thromb Haemost. 2014;112(6):1159–1166. doi: 10.1160/TH14-04-0391. [DOI] [PubMed] [Google Scholar]
- 48.Rosenberg JB, Haberichter SL, Jozwiak MA, et al. The role of the D1 domain of the von Willebrand factor propeptide in multimerization of VWF. Blood. 2002;100(5):1699–1706. doi: 10.1182/blood-2002-03-0789. [DOI] [PubMed] [Google Scholar]
- 49.Haberichter SL, Allmann AM, Jozwiak MA, Montgomery RR, Gill JC. Genetic alteration of the D2 domain abolishes von Willebrand factor multimerization and trafficking into storage. J Thromb Haemost. 2009;7(4):641–650. doi: 10.1111/j.1538-7836.2009.03290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haberichter SL, Budde U, Obser T, Schneppenheim S, Wermes C, Schneppenheim R. The mutation N528S in the von Willebrand factor (VWF) propeptide causes defective multimerization and storage of VWF. Blood. 2010;115(22):4580–4587. doi: 10.1182/blood-2009-09-244327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baronciani L, Federici AB, Cozzi G, et al. Expression studies of missense mutations p.D141Y, p.C275S located in the propeptide of von Willebrand factor in patients with type 3 von Willebrand disease. Haemophilia. 2008;14(3):549–555. doi: 10.1111/j.1365-2516.2008.01682.x. [DOI] [PubMed] [Google Scholar]
- 52.Holmberg L, Karpman D, Isaksson C, Kristoffersson AC, Lethagen S, Schneppenheim R. Ins405AsnPro mutation in the von Willebrand factor propeptide in recessive type 2A (IIC) von Willebrand’s disease. Thromb Haemost. 1998;79(4):718–722. [PubMed] [Google Scholar]
- 53.Mohlke KL, Nichols WC, Rehemtulla A, et al. A common frameshift mutation in von Willebrand factor does not alter mRNA stability but interferes with normal propeptide processing. Br J Haematol. 1996;95(1):184–191. doi: 10.1046/j.1365-2141.1996.7572377.x. [DOI] [PubMed] [Google Scholar]
- 54.Journet AM, Saffaripour S, Cramer EM, Tenza D, Wagner DD. von Willebrand factor storage requires intact prosequence cleavage site. Eur J Cell Biol. 1993;60(1):31–41. [PubMed] [Google Scholar]
- 55.James PD, Notley C, Hegadorn C, et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood. 2007;109(1):145–154. doi: 10.1182/blood-2006-05-021105.. [DOI] [PubMed] [Google Scholar]
- 56.Allen S, Abuzenadah AM, Hinks J, et al. A novel von Willebrand disease-causing mutation (Arg273Trp) in the von Willebrand factor propeptide that results in defective multimerization and secretion. Blood. 2000;96(2):560–568. [PubMed] [Google Scholar]
- 57.Inouye M. Intramolecular chaperone: the role of the pro-peptide in protein folding. Enzyme. 1991;45(5-6):314–321. doi: 10.1159/000468904. [DOI] [PubMed] [Google Scholar]
- 58.Klionsky DJ, Banta LM, Emr SD. Intracellular sorting and processing of a yeast vacuolar hydrolase: proteinase A propeptide contains vacuolar targeting information. Mol Cell Biol. 1988;8(5):2105–2116. doi: 10.1128/mcb.8.5.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCracken AA, Kruse KB. Intracellular transport of rat serum albumin is altered by a genetically engineered deletion of the propeptide. J Biol Chem. 1989;264(35):20843–20846. [PubMed] [Google Scholar]
- 60.Takagi J, Sekiya F, Kasahara K, Inada Y, Saito Y. Inhibition of platelet-collagen interaction by propolypeptide of von Willebrand factor. J Biol Chem. 1989;264(11):6017–6020. [PubMed] [Google Scholar]
- 61.Takagi J, Fujisawa T, Sekiya F, Saito Y. Collagen-binding domain within bovine propolypeptide of von Willebrand factor. J Biol Chem. 1991;266(9):5575–5579. [PubMed] [Google Scholar]
- 62.Usui T, Takagi J, Saito Y. Propolypeptide of von Willebrand factor serves as a substrate for factor XIIIa and is cross-linked to laminin. J Biol Chem. 1993;268(17):12311–12316. [PubMed] [Google Scholar]
- 63.Isobe T, Hisaoka T, Shimizu A, et al. Propolypeptide of von Willebrand factor is a novel ligand for very late antigen-4 integrin. J Biol Chem. 1997;272(13):8447–8453. doi: 10.1074/jbc.272.13.8447. [DOI] [PubMed] [Google Scholar]
- 64.Madabhushi SR, Shang C, Dayananda KM, et al. von Willebrand factor (VWF) propeptide binding to VWF D′D3 domain attenuates platelet activation and adhesion. Blood. 2012;119(20):4769–4778. doi: 10.1182/blood-2011-10-387548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borchiellini A, Fijnvandraat K, ten Cate JW, et al. Quantitative analysis of von Willebrand factor propeptide release in vivo: effect of experimental endotoxemia and administration of 1-deamino-8-D-arginine vasopressin in humans. Blood. 1996;88(8):2951–2958. [PubMed] [Google Scholar]
- 66.Vischer UM, Ingerslev J, Wollheim CB, et al. Acute von Willebrand factor secretion from the endothelium in vivo: assessment through plasma propeptide (vWf:AgII) levels. Thromb Haemost. 1997;77(2):387–393. [PubMed] [Google Scholar]
- 67.Vischer UM, Emeis JJ, Bilo HJ, et al. von Willebrand factor (vWf) as a plasma marker of endothelial activation in diabetes: improved reliability with parallel determination of the vWf propeptide (vWf:AgII). Thromb Haemost. 1998;80(6):1002–1007. [PubMed] [Google Scholar]
- 68.van Mourik JA, Boertjes R, Huisveld IA, et al. von Willebrand factor propeptide in vascular disorders: a tool to distinguish between acute and chronic endothelial cell perturbation. Blood. 1999;94(1):179–185. [PubMed] [Google Scholar]
- 69.van Mourik JA, Romani de Wit T. Von Willebrand factor propeptide in vascular disorders. Thromb Haemost. 2001;86(1):164–171. [PubMed] [Google Scholar]
- 70.Brott DA, Katein A, Thomas H, et al. Evaluation of von Willebrand factor and von Willebrand factor propeptide in models of vascular endothelial cell activation, perturbation, and/or injury. Toxicol Pathol. 2014;42(4):672–683. doi: 10.1177/0192623313518664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Mast Q, Groot E, Lenting PJ, et al. Thrombocytopenia and release of activated von Willebrand factor during early plasmodium falciparum malaria. J Infect Dis. 2007;196(4):622–628. doi: 10.1086/519844. [DOI] [PubMed] [Google Scholar]
- 72.Hollestelle MJ, Donkor C, Mantey EA, et al. von Willebrand factor propeptide in malaria: evidence of acute endothelial cell activation. Br J Haematol. 2006;133(5):562–569. doi: 10.1111/j.1365-2141.2006.06067.x. [DOI] [PubMed] [Google Scholar]
- 73.Phiri HT, Bridges DJ, Glover SJ, et al. Elevated plasma von Willebrand factor and propeptide levels in Malawian children with malaria. PLoS One. 2011;6(11):e25626. doi: 10.1371/journal.pone.0025626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Conroy AL, Phiri H, Hawkes M, et al. Endothelium-based biomarkers are associated with cerebral malaria in Malawian children: a retrospective case-control study. PLoS One. 2010;5(12):e15291. doi: 10.1371/journal.pone.0015291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Verrotti A, Greco R, Basciani F, Morgese G, Chiarelli F. von Willebrand factor and its propeptide in children with diabetes. Relation between endothelial dysfunction and microalbuminuria. Pediatr Res. 2003;53(3):382–386. doi: 10.1203/01.PDR.0000049509.65496.BF. [DOI] [PubMed] [Google Scholar]
- 76.Nossent AY, Ellenbroek JH, Frölich M, Bertina RM, Knoers NV, Eikenboom JC. Plasma levels of von Willebrand factor, von Willebrand factor propeptide and factor VIII in carriers and patients with nephrogenic diabetes insipidus. Thromb Res. 2010;125(6):554–556. doi: 10.1016/j.thromres.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 77.Chen SF, Xia ZL, Han JJ, et al. Increased active von Willebrand factor during disease development in the aging diabetic patient population. Age (Dordr) 2013;35(1):171–177. doi: 10.1007/s11357-011-9335-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Trappenburg MC, van Schilfgaarde M, Frerichs FC, et al. Chronic renal failure is accompanied by endothelial activation and a large increase in microparticle numbers with reduced procoagulant capacity. Nephrol Dial Transplant. 2012;27(4):1446–1453. doi: 10.1093/ndt/gfr474. [DOI] [PubMed] [Google Scholar]
- 79.Hollestelle MJ, Loots CM, Squizzato A, et al. Decreased active von Willebrand factor level owing to shear stress in aortic stenosis patients. J Thromb Haemost. 2011;9(5):953–958. doi: 10.1111/j.1538-7836.2011.04247.x. [DOI] [PubMed] [Google Scholar]
- 80.van Schie MC, DE Maat MP, Dippel DW, et al. von Willebrand factor propeptide and the occurrence of a first ischemic stroke. J Thromb Haemost. 2010;8(6):1424–1426. doi: 10.1111/j.1538-7836.2010.03863.x. [DOI] [PubMed] [Google Scholar]
- 81.Hollestelle MJ, Sprong T, Bovenschen N, et al. von Willebrand factor activation, granzyme-B and thrombocytopenia in meningococcal disease. J Thromb Haemost. 2010;8(5):1098–1106. doi: 10.1111/j.1538-7836.2010.03811.x. [DOI] [PubMed] [Google Scholar]
- 82.Djamiatun K, van der Ven AJ, de Groot PG, et al. Severe dengue is associated with consumption of von Willebrand factor and its cleaving enzyme ADAMTS-13. PLoS Negl Trop Dis. 2012;6(5):e1628. doi: 10.1371/journal.pntd.0001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scheja A, Akesson A, Geborek P, et al. Von Willebrand factor propeptide as a marker of disease activity in systemic sclerosis (scleroderma). Arthritis Res. 2001;3(3):178–182. doi: 10.1186/ar295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van der Land V, Peters M, Biemond BJ, Heijboer H, Harteveld CL, Fijnvandraat K. Markers of endothelial dysfunction differ between subphenotypes in children with sickle cell disease. Thromb Res. 2013;132(6):712–717. doi: 10.1016/j.thromres.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 85.Hulstein JJ, van Runnard Heimel PJ, Franx A, et al. Acute activation of the endothelium results in increased levels of active von Willebrand factor in hemolysis, elevated liver enzymes and low platelets (HELLP) syndrome. J Thromb Haemost. 2006;4(12):2569–2575. doi: 10.1111/j.1538-7836.2006.02205.x. [DOI] [PubMed] [Google Scholar]
- 86.Johansson MW, Kruger SJ, Schiebler ML, et al. Markers of vascular perturbation correlate with airway structural change in asthma. Am J Respir Crit Care Med. 2013;188(2):167–178. doi: 10.1164/rccm.201301-0185OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jacobi PM, Kanaji S, Gehrand A, Jakab D, Haberichter SL. Von Willebrand factor propeptide (VWFpp): a marker useful for identifying adverse platelet activation in murine blood phlebotomy samples. J Thromb Haemost. 2013;11(suppl 2) Abstract PA 2.09-2. [Google Scholar]
- 88.Schooten CJ, Tjernberg P, Westein E, et al. Cysteine-mutations in von Willebrand factor associated with increased clearance. J Thromb Haemost. 2005;3(10):2228–2237. doi: 10.1111/j.1538-7836.2005.01571.x. [DOI] [PubMed] [Google Scholar]
- 89.Haberichter SL, Balistreri M, Christopherson P, et al. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood. 2006;108(10):3344–3351. doi: 10.1182/blood-2006-04-015065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haberichter SL, James PD, Christopherson P, Lillicrap D, Montgomery RR. Reduced VWF survival in two large cohorts of type 1 VWD patients from the Canadian type 1 VWD and US VWD (ZPMCB-VWD) studies. J Thromb Haemost. 2009;7(suppl 2) OC-WE-133. [Google Scholar]
- 91.Nossent AY, VAN Marion V, VAN Tilburg NH, et al. von Willebrand factor and its propeptide: the influence of secretion and clearance on protein levels and the risk of venous thrombosis. J Thromb Haemost. 2006;4(12):2556–2562. doi: 10.1111/j.1538-7836.2006.02273.x. [DOI] [PubMed] [Google Scholar]
- 92.Eikenboom JC, Tjernberg P, Van Marion V, Heering KJ. Acquired von Willebrand syndrome: diagnostic problems and therapeutic options. Am J Hematol. 2007;82(1):55–58. doi: 10.1002/ajh.20760. [DOI] [PubMed] [Google Scholar]
- 93.Eikenboom J, Federici AB, Dirven RJ, et al. MCMDM-1VWD Study Group. VWF propeptide and ratios between VWF, VWF propeptide, and FVIII in the characterization of type 1 von Willebrand disease. Blood. 2013;121(12):2336–2339. doi: 10.1182/blood-2012-09-455089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sanders YV, Groeneveld D, Meijer K, et al. WiN study group. von Willebrand factor propeptide and the phenotypic classification of von Willebrand disease. Blood. 2015;125(19):3006–3013. doi: 10.1182/blood-2014-09-603241. [DOI] [PubMed] [Google Scholar]
- 95.Fabris F, Betterle C, Casonato A, De Marco L, Girolami A. Coagulation factors and blood platelets. (Presence and location of factor VIII and of fibrinogen in normal human platelets and in patients with coagulation disorders, using an immunofluorescence technic). Ric Clin Lab. 1979;9(suppl 2):51–63. [PubMed] [Google Scholar]
- 96.Sztukowska M, Gallinaro L, Cattini MG, et al. Von Willebrand factor propeptide makes it easy to identify the shorter Von Willebrand factor survival in patients with type 1 and type Vicenza von Willebrand disease. Br J Haematol. 2008;143(1):107–114. doi: 10.1111/j.1365-2141.2008.07311.x. [DOI] [PubMed] [Google Scholar]
- 97.Casonato A, Gallinaro L, Cattini MG, et al. Reduced survival of type 2B von Willebrand factor, irrespective of large multimer representation or thrombocytopenia. Haematologica. 2010;95(8):1366–1372. doi: 10.3324/haematol.2009.019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Casonato A, Daidone V, Padrini R. Assessment of von Willebrand factor propeptide improves the diagnosis of von Willebrand disease. Semin Thromb Hemost. 2011;37(5):456–463. doi: 10.1055/s-0031-1281029. [DOI] [PubMed] [Google Scholar]
- 99.Casonato A, Pontara E, Sartorello F, et al. Reduced von Willebrand factor survival in type Vicenza von Willebrand disease. Blood. 2002;99(1):180–184. doi: 10.1182/blood.v99.1.180. [DOI] [PubMed] [Google Scholar]
- 100.Gallinaro L, Cattini MG, Sztukowska M, et al. A shorter von Willebrand factor survival in O blood group subjects explains how ABO determinants influence plasma von Willebrand factor. Blood. 2008;111(7):3540–3545. doi: 10.1182/blood-2007-11-122945. [DOI] [PubMed] [Google Scholar]
- 101.Haberichter SL, Christopherson P, Flood VH, et al. Critical importance of VWF propeptide (VWFpp) in the diagnosis of von Willebrand disease (VWD). Blood. 2013;122(21):331. [Google Scholar]
- 102.Tiede A, Priesack J, Werwitzke S, et al. Diagnostic workup of patients with acquired von Willebrand syndrome: a retrospective single-centre cohort study. J Thromb Haemost. 2008;6(4):569–576. doi: 10.1111/j.1538-7836.2008.02909.x. [DOI] [PubMed] [Google Scholar]