

Enhancing TCR signal strength can attenuate TGFβ-mediated transcriptional changes and functions in CD8+ T cells, independent of changes to canonical TGFβ signaling.
Keywords: Diacylglycerol, diacylglycerol kinase ζ, Smad2
Abstract
DGK-ζ is a negative regulator of TCR signaling that causes degradation of the second messenger DAG, terminating DAG-mediated activation of Ras and PKCθ. Cytotoxic T cells deficient in DGK-ζ demonstrate enhanced effector functions in vitro and antitumor activity in vivo, perhaps because of insensitivity to inhibitory cytokines. We sought to determine whether the enhanced responsiveness of DGK-ζ-deficient T cells renders them insensitive to the inhibitory cytokine TGF-β and to determine how the loss of DGK-ζ facilitates this insensitivity. We identified decreased transcriptional and functional responses to TGF-β in CD8+ DGK-ζ−/− T cells but preserved TGF-β-mediated conversion of naïve DGK-ζ−/− CD4+ T cells to a regulatory T cell phenotype. Decreased CD8+ T cell responsiveness to TGF-β did not result from impaired canonical TGF-β signal transduction, because similar levels of TGF-β-R and intracellular Smad components were identified in WT and DGK-ζ−/− CD8+ T cells, and TGF-β-mediated activation of Smad2 was unchanged. Instead, an enhanced TCR signal strength was responsible for TGF-β insensitivity, because (i) loss of DGK-ζ conferred resistance to TGF-β-mediated inhibition of Erk phosphorylation, (ii) TGF-β insensitivity could be recapitulated by exogenous addition of the DAG analog PMA, and (iii) TGF-β sensitivity could be observed in DGK-ζ-deficient T cells at limiting dilutions of TCR stimulation. These data indicate that enhanced TCR signal transduction in the absence of DGK-ζ makes T cells relatively insensitive to TGF-β, in a manner independent of Smads, a finding with practical implications in the development of immunotherapies that target TGF-β.
Introduction
TGF-β is a pleiotropic cytokine that acts to inhibit or activate cells in virtually every tissue and organ system [1]. In the immune system, TGF-β acts as a potent suppressor, helping facilitate the termination of ongoing immune reactions and preventing the generation of new responses [2]. TGF-β acts on both the innate immune system, by suppressing effector functions of neutrophils, macrophages, and NK cells, and the adaptive immune system, by interfering with the function of cytotoxic CD8+ T cells and Th1 cells, a subtype of CD4+ T cells that provide support for cytotoxic T cells [3, 4]. TGF-β also induces the generation of Tregs, an immunosuppressive CD4+ T cell and facilitates recruitment of Tregs and MDSCs, another immunosuppressive cell type, to sites of immunologic activity [5]. During the establishment and progression of malignant tumor growth, TGF-β plays a central role in generating a permissive environment for tumor growth [6–8]. Although TGF-β is capable of direct effects on tumor cells that can either lead to suppressed or enhanced tumor cell growth, its presence uniformly leads to impaired immune cell responses against tumor cells [9]. In several tumor types, the presence of TGF-β correlates with both a poorer overall prognosis and decreased cytotoxic T cell infiltration of tumor stroma [10, 11].
TGF-β signaling is initiated by binding of TGF-β ligand to the cell surface receptor TGF-β-RII resulting in phosphorylation and activation of the serine/threonine kinase domain of TGF-β-RI, and formation of a heterotetrameric complex containing two subunits each of TGF-β-RI and TGF-β-RII. Once activated, TGF-β-RI directly phosphorylates transcription factors Smad2 and Smad3, resulting in formation of a cytoplasmic complex of Smad2, Smad3, and Smad4 that subsequently translocates to the nucleus to direct repression or transactivation of target genes [12, 13]. Smad2/3 phosphorylation and nuclear translocation are inhibited by Smad7, a protein that competes with Smad2/3 as a substrate for phosphorylation and DNA binding [14, 15]. Binding of TGF-β to its receptor also induces Smad-independent signaling events, referred to as “noncanonical” signaling, that are cell-type dependent and poorly characterized in T cells [16, 17]. Noncanonical TGF-β-dependent signaling is likely to be important in T cells, because Smad3-deficient mice partially phenocopy TGF-β-deficient mice but do not develop the profound autoimmunity observed in mice deficient in TGF-β or its receptor [18, 19].
Diacylglycerol kinases (DGKs), of which two isoforms, α and ζ, are known to play a role in TCR signal transduction, initiate degradation of the second messenger DAG, thereby terminating DAG-mediated activation of Ras and PKCθ downstream of TCR in T cells and other receptors that generate DAG in cells of hematopoietic lineage [20, 21]. A deficiency of either isoform of DGK results in T cells with increased generation of DAG, enhanced Ras and PKCθ signaling, and hyperresponsive effector functions, including enhanced activity against murine tumor in vivo [11, 22–24]. Using a model system of CAR-transduced T cells, we recently observed that cytotoxic T lymphocytes deficient in DGK-ζ, appear less sensitive to TGF-β than do WT CAR T cells, which might have contributed to the improved in vivo activity of DGK-ζ-deficient CAR-T cells against tumors [25]. The purpose of the present studies was to characterize the effect of DGK-ζ deficiency on T cell sensitivity to TGF-β. We observed that the ability of TGF-β to inhibit TCR-mediated effector functions and induce transcriptional changes in cytotoxic T cells was impaired in the absence of DGK-ζ but that conversion of naïve CD4+ T cells to a Treg phenotype was not affected. We further determined that TGF-β insensitivity of DGK-ζ-deficient CD8+ T cells did not result from direct inhibition of TGF-β-dependent signal transduction but, instead, resulted from enhanced TCR signal strength downstream of DAG. These data identify DGK-ζ as a novel target for improving cytotoxic T cell activity in areas rich in TGF-β, such as the tumor microenvironment.
MATERIALS AND METHODS
Mice
Mice deficient in DGK-ζ backcrossed to C57BL/6 have been described previously [22]. C57BL/6 mice expressing FoxP3 and EGFP from a bicistronic construct in the FoxP3 locus (“foxP3 reporter mice”) [26] were purchased from The Jackson Laboratory (Bar Harbor, ME). DGK-ζ-deficient FoxP3 reporter mice were generated by crossing these two strains. All experiments were performed in 5–8-wk-old mice. Animal maintenance and experimentation were performed in accordance with the Institutional Animal Care and Use Committees at the Medical College of Wisconsin and University of Pennsylvania.
Measurement of T cell proliferation, IFN-γ production, and granzyme B upregulation
CD8+ T cells were magnetically isolated by negative selection using a kit and instructions provided by the manufacturer (Miltenyi Biotec, Bergisch Gladbach, Germany). The cells were stained with CFSE (Molecular Probes, Eugene, OR), as described previously [25], and the dilution of CFSE was assessed after incubation of 1 × 105 cells/well in a 96-well plate precoated with 1 μg/ml α−CD3 (clone 2C11; BD Pharmingen, San Diego, CA) 72 h in the presence or absence of TGF-β (5 ng/ml; R&D Systems, Minneapolis, MN), or PMA (Sigma-Aldrich, St. Louis, MO). The cells were analyzed by flow cytometry by gating for live cells using a live/dead stain (Molecular Probes) and a fluorochrome-conjugated antibody to CD8 (BD Pharmingen), with an LSR II flow cytometer (BD Pharmingen) and FlowJo analysis software (Ashland, OR). IFN-γ was measured from cell culture supernatants using an ELISA kit (BioLegend, San Diego, CA) and instructions from the manufacturer. Statistics were calculated using algorithms from Prism graphing software (GraphPad Software, Inc., San Diego, CA). To identify granzyme B up-regulation, the cells were stimulated as above and, 72 h later, incubated with fluorescently labeled antibodies against cell surface markers, permeabilized using a foxP3 staining kit (eBioscience Inc., San Diego, CA), and incubated with fluorescently labeled antibody against granzyme B (Invitrogen, Carlsbad, CA), using protocols supplied by the manufacturer.
Regulatory T cell conversion assay
Naïve (CD45RBhi) CD4+ EGFP-negative cells were flow sorted from the spleens of mice using a FACSAria Cell Sorter (BD Biosciences, Franklin Lakes, NJ), and 1.5 × 105 cells were incubated with plate-bound anti-CD3 (clone 2C11) at 2.5 μg/ml or the indicated concentrations along with plate-bound anti-CD28 (eBioscience) at 1 μg/ml in the presence of 5 ng/ml TGF-β. After 72 h, the cells were assessed for EGFP expression and surface CD25 using flow cytometry [27].
Immunoblotting
CD8+ T cells were isolated just as in T cell proliferation assays, and 1–2 × 106 T cells were stimulated with soluble anti-CD3 (clone 500A2; BD Pharmingen) at a final concentration of 2.5 μg/ml in RPMI 1640 medium with glutamine and 10% FCS. TGF-β was used to stimulate T cells at a concentration of 10 ng/ml in a final volume of 200 μl. The reactions were terminated at the indicated time points with the addition of 1 ml of ice-cold PBS. The cells were centrifuged and resuspended in 1% Nonidet P-40 containing PMSF, NaF, and mixes of phosphatase (Thermo Fisher Scientific, Wilmington, DE) and protease (Sigma-Aldrich) inhibitors. After 10 min on ice, the cells were centrifuged for 10 min at 14,000 rpm to remove debris, and lysate was added to the sample buffer and boiled. A small aliquot of lysate was reserved for a BCA assay (Pierce, Rockford, IL) to normalize the protein content before SDS-PAGE electrophoresis, transfer to polyvinylidene difluoride, and incubation with primary and secondary antibodies. The primary antibodies were from Cell Signaling Technology (Beverly, MA), except for antibody to Smad7 (Santa Cruz Biotechnology, Santa Cruz, CA). For evaluation of proteins after fractionation, nuclear and cytosolic lysates were obtained from 4 × 106 T cells (Smad2) or 10 × 107 T cells (Smad7) for each condition using a commercial kit (Thermo Fisher Scientific). The integrity of fractionation was confirmed using nuclear HDAC as a control.
Quantitative PCR
RT-PCR analysis of mRNA levels was performed on total RNA extracted from 1 × 106 cells using RNA binding columns (Qiagen, Hilden, Germany). Next, 500 ng of RNA was used for cDNA synthesis in accordance with the manufacturer’s protocol (Life Technologies, Carlsbad, CA). The gene expression level was quantified using validated TaqMan primer sets (Integrated DNA Technologies, Coralville, IA), Brilliant II QPCR Master Mix (Agilent Technologies, Palo Alto, CA), on a 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA). The values in each assay set were normalized to β-actin.
RESULTS
TCR-stimulated DGK-ζ-deficient CD8+ T cells show insensitivity to TGF-β
We have previously demonstrated that DGK-ζ, a protein responsible for metabolism of the second messenger DAG, limits the antitumor responses of CAR-expressing T cells [25]. During the course of these studies, we observed that DGK-ζ-deficient CAR-T cells also appeared relatively insensitive to TGF-β. Thus, We sought to characterize the effect of loss of DGK-ζ on T cell insensitivity to TGF-β. Toward this, we first evaluated the proliferation of WT and DGK-ζ-deficient cytotoxic T cells in the presence or absence of TGF-β. Magnetically purified CFSE-labeled CD8+ T cells from the spleens of WT or DGK-ζ-deficient mice were stimulated through the TCR signaling complex with plate-bound antibody against CD3 in the presence or absence of TGF-β, and cell proliferation, as measured by CFSE dilution, was assessed after 72 h. Although the addition of TGF-β to cell culture medium decreased proliferation of WT CD8+ T cells, DGK-ζ-deficient CD8+ T cells were unaffected by the addition of this cytokine (Fig. 1A). In addition to assaying for changes in proliferation, we assessed the effect of DGK-ζ deficiency on cytokine production by CD8+ T cells in the presence or absence of TGF-β. Thus, supernatants from cell proliferation experiments were subjected to IFN-γ ELISA. Although IFN-γ production by WT CD8+ T cells was significantly inhibited in the presence of TGF-β, CD8+ T cells lacking DGK-ζ were less inhibited to only approximately 50% in the presence of TGF-β (Fig. 1B). Similar results were obtained from naïve CD8+ cells (CD44lo) isolated by flow cytometry instead of by magnetic bead labeling (data not shown). These data indicate that DGK-ζ deficiency decreases the sensitivity of CD8+ T cells to TGF-β downstream of TCR.
Figure 1. DGK-ζ-deficient CD8+ T cells demonstrate decreased sensitivity to TGF-β.

Splenic CD8+ T cells from WT or DGK-ζ−/− mice were labeled with CFSE and incubated in 96-well plates (105 cells/well) precoated with α-CD3 (2C11; 1 μg/ml) for 72 h in the presence or absence of TGF-β. (A) Cells were assessed for proliferation as determined by dilution of CFSE by flow cytometry from representative flow cytometry plots (A, top, without TGF-β [unshaded] or with TGF-β [shaded]) and quantified in the number of cell divisions from replicate wells (A, bottom). Data from one of three representative experiments are shown. (B) Cell culture supernatant from (A) was assayed for the presence of IFN-γ by ELISA, and the amount of IFN-γ (left) or fold-change resulting from the addition of TGF-β was quantified. The mean and standard error are depicted from data pooled from the average values determined in six independent experiments.
TGF-β-dependent conversion of DGK-ζ-deficient CD4+ T cells into a Treg phenotype is not impaired
To determine whether DGK-ζ-deficient CD4+ T cells are similarly insensitive to TGF-β, we tested the ability of TGF-β to “skew” naïve CD4+ T cells into a regulatory phenotype (Tregs), a well-established functional effect of TGF-β on naïve CD4+ T cells [18, 28]. Tregs, a CD4+ T cell subtype that plays an essential role in limiting immune activity in vivo, are characterized by expression of the transcription factor FoxP3 and high levels of surface expression of the high-affinity IL-2 receptor, CD25. To evaluate Treg skewing, we first bred DGK-ζ-deficient mice with FoxP3 reporter mice that express EGFP from the FoxP3 gene locus, which enables identification of Tregs based on intracellular expression of this fluorescent reporter. We then isolated naïve CD4+ T cells from these mice and incubated the cells with plate-bound anti-CD3 and anti-CD28 and/or IL-2 in the presence or absence of TGF-β for 72 h, after which Treg formation was assessed using flow cytometric determination of EGFP fluorescence and expression of the high affinity IL-2 receptor CD25. Both WT and DGK-ζ-deficient CD4+ T cells robustly converted into Tregs in the presence of TGF-β, under all stimulatory conditions tested, with a trend toward enhanced conversion in DGK-ζ-deficient CD4+ T cells (Fig. 2A). Because DGK-ζ-deficient T cells are known to secrete increased amounts of IL-2 on stimulation with anti-CD3 and anti-CD28 in a manner that could affect Treg skewing, we repeated our conversion assay by coincubating naïve CD4+ T cells of each genotype under IL2 stimulatory conditions. Similar to our initial experiment, DGK-ζ-deficient CD4+ T cells did not demonstrate impaired conversion into a Treg phenotype (Fig. 2B). Collectively, these data indicate that TGF-β insensitivity is differentially regulated in DGK-ζ-deficient T cells.
Figure 2. DGK-ζ-deficient CD4+ T cells are not impaired in Treg conversion.

Naïve (CD45RBhi, EGFP-negative) CD4+ T cells were isolated from the spleens of WT or DGK-ζ−/− FoxP3-EGFP mice by flow cytometry, and the cells were incubated with soluble α-CD28 (1 μg/ml) and 2.5 μg plate-bound α-CD3 with or without TGF-β (5 ng/ml) and IL-2 (1000 U/ml). At 72 h, expression of regulatory T cell markers (CD25 and EGFP) was assessed by flow cytometry. (A) Naïve T cells of each genotype were cultured independently. (B) Cells from each genotype were cultured together. Each line indicates a single experiment comparing WT (white circles) to DGK-ζ-deficient (black circles) T cells.
TGF-β-induced transcriptional changes are impaired in DGK-ζ-deficient T cells
To better characterize the nature of TGF-β insensitivity of CD8+ T cells, we next sought to determine whether TGF-β-mediated transcriptional changes were blunted in cells lacking DGK-ζ, because stimulation with TGF-β results in well-established alterations in transcription in CD8+ T cells stimulated with TCR [7]. Using published microarray data, we selected five genes known to be strongly up-regulated by TGF-β in CD8+ T cells and two gene changes known to be strongly down-regulated by TGF-β [7]. To determine whether the ability of TGF-β to alter the expression of these genes was affected by the loss of DGK-ζ in T cells, we measured the mRNA transcript levels for each of these genes in T cells stimulated with anti-CD3 in the presence or absence of TGF-β. We found that loss of DGK-ζ diminished TGF-β-induced changes in a subset of genes, such as TGF-β-RI, TGF-β-RII, Smad7, IFN-γ, and granzyme B, but not other genes, such as IFN-activated gene 203 (IFI203), or integrin-αe (ITGAE)in TCR-stimulated CD8+ T cells (Fig. 3). These data indicate that the loss of DGK-ζ impacts only a portion of the transcriptional program generated by TGF-β binding to its receptor in TCR-stimulated CD8+ T cells.
Figure 3. Loss of DGK-ζ attenuates a subset of TGF-β-mediated transcriptional changes in cytotoxic T cells.

WT or DGK-ζ-deficient CD8+ T cells were stimulated for 72 h with plate-bound α-CD3 (1 μg/ml) in the presence or absence of TGF-β (5 ng/ml), and changes in the mRNA levels of selected genes resulting from the presence of TGF-β was determined by RT-PCR. Data shown as the mean and standard error bars generated from data pooled from four independent experiments. The selected genes included TGF-β-RI and -RII, Smad7, INF activated gene 203 (IFI203), integrin-αe (ITGAE), IFN-γ, and granzyme B (GZMB). *P = 0.035, **P = 0.001, ***P < 0.001.
TGF-β-dependent signal transduction is unaffected by loss of DGK-ζ in CD8+ T cells
The finding that TGF-β-mediated functional and transcriptional changes were attenuated only in some instances in T cells from DGK-ζ-deficient mice suggested that loss of DGK-ζ did not act to globally inhibit canonical TGF-β signal transduction. Canonical TGF-β-dependent signal transduction is regulated in multiple ways in T cells, including regulation of TGF-β receptor subunits at both the mRNA and the protein levels, phosphorylation and nuclear translocation of Smad signaling mediators, and expression levels of inhibitory Smad proteins, such as Smad7 (for review, see Ref. 29). To determine whether canonical TGF-β-dependent signaling was disrupted in DGK-ζ-deficient CD8+ T cells, we first evaluated the mRNA and cell surface expression levels of the TGF-β receptor subunits TGF-β-RI and TGF-β-RII. We observed similar levels of transcripts for either component of the receptor heterodimer (Fig. 4A) and observed no difference in the level of surface expression of TGF-β-RII (Fig. 4B) between WT and DGK-ζ−/− CD8+ T cells. TGF-β binding to its receptor results in activation of TGF-β-RI and TGF-β-RII kinase activity and subsequent phosphorylation of receptor-associated transcription factors Smad2 and Smad3, leading to the nuclear translocation of Smad2/3 in complex with Smad4. Thus, we next evaluated whether activation and nuclear translocation of Smad was decreased in DGK-ζ-deficient CD8+ T cells. We performed these experiments in the presence or absence of concurrent T cell receptor stimulation, because this has been suggested to enhance the biochemical changes induced by TGF-β stimulation [15]. Nuclear extracts were obtained from WT and DGK-ζ-deficient cells that had been left untreated or treated with TGF-β and/or anti-CD3/CD28 for 1 h, and similar amounts of phosphorylated Smad2 were observed in the nucleus (Fig. 4C) of cells treated with TGF-β in the presence or absence of TCR. Moreover, similar increases in total nuclear Smad2 were observed between WT and DGK-ζ-deficient cells of each genotype under these treatment conditions (Fig. 4C). The purity of fractionation, assessed by immunoblotting for nuclear protein HDAC, was similar between the WT and DGK-ζ-deficient CD8+ T cells (data not shown).
Figure 4. Smad signal transduction downstream of TGF-β is unaffected by the loss of DGK-ζ in T cells.
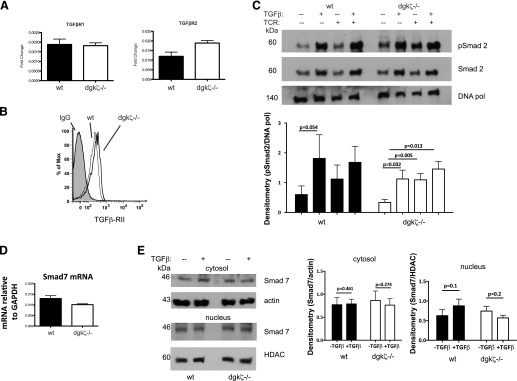
CD8+ T cells were isolated from spleens of WT or DGK-ζ-deficient mice and assayed either for TGF-β-RI or TGF-β-RII mRNA levels by RT-PCR (A) or cell surface expression levels of TGF-β-RII by flow cytometry (B). (C) CD8+ T cells from the mice of each genotype were stimulated for 1 h with or without TGF-β (10 ng/ml) and harvested (without TCR) or stimulated for an additional 30 min (with TCR) with 2.5 g/ml soluble α-CD3 (TCR). Fractionated lysates were assessed for nuclear levels of phosphorylated Smad2, total Smad2, or DNA polymerase (DNA pol) Representative data (top) and densitometry pooled data from four experiments (bottom) is shown. (D) mRNA levels of Smad7 were determined in WT or DGK-ζ-deficient CD8+ T cells was determined by RT-PCR averaged from three experiments. (E) Protein levels of Smad7 from cytosol or nucleus were determined in WT or DGK-ζ-deficient CD8+ T cells untreated or treated with 10 ng/ml TGF-β for 1 h. Representative data (left) and data pooled from six experiments (right) are depicted.
Smad7, an inhibitory Smad that provides negative feedback regulation of TGF-β signaling, is known to be upregulated by strong TCR-mediated signaling through PKCθ [15]. Additionally, others have reported that TGF-β signaling is inhibited in T cells deficient in CBL-B [14], an E3 ubiquitin ligase that, similar to DGK-ζ, serves to negatively regulate TCR signal transduction. The mechanism of loss of TGF-β sensitivity is postulated to result from decreased CBL-B-mediated degradation and in increased protein levels of Smad7 (for review, see [30]). To specifically address whether Smad7 levels were increased in DGK-ζ-deficient CD8+ T cells, we probed WT and DGK-ζ-deficient cells for mRNA and protein expression of Smad7. Consistent with the lack of Smad2 signaling changes observed in DGK-ζ-deficient CD8+ T cells, we observed insignificant differences in mRNA levels of Smad7 (Fig. 4D) or cytoplasmic or nuclear protein levels of Smad7 (Fig. 4E) between WT and DGK-ζ−/− CD8+ T cells. Collectively, these data indicate that TGF-β insensitivity does not result from altered Smad signaling in DGK-ζ-deficient CD8+ T cells.
TCR-mediated signal transduction in the presence of TGF-β is enhanced by loss of DGK-ζ in T cells
Because TGF-β insensitivity in DGK-ζ-deficient T cells could not be explained by defective TGF-β-dependent signaling, we hypothesized that the enhanced TCR signal transduction downstream of DAG that is observed with loss of DGK-ζ could be responsible for our results. Previously, it has been reported that TGF-β directly interferes with TCR signal transduction just upstream of PLC-γ1, the enzyme responsible for DAG generation [31], such that Erk1/2 phosphorylation and calcium flux, which are downstream of PLC-γ1, are diminished after TCR stimulation in T cells treated with TGF-β. In contrast, signaling events upstream of PLC-γ1, such as phosphorylation of Lck or Zap-70, were unaffected by the presence of TGF-β [28]. Consistent with this report, we observed that TCR stimulation in the presence of TGF-β led to a moderate, but reproducible, decrease in the phosphorylation of Erk1/2 (Fig. 5A) in the presence of TGF-β relative to its absence. In contrast, T cells deficient in DGK-ζ demonstrated enhanced phosphorylation of ERK in the absence or presence of TGF-β, such that the levels of protein phosphorylation in stimulated DGK-ζ-deficient CD8+ T cells incubated with TGF-β exceeded the levels of stimulated WT cells in the absence of TGF-β. These data support a hypothesis that increased TCR signal strength downstream of DAG is responsible for insensitivity to TGF-β.
Figure 5. TCR activation of DGK-ζ-deficient CD8+ T cells results in enhanced Erk and S6 phosphorylation in the presence or absence of TGF-β.

(A) WT or DGK-ζ-deficient CD8+ T cells were stimulated with soluble α-CD3 (2.5 μg/ml) with or without TGF-β (10 ng/ml) for 15 min, and lysates were probed for levels of phosphorylated or total Erk by immunoblotting (above dotted line). Because of artifactual photobleaching, total Erk appears diminished in blots previously probed for phosphorylated Erk. Subsequent immunoblotting of samples for total Erk without previous blotting for phosphorylated Erk demonstrated similar levels of total Erk proteins (below dotted line). Densitometry of phosphorylated Erk normalized to actin was calculated over six experiments (graph). (B) Splenic CD8+ T cells were isolated from WT and DGK-ζ-deficient mice, labeled with CFSE, and incubated with plate-bound α-CD3 (2.5 μg/ml) for 72 h in the presence of indicated concentrations of DAG analog (PMA). Proliferation, as assessed by dilution of CFSE, was determined by flow cytometry after gating on live CD8+ T cells. The data from one of three representative experiments are shown.
TCR signal strength determines sensitivity of CD8+ T cells to TGF-β-mediated inhibition
To the extent that increased TCR signal strength downstream of DAG is responsible for TGF-β insensitivity in DGK-ζ-deficient CD8+ T cells, TGF-β sensitivity should also be modulated in WT T cells by increasing DAG concentrations. To test this notion, we evaluated the ability of high concentrations of DAG to overcome the inhibitory effect of TGF-β on the proliferation of WT T cells. Although DAG itself is largely insoluble and has limited utility in experiments with intact cell surface membranes, the DAG analog PMA is widely used in cellular experiments, including T cells. Thus, we monitored proliferation of CFSE-labeled WT cells in the presence or absence of TGF-β with increasing concentrations of PMA. Although low doses of PMA (0.05 ng/μl) did not interfere with the ability of TGF-β to inhibit proliferation, increasing amounts of PMA abrogated TGF-β-mediated inhibition of proliferation (Fig. 5B). This finding is consistent with the conclusion that strong TCR signals downstream of DAG result in insensitivity to TGF-β.
We next evaluated the ability of low levels of TCR stimulation to render DGK-ζ-deficient CD8+ T cells sensitive to the inhibitory effects of TGF-β. TGF-β inhibited WT, but not DGK-ζ-deficient, CD8+ T cell responses to the concentrations of anti-CD3 normally used in T cell stimulation assays (e.g., 1 μg/ml), as assessed by proliferation (Fig. 6A) or intracellular generation of the effector molecule granzyme B (Fig. 6B). However, relative to their WT counterparts, DGK-ζ-deficient CD8+ T cells exhibited heightened responses to lower doses of anti-CD3, and TGF-β efficiently inhibited proliferation (Fig. 6A) and granzyme B generation (Fig. 6B). These data have further demonstrated that enhanced TCR signal strength is responsible for the blunted TGF-β responsiveness of DGK-deficient CD8+ T cells.
Figure 6. DGK-ζ-deficient CD8+ T cells are sensitive to TGF-β in the presence of low levels of TCR stimulation.

Splenic CD8+ T cells from WT and DGK-ζ-deficient mice were labeled with CFSE and stimulated with the indicated concentration of plate-bound α-CD3 for 72 h in the presence or absence of 5 ng/ml TGF-β. (A) T cell proliferation of live CD8+ T cells was determined with dilution of CFSE by flow cytometry for cells left untreated (unshaded) or treated (shaded) with TGF-β. (B) Intracellular protein levels of granzyme B was assessed in live CD8+ T cells after fixation and permeabilization. Data from one of three representative experiments are shown. Induction of granzyme B was not observed in unstimulated cells of either genotype (data not shown; n = 2).
DISCUSSION
Previously, we observed that deletion of DGK-ζ, a protein expressed primarily in cells of hematopoietic origin, leads to enhanced T cell-mediated clearance of tumor cells in vivo [25]. As a component of these studies, we cursorily examined the ability of T cells transduced with CARs, artificial proteins engineered to contain extracellular domains specific for tumor fused to intracellular domains capable of activating T cells, to respond to CAR stimulation in the presence of TGF-β, a cytokine known to potently inhibit CD8+ T cells specifically within the tumor microenvironment [6]. We determined that DGK-ζ-deficient CAR-T cells remained responsive to CAR antigen under certain conditions in the presence of TGF-β [25]. To establish that DGK-ζ-deficient T cells were insensitive to TGF-β after stimulation through TCR, we measured the ability of CD8+ T cells to proliferate and generate cytokines in response to plate-bound anti-CD3. We found that DGK-ζ-deficient CD8+ T cells demonstrated marked insensitivity to TGF-β. Moreover, we found that this was not a global feature of T cells, because DGK-ζ-deficient naïve CD4+ T cells could efficiently skew toward a Treg phenotype, an event highly dependent on the activity of TGF-β [2]. We also sought to explain how the loss of DGK-ζ conferred insensitivity to TGF-β in CD8+ T cells. Our initial perfunctory experiments with CAR-T cells and TGF-β also used two other potent inhibitors of CD8+ T cell function, adenosine, and PGE2 after exposure of T cells to CAR antigen [25]. In contrast to the insensitivity observed with TGF-β, DGK-ζ-deficient CAR-T cells remained equally sensitive to WT cells treated with adenosine and PGE2 after exposure of T cells to CAR antigen. This suggested that a unique relationship exists between TGF-β and TCR signal transduction pathways that is influenced by DGK-ζ.
The loss of DGK-ζ could have resulted either from direct inhibition of TGF-β signal transduction or through an alternate mechanism, such as enhanced TCR signaling. Although we hypothesized that DGK-ζ did not directly impact TGF-β signaling, because Treg skewing was unaffected in DGK-ζ-deficient mice and because not all TGF-β-induced gene changes were affected in DGK-ζ-deficient CD8+ T cells, recent data from two studies suggested a connection between TCR signal transduction and canonical TGF-β signaling. First, activation of TCR signaling has been demonstrated to induce upregulation of Smad7, with subsequent inhibition of effector Smad activation in a manner dependent on PKCθ, a TCR effector protein known, in part, to be regulated by DAG [15]. Second, deletion of CBL-B, an E3 ubiquitin ligase that plays an important role in terminating TCR signaling events by degrading critical signaling mediators (for review, see Ref. 30), results in TGF-β insensitivity of T cells through decreased degradation and increased protein levels of Smad7 [14]. In contrast to these studies, our results demonstrated that Smad7 protein levels are not increased in T cells deficient in DGK-ζ (Fig. 4D and E). Moreover, effector Smad2 phosphorylation and nuclear translation did not significantly differ between WT and DGK-ζ-deficient CD8+ T cells (Fig. 4C), indicating that the loss of DGK-ζ does not contribute to alterations in canonical TGF-β signaling.
As an alternative hypothesis, we tested whether the enhanced TCR signal transduction downstream of DAG that results from the loss of DGK-ζ could explain decreased sensitivity to TGF-β. The suppression of TCR signaling induced by TGF-β has been described previously by other groups, including direct evidence that phosphorylation of Erk1/2 is reduced after TCR stimulation in the presence of TGF-β [31]. Because DGK-ζ-deficient T cells have been well established to generate enhanced Erk1/2 activation after TCR stimulation [22, 23], it is plausible that the loss of DGK-ζ could “rescue” TGF-β-mediated reductions in Erk1/2 activation. Consistent with this notion, we observed that biochemical activation of Erk1/2 was increased in TGF-β-treated TCR-stimulated DGK-ζ-deficient T cells relative to TGF-β-untreated TCR-stimulated WT T cells (Fig. 5A). To further support the hypothesis that enhanced DAG-mediated signaling is responsible for TGF-β insensitivity in DGK-ζ-deficient T cells, we made two other predictions. First, we hypothesized that the addition of exogenous DAG should recapitulate TGF-β insensitivity, a prediction that we validated with an analysis of WT cell proliferation in the presence or absence of TGF-β after titration of the DAG analog PMA (Fig. 5B). Second, we predicted that reducing TCR signal strength with suboptimal stimulation would uncover conditions under which DGK-ζ-deficient T cells are sensitive to TGF-β, a prediction we also confirmed (Fig. 6). These data suggest that noncanonical TGF-β signaling results in direct inhibition of TCR signaling in a physiologically important manner and supports key discrepancies in the published data, including the observation that mice with T cells deficient in Smad3, an effector Smad critical for canonical TGF-β signaling, demonstrate an attenuated phenotype relative to mice deficient in TGF-β1 [19, 32, 33]. Additionally, these data indicate that pathways independent of Smad2/3 are important in facilitating TGF-β signaling in T cells. A critical question posed by our results is which signal transduction modules facilitate noncanonical TGF-β signal transduction and the resultant inhibition of TCR signaling. One potential model is that activation of TGF-β results in phosphorylation of ITIMs that facilitate recruitment of SHP-1 and/or SHP-2, inhibitory protein phosphatases that oppose proximal activating kinases such as Lck in T cells, resulting in attenuation of TCR signaling. Given that the TGF-β-RI is a serine/threonine kinase and that ITIMs are substrates for tyrosine kinases, however, this type of crosstalk would likely require multiple steps, including phosphorylation of a serine/threonine site by TGF-β-RI that subsequently opens an ITIM site for phosphorylation by a tyrosine kinase. The presence of an additional noncanonical TGF-β pathway in T cells could also explain why only a subset of TGF-β-mediated gene transcription is affected in DGK-ζ-deficient CD8+ T cells and why some functions of T cells (e.g., conversion of Treg) are unaffected by loss of DGK-ζ. Ongoing work in the laboratory has focused on attempting to identify the key steps that facilitate TGF-β crosstalk with TCR signaling.
TGF-β has been well established as an inhibitor of CD8+ T cells and Th1 CD4+ T cells and is an important method by which tumors evade effector immune responses [6, 7]. Thus, significant interest has developed toward targeting TGF-β signal transduction as a method to augment immune reactivity against tumor. These include anti-sense RNA or antibodies directed against TGF-β, antibodies directed against TGF-β-RI or -RII, inhibitors of TGF-β-RI or -RII kinase activity, and peptide aptamers against Smads (for review, see [34]). However, because TGF-β plays an important role in virtually every organ system, disrupting TGF-β systemically results in significant off-target effects, limiting its utility as a target for cancer therapy. In fact, in some tumor types, TGF-β is thought to play an important role in directly suppressing tumor cell growth [5]. Thus, the molecules required for TGF-β signaling selectively within T cells or cells of hematopoietic origin, such as DGK-ζ, represent an attractive method to selectively augment immune responses against tumor.
AUTHORSHIP
V.A., T.B., E.W., S.M., and M.J.R. conceived and designed the study. V.A., T.B., E.W., A.M.S., and M.J.R. acquired the data. V.A., T.B., E.W., A.M.S., T.K., S.M., and M.J.R. analyzed and interpreted the data. V.A., and M.J.R. drafted the manuscript. V.A., S.M., and M.J.R. performed the critical revision.
ACKNOWLEDGMENTS
This work was supported by the U.S. National Institutes of Health (Grant K08-CA151893); an Institutional Award from the American Cancer Society; and the Kathy Duffey Fogarty Award for Breast Cancer Research from the Medical College of Wisconsin Cancer Center (to M.J.R.). The authors thank Debra Newman (Blood Research Institute, Blood Center of Wisconsin, Milwaukee, WI) for scientific advice and their critical review of the manuscript. The authors also thank Demin Wang and Sridhar Rao (Blood Research Institute, Blood Center of Wisconsin) for their critical review of the manuscript.
Glossary
- CAR
chimeric-antigen receptor
- DAG
diacylglycerol
- DGK-ζ
diacylglycerol kinase-ζ
- EGFP
enhanced GFP
- HDAC
histone deacetylase
- MDSCs
myeloid-derived suppressor cells
- PLC-γ1
phospholipase C-γ1
- TGF-βR
TGF-β receptor
- Tregs
regulatory T cells
- SHP
Src homology region 2 domain-containing phosphatase
- WT
wild-type
Footnotes
SEE CORRESPONDING EDITORIAL ON PAGE 685
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1.Massagué J. (2012) TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wan Y. Y., Flavell R. A. (2007) “Yin-Yang” functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol. Rev. 220, 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oh S. A., Li M. O. (2013) TGF-β: guardian of T cell function. J. Immunol. 191, 3973–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flavell R. A., Sanjabi S., Wrzesinski S. H., Licona-Limón P. (2010) The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 10, 554–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bierie B., Moses H. L. (2010) Transforming growth factor beta (TGF-beta) and inflammation in cancer. Cytokine Growth Factor Rev. 21, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorelik L., Flavell R. A. (2001) Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat. Med. 7, 1118–1122. [DOI] [PubMed] [Google Scholar]
- 7.Thomas D. A., Massagué J. (2005) TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380. [DOI] [PubMed] [Google Scholar]
- 8.di Bari M. G., Lutsiak M. E. C., Takai S., Mostböck S., Farsaci B., Semnani R. T., Wakefield L. M., Schlom J., Sabzevari H. (2009) TGF-beta modulates the functionality of tumor-infiltrating CD8+ T cells through effects on TCR signaling and Spred1 expression. Cancer Immunol. Immunother. 58, 1809–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Principe D. R., Doll J. A., Bauer J., Jung B., Munshi H. G., Bartholin L., Pasche B., Lee C., Grippo P. J. (2014) TGF-β: duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 106, djt369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wikström P., Stattin P., Franck-Lissbrant I., Damber J. E., Bergh A. (1998) Transforming growth factor beta1 is associated with angiogenesis, metastasis, and poor clinical outcome in prostate cancer. Prostate 37, 19–29. [DOI] [PubMed] [Google Scholar]
- 11.Zhong Z., Carroll K. D., Policarpio D., Osborn C., Gregory M., Bassi R., Jimenez X., Prewett M., Liebisch G., Persaud K., Burtrum D., Wang S., Surguladze D., Ng S., Griffith H., Balderes P., Doody J., Schwartz J. D., Youssoufian H., Rowinsky E. K., Ludwig D. L., Witte L., Zhu Z., Wu Y. (2010) Anti-transforming growth factor beta receptor II antibody has therapeutic efficacy against primary tumor growth and metastasis through multieffects on cancer, stroma, and immune cells. Clin. Cancer Res. 16, 1191–1205. [DOI] [PubMed] [Google Scholar]
- 12.Mehra A., Wrana J. L. (2002) TGF-beta and the Smad signal transduction pathway. Biochem. Cell Biol. 80, 605–622. [DOI] [PubMed] [Google Scholar]
- 13.Moustakas A., Heldin C.-H. (2009) The regulation of TGFbeta signal transduction. Development 136, 3699–3714. [DOI] [PubMed] [Google Scholar]
- 14.Gruber T., Hinterleitner R., Hermann-Kleiter N., Meisel M., Kleiter I., Wang C. M., Viola A., Pfeifhofer-Obermair C., Baier G. (2013) Cbl-b mediates TGFβ sensitivity by downregulating inhibitory SMAD7 in primary T cells. J. Mol. Cell Biol. 5, 358–368. [DOI] [PubMed] [Google Scholar]
- 15.Giroux M., Delisle J.-S., O’Brien A., Hébert M.-J., Perreault C. (2010) T cell activation leads to protein kinase C theta-dependent inhibition of TGF-beta signaling. J. Immunol. 185, 1568–1576. [DOI] [PubMed] [Google Scholar]
- 16.Zi Z., Chapnick D. A., Liu X. (2012) Dynamics of TGF-β/Smad signaling. FEBS Lett. 586, 1921–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L., Yu Z., Muranski P., Palmer D. C., Restifo N. P., Rosenberg S. A., Morgan R. A. (2013) Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. 20, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takimoto T., Wakabayashi Y., Sekiya T., Inoue N., Morita R., Ichiyama K., Takahashi R., Asakawa M., Muto G., Mori T., Hasegawa E., Saika S., Hara T., Nomura M., Yoshimura A., Yoshimura A. (2010) Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J. Immunol. 185, 842–855. [DOI] [PubMed] [Google Scholar]
- 19.Delisle J.-S., Giroux M., Boucher G., Landry J.-R., Hardy M.-P., Lemieux S., Jones R. G., Wilhelm B. T., Perreault C. (2013) The TGF-β-Smad3 pathway inhibits CD28-dependent cell growth and proliferation of CD4 T cells. Genes Immun. 14, 115–126. [DOI] [PubMed] [Google Scholar]
- 20.Joshi R. P., Koretzky G. A. (2013) Diacylglycerol kinases: regulated controllers of T cell activation, function, and development. Int. J. Mol. Sci. 14, 6649–6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krishna S., Zhong X. (2013) Role of diacylglycerol kinases in T cell development and function. Crit. Rev. Immunol. 33, 97–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong X.-P., Hainey E. A., Olenchock B. A., Jordan M. S., Maltzman J. S., Nichols K. E., Shen H., Koretzky G. A. (2003) Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat. Immunol. 4, 882–890. [DOI] [PubMed] [Google Scholar]
- 23.Riese M. J., Grewal J., Das J., Zou T., Patil V., Chakraborty A. K., Koretzky G. A. (2011) Decreased diacylglycerol metabolism enhances ERK activation and augments CD8+ T cell functional responses. J. Biol. Chem. 286, 5254–5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prinz P. U., Mendler A. N., Masouris I., Durner L., Oberneder R., Noessner E. (2012) High DGK-α and disabled MAPK pathways cause dysfunction of human tumor-infiltrating CD8+ T cells that is reversible by pharmacologic intervention. J. Immunol. 188, 5990–6000. [DOI] [PubMed] [Google Scholar]
- 25.Riese M. J., Wang L.-C. S., Moon E. K., Joshi R. P., Ranganathan A., June C. H., Koretzky G. A., Albelda S. M. (2013) Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res. 73, 3566–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haribhai D., Lin W., Relland L. M., Truong N., Williams C. B., Chatila T. A. (2007) Regulatory T cells dynamically control the primary immune response to foreign antigen. J. Immunol. 178, 2961–2972. [DOI] [PubMed] [Google Scholar]
- 27.Zou T., Caton A. J., Koretzky G. A., Kambayashi T. (2010) Dendritic cells induce regulatory T cell proliferation through antigen-dependent and -independent interactions. J. Immunol. 185, 2790–2799. [DOI] [PubMed] [Google Scholar]
- 28.Chen W., Jin W., Hardegen N., Lei K.-J., Li L., Marinos N., McGrady G., Wahl S. M. (2003) Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rubtsov Y. P., Rudensky A. Y. (2007) TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat. Rev. Immunol. 7, 443–453. [DOI] [PubMed] [Google Scholar]
- 30.Wallner S., Gruber T., Baier G., Wolf D. (2012) Releasing the brake: targeting Cbl-b to enhance lymphocyte effector functions. Clin. Dev. Immunol. 2012, 692639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen C.-H., Seguin-Devaux C., Burke N. A., Oriss T. B., Watkins S. C., Clipstone N., Ray A. (2003) Transforming growth factor beta blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J. Exp. Med. 197, 1689–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kulkarni A. B., Huh C. G., Becker D., Geiser A., Lyght M., Flanders K. C., Roberts A. B., Sporn M. B., Ward J. M., Karlsson S. (1993) Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 90, 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shull M. M., Ormsby I., Kier A. B., Pawlowski S., Diebold R. J., Yin M., Allen R., Sidman C., Proetzel G., Calvin D., Annunziata N., Doetschman T. (1992) Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 359, 693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith A. L., Robin T. P., Ford H. L. (2012) Molecular pathways: targeting the TGF-β pathway for cancer therapy. Clin. Cancer Res. 18, 4514–4521. [DOI] [PubMed] [Google Scholar]