

ABSTRACT
In all three domains of life, organisms use nonspecific DNA-binding proteins to compact and organize the genome as well as to regulate transcription on a global scale. Histone is the primary eukaryotic nucleoprotein, and its evolutionary roots can be traced to the archaea. However, not all archaea use this protein as the primary DNA-packaging component, raising questions regarding the role of histones in archaeal chromatin function. Here, quantitative phenotyping, transcriptomic, and proteomic assays were performed on deletion and overexpression mutants of the sole histone protein of the hypersaline-adapted haloarchaeal model organism Halobacterium salinarum. This protein is highly conserved among all sequenced haloarchaeal species and maintains hallmark residues required for eukaryotic histone functions. Surprisingly, despite this conservation at the sequence level, unlike in other archaea or eukaryotes, H. salinarum histone is required to regulate cell shape but is not necessary for survival. Genome-wide expression changes in histone deletion strains were global, significant but subtle in terms of fold change, bidirectional, and growth phase dependent. Mass spectrometric proteomic identification of proteins from chromatin enrichments yielded levels of histone and putative nucleoid-associated proteins similar to those of transcription factors, consistent with an open and transcriptionally active genome. Taken together, these data suggest that histone in H. salinarum plays a minor role in DNA compaction but important roles in growth-phase-dependent gene expression and regulation of cell shape. Histone function in haloarchaea more closely resembles a regulator of gene expression than a chromatin-organizing protein like canonical eukaryotic histone.
IMPORTANCE
Histones comprise the major protein component of eukaryotic chromatin and are required for both genome packaging and global regulation of expression. The current paradigm maintains that archaea whose genes encode histone also use these proteins to package DNA. In contrast, here we demonstrate that the sole histone encoded in the genome of the salt-adapted archaeon Halobacterium salinarum is both unessential and unlikely to be involved in DNA compaction despite conservation of residues important for eukaryotic histones. Rather, H. salinarum histone is required for global regulation of gene expression and cell shape. These data are consistent with the hypothesis that H. salinarum histone, strongly conserved across all other known salt-adapted archaea, serves a novel role in gene regulation and cell shape maintenance. Given that archaea possess the ancestral form of eukaryotic histone, this study has important implications for understanding the evolution of histone function.
INTRODUCTION
Cells of known organisms across the three domains of life compact their genomes, since the extended length of genetic material exceeds the volume of the cell or nucleus. Eukaryotic genomes are primarily packaged into nucleosome particles consisting of a histone protein octamer and approximately 150 bp of DNA (1). Histone proteins generally contain a core histone fold domain consisting of three α-helices and an N-terminal tail. Covalent modifications of the histone tail, and in some cases the histone core, change the strength of the association of the histone octamer with DNA. Modification leads to passive regulation of gene expression by modulating accessibility to the basal transcriptional apparatus (2, 3). Like histones, bacterial nucleoid-associated proteins (NAPs; e.g., HU, H-NS, and Fis) bind DNA nonspecifically, causing compaction (4, 5). These DNA-NAP interactions are dynamic throughout the growth curve and affect gene expression (5, 6).
Much research on archaeal chromatin focuses on the role of histones in DNA packaging. The studies that have been conducted on archaeal histones demonstrated that proteins homologous to eukaryotic histones H3 and H4 are found in the genomes of most Euryarchaea and members of the early-branching phyla Nanoarchaea, Korarchaeota, and Thaumarchaeota but only some Crenarchaea (7, 8). Euryarchaeal histone crystal structures from Methanothermus fervidus and Methanopyrus kandleri strongly resemble those of the eukaryotic tetramer, an intermediate in nucleosome assembly (9, 10). In vitro experiments have confirmed that archaeal histones compact DNA into nucleosome-like particles, which form tetramers rather than octamers (9, 11, 12). Although in vitro evidence suggests that archaeal nucleosomes may inhibit transcription in a manner similar to that of eukaryotic tetramers (13–15), in vivo gene expression experiments have yielded variable results in different organisms (16, 17).
Several attributes of archaeal histone proteins complicate the interpretation of their function in genome organization and gene expression. First, archaeal histones lack both N-terminal tails and posttranslational modifications of the core domain (7, 18). Second, in addition to eukaryotic-type histone proteins, most archaeal genomes encode a multitude of NAPs, including bacterial packaging proteins such as DpsA and archaeon-specific packaging proteins such as Alba, Sul7d, and MC1 (8). MC1, a small nonspecific DNA-binding protein, is preferentially used over histone as the principal chromatin-packaging protein in Methanosarcina species (17, 19, 20).
According to whole-genome sequence data (21, 22), genomes of members of the haloarchaeal clade of Euryarchaeota encode a single histone protein. Histones from these hypersaline-adapted organisms (and a single methanogenic species, M. kandleri [10]) contain two histone fold domains fused together with a linker region, which has been suggested as an evolutionary intermediate between archaeal and eukaryotic histones (7, 8, 23). The haloarchaeal histone protein is primarily comprised of acidic amino acids (pI, 4.4) as an adaptation to high intracellular salt (22, 24), unlike all other histones and NAPs. For example, histone proteins are typically among the most basic proteins in a eukaryotic cell (the pI of Homo sapiens H3 is 11.71; that of Saccharomyces cerevisiae H3 is 11.90) and other archaea (M. fervidus HMfA, 9.21).
Despite these intriguing differences, the physiological function of histone proteins in halophilic archaea remains to be investigated. The few studies that have been done on haloarchaeal chromatin suggest nucleosome formation. Electron micrographs of sucrose gradients from the model haloarchaeal species Halobacterium salinarum have demonstrated the growth-dependent presence of two different populations of DNA: protein-bound fractions with a “beads-on-a-string” appearance predominate in stationary phase and protein-free fractions predominate in log phase (25, 26). More recent micrococcal nuclease sequencing (MNase-seq) experiments in Haloferax volcanii suggest regular phasing in gene expression and protein occupancy surrounding transcriptional start sites (27). However, the identity of proteins causing these patterns and the physiological function of haloarchaeal histones remain unclear.
In order to shed light on the function of histone protein in the haloarchaea, we constructed deletion and overexpression mutants of hpyA (VNG0134G), encoding the sole histone of H. salinarum NRC-1. Histone deletion and overexpression mutants are viable, and hpyA deletion increases the growth rate in batch culture without affecting susceptibility to several types of stress. Unexpectedly, histone dosage appears to be linked to cell shape and size, a function not previously observed in the archaea. Genome-wide expression profiling in histone mutants suggests that histone has a bifunctional, growth-phase-specific effect on transcription. Identification of proteins from sucrose-fractionated DNA yielded no evidence to support the existence of eukaryotic-type chromatin. Instead, proteomics and gene expression analysis support a model in which a dynamic population of low-abundance NAPs functions in open chromatin throughout growth. Together, our experimental results suggest that the histone protein of H. salinarum functions primarily as a modulator of transcription required for wild-type morphology and growth rate.
RESULTS
HpyA, a fused histone heterodimer, is conserved throughout the halophilic archaea.
A single histone homolog gene, VNG0134G (hpyA), is detectable in the genome of Halobacterium salinarum (22). The protein consists of two histone fold domains arranged in tandem within a single open reading frame (ORF). To determine the extent of homology with canonical histones, the amino acid sequences for each of the N- and C-terminal domains of HpyA were compared with well-characterized histone proteins from Homo sapiens, Saccharomyces cerevisiae, and Methanothermus fervidus (Fig. 1A). Because archaeal histones lack N-terminal tails (7), these tail domains were removed from the eukaryotic histones prior to alignment. As expected, the HpyA amino acid sequence resembles that of canonical eukaryotic histones (e.g., N- and C-terminal domains are 23.6% and 27.9% identical to yeast H3, respectively). Importantly, the majority of core residues thought to be essential for cell viability and for the structure of the histone fold were conserved in both domains of HpyA (Fig. 1A) (9, 28–30). To investigate conservation of histones across the haloarchaeal order Halobacteriales, the amino acid sequences of histones from nearly 70 haloarchaeal genomes currently curated in the NCBI database were aligned (21) (see Text S1 in the supplemental material). While the linker region between the two domains is highly degenerate, the fused histone heterodimer conformation and essential residues are conserved across all haloarchaeal sequences examined (Fig. 1B).
FIG 1 .

Haloarchaeal species encode a single conserved histone heterodimer. (A) Alignment of histone protein sequences: the eukaryotic H3 core domains from Homo sapiens and Saccharomyces cerevisiae, HMfA from Methanothermus fervidus, and each of two histone fold domains of H. salinarum HpyA. Residues essential for histone function or cell viability in the Eukarya and/or Archaea are highlighted in purple. Gray bars indicate the locations of the three alpha-helical regions that comprise the histone fold domain of the canonical eukaryotic H3. Asterisks, fully conserved residues; colons, strongly similar residues; periods, weakly similar residues. (B) Consensus logo representing histone sequence conservation between haloarchaeal species. The linker region between histone fold domains (yellow-shaded residues) is variable in length and sequence. Residue colors are as follows: red, acidic; black, uncharged; blue, basic. (C) Space-filling representation of the surface charge of H. salinarum HpyA and in silico fusion of the S. cerevisiae H3 to H4 dimer modeled onto the M. kandleri histone HMk crystal structure (10). Blue indicates a positive charge, whereas red indicates a negative charge (see scale at top).
An unusual feature of haloarchaeal histones is the preponderance of acidic amino acids relative to the histones of eukaryotes and to the histones of other archaea. This acidity is approximately evenly distributed between the two histone fold domains, although the N-terminal domain is significantly more basic on average (4.62 versus 4.36, P = 0.005). In order to predict whether this acidity could affect histone function, we projected the amino acid sequence of HpyA onto the only other known fused histone heterodimer crystal structure from Methanopyrus kandleri HMk (10) and calculated the surface electrostatic potential. As a comparison to the eukaryotic histone dimer, S. cerevisiae H3 and H4 were also modeled onto the HMk structure (see Materials and Methods). Surprisingly, unlike eukaryotic histones, HpyA is a highly polarized molecule. Our model illustrates that although HpyA is a highly acidic protein, it also contains an isolated basic region largely consisting of arginine residues from both histone fold domains (Fig. 1C). These analyses suggest that, despite its clear conservation with the eukaryotic H3 protein sequence, evolutionary adaptations for high salt have made fundamental structural changes that may affect the function of HpyA.
Phenotypic characterization of HpyA function in H. salinarum physiology.
To gain insight into the function of the haloarchaeal histone protein, a strain deleted in frame for hpyA was constructed (VNG0134G; Δura3 ΔhpyA; see Materials and Methods; strain list in Table 1). Out of 110 clones screened, three were heterozygous (H. salinarum is highly polyploid [31]), and three were deleted for the hpyA ORF as confirmed by PCR and Southern blotting (Fig. 2A and B). This rate of isolation of deletion mutants is consistent with those of known nonessential genes (32). Clonal isolates of a single, in-frame, homozygous deletion mutant were used in all subsequent experiments. To explore the effects of altered histone dosage on cell physiology and gene expression, a histone overexpression strain was generated in which a strong constitutive promoter drives ectopic hpyA expression in the Δura3 parent background (Δura3/pKAD02). Histone overexpression strains were obtained for all clones screened. To examine the phenotypic effects of deletion and overexpression of the histone coding gene in H. salinarum, growth and survival were measured under optimum and stress conditions. Unexpectedly, the histone mutant growth rate under optimum growth conditions is significantly higher (n = 27, P = 0.0009) than that of the parent strain (Fig. 2C, left). In contrast, the growth rate of the Δura3/pKAD02 histone overexpression strain is not significantly different (n = 3, P = 0.98) from that of the Δura3 strain containing the empty vector control (Fig. 2C, right). No significant differences in survival were observed between the Δura3 ΔhpyA strain and the parent strain under a variety of stress conditions, including oxidative stress, UV, shear stress, bacitracin, and novobiocin (see Text S2 and Fig. S1 in the supplemental material). Taken together, these results suggest that, although hpyA encodes the sole histone in this organism, it is not essential.
TABLE 1 .
Plasmids and strains used in this study
| Strain or plasmid | Description or genotype | Purpose or description | Reference or source |
|---|---|---|---|
| Plasmids | |||
| pMTFCHA | Pfdx Mevr | Expression vector | 52 |
| pRSK01 | pNBK07::attB1,attB2, Mevr Ura+ | Genomic integration vector | 52 |
| pKAD01 | pRSK01::ΔhpyA | Deletion of hpyA | This study |
| pKAD02 | pMTFCHA::hpyA | Overexpression of hpyA | This study |
| pKAD03 | pMTFCHA::Prpa200::hpyA | Complementation of ΔhpyA | This study |
| Strains | |||
| MDK407 | Δura3 | Parent strain, Ura− | 53 |
| KAD100 | Δura3 ΔhpyA | Histone null mutant, Ura− | This study |
| KAD101 | Δura3/pMTFCHA | Ura− Mevr | This study |
| KAD102 | Δura3/pKAD02 | hpyA overexpression, Ura− Mevr | This study |
| KAD103 | Δura3 ΔhpyA/pKAD03 | ΔhpyA complementation, Ura− Mevr | This study |
FIG 2 .
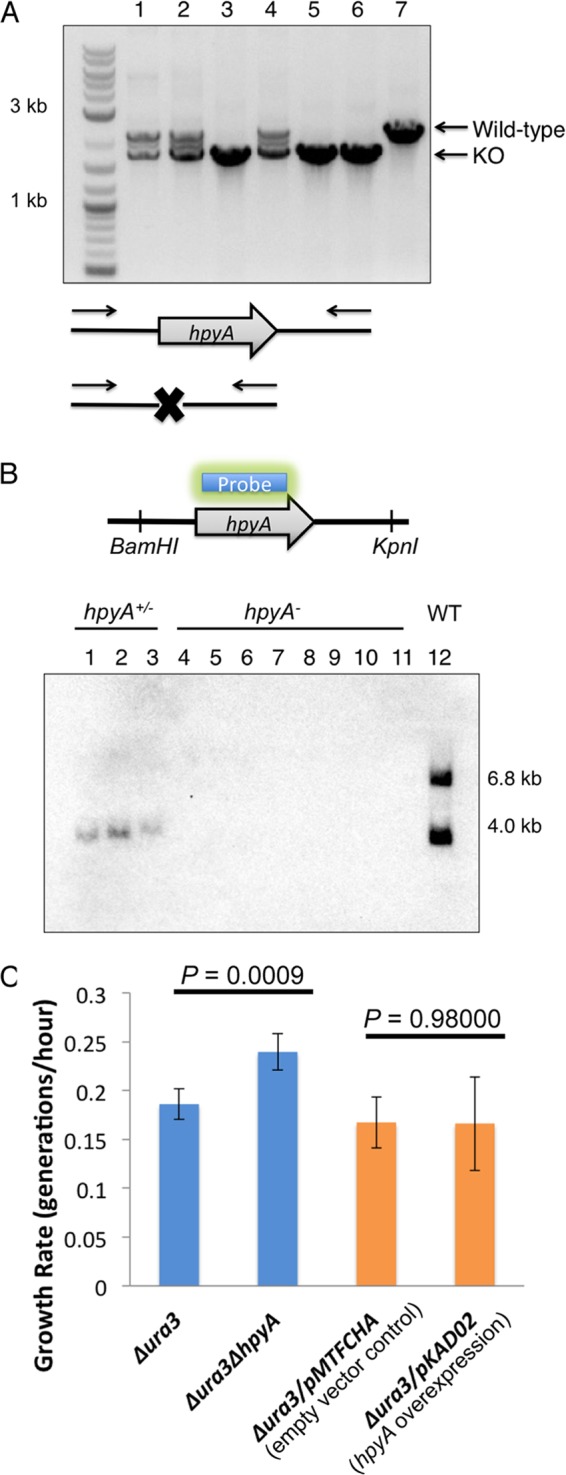
Histone knockout mutants are readily generated and viable. (A) PCR validation of transformants: lanes 1, 2, and 4 are heterozygous at the hpyA locus; lanes 3, 5, and 6 are deletion mutants; and lane 7 corresponds to the Δura3 parent strain. Wild-type amplicon, 2.1 kb; knockout (KO), 1.7 kb. (B) Southern blotting of restriction enzyme-digested genomic DNA using a probe internal to the hpyA ORF confirms that knockouts are homozygous. Lanes 1, 2, and 3 correspond to the heterozygous mutants; lanes 4 to 9 are biological replicates used in this study; lanes 10 and 11 are independently isolated histone deletion mutant clones; and lane 12 is a partial digest control of Δura3 parent strain genomic DNA. Primer sequences for both screens are listed in Table S5 in the supplemental material. WT, wild type. (C) The Δura3 ΔhpyA histone deletion strain grows significantly faster than the parent Δura3 strain (blue bars), while the Δura3/pKAD02 hpyA overexpression strain is indistinguishable from the Δura3/pMTFCHA empty vector control (orange bars). Error bars represent standard deviations from the means for three biological replicate cultures, each with three technical replicates.
In order to determine the morphological effects of histone deletion, all four strains described above were visualized using phase-contrast microscopy during log (optical density at 600 nm [OD600] of 0.3 to 0.5) and stationary (OD600 of ≥1.3) phases of growth. As expected, the Δura3 parent strain and the Δura3/pMTFCHA empty vector control are rod shaped regardless of growth phase (Fig. 3). Surprisingly, histone mutant cells are pleomorphic: cells range from wild-type rods to triangular, circular, and irregular forms (Fig. 3A and B). As a proxy for the measurement of pleomorphism in the population, the width/length ratio was calculated for individual cells. The histone knockout mutant cells exhibited a significantly higher width/length ratio than did the parent strain in both growth phases tested (log-phase mean value for parent is 0.23; log-phase mean value for mutant is 0.40; P = 1.3 × 10−13) (see also Table S1 in the supplemental material for P values of t test comparisons for all strains and growth phases), even though the parent strain exhibited a slightly increased width/length ratio in stationary phase. Ectopic expression of hpyA under the native promoter (levels 0.2-fold lower than those of endogenous hpyA in log phase and 8-fold higher in stationary phase [see Fig. S2]) significantly complements the deletion strain morphological phenotype (Δura3 parent versus ΔhpyA/pKAD03 complemented strain, P = 5.04 × 10−2; ΔhpyA strain versus complemented strain, P = 1.65 × 10−8), ruling out polar effects on surrounding genes or second-site mutations (Fig. 3A and B; see Materials and Methods and also Table S1). In contrast to Δura3 ΔhpyA cells, Δura3/pKAD02 cells overexpressing hpyA exhibited a dramatic increase in length in log phase (11.5 ± 1.16 µm, standard error) relative to the empty vector control (5.8 ± 0.26 µm, standard error). Unlike the Δura3 ΔhpyA pleomorphic phenotype, enlargement of the Δura3/pKAD02 strain is largely ameliorated during stationary phase (Fig. 3C and D). Wild-type dimensions are restored in strains cured of the hpyA overexpression plasmid (Fig. 3C and D), confirming that the observed phenotypes are due to overexpression alone. Taken together, these phenotypic analyses suggest that, although histones are not essential for cell viability, batch culture growth, or resistance to the stressors tested here, they are required to regulate cell morphology in H. salinarum.
FIG 3 .

Histone dosage regulates cell shape and size. (A) Δura3 ΔhpyA cells are pleomorphic. Phase-contrast light microscopy images are shown for the Δura3 parent strain versus the Δura3 ΔhpyA histone deletion mutant and the Δura3/pMTFCHA control strain versus the Δura3 ΔhpyA/pKAD03 complemented strain in log or stationary phase of growth. (B) Individual cells were fitted as ellipses, and the width/length ratio was calculated. Error bars represent the first and fourth quartiles. For quantitation of log-phase cultures, n was ≥113 cells for each biological replicate sample. For stationary-phase cultures, n was ≥227 cells. Bar, 10 µm. (C) hpyA overexpression cells are larger than isogenic parent cells during logarithmic growth. Phase-contrast light microscopy images of the Δura3/pMTFCHA strain versus the Δura3/pKAD02 strain (histone overexpression) at mid-log or stationary phase of growth are shown. (D) Individual cells were fitted as ellipses, and area (length × width) was calculated. Error bars are as described for panel B. For quantitation of log-phase cultures, n was ≥76 cells. For stationary-phase cultures, n was ≥272 cells.
Histone deletion results in growth-phase-specific effects on gene expression.
To investigate whether histone regulates gene expression in H. salinarum, we compared genome-wide mRNA expression profiles between the Δura3 parent strain and the Δura3 ΔhpyA strain at mid-logarithmic and stationary phases of growth. Thirty-seven genes (1.5% of predicted ORFs in the genome) exhibited significantly altered expression in logarithmic phase, whereas 220 (6.5%) exhibited significantly altered expression in stationary phase (Fig. 4A and B). Strikingly, only seven genes were regulated by histone at both time points tested (see Table S2 in the supplemental material). Expression data were corroborated using reverse transcription quantitative PCR (RT-qPCR) (Pearson correlation of microarray and PCR data = 0.78 [see Fig. S2]). In log phase, we observed only downregulation of gene expression in the histone mutant compared to its isogenic parent strain, suggesting that histone activates gene expression during this phase. In contrast, during stationary phase the majority of significantly regulated genes are repressed in the histone deletion strain relative to the parent strain (66%), while the remainder are overexpressed. This pattern suggests a bifunctional regulatory role for histone during this phase with a tendency toward activation. Together, these results suggest that histone is required for wild-type gene expression levels and that the mechanism of its action is phase specific.
FIG 4 .
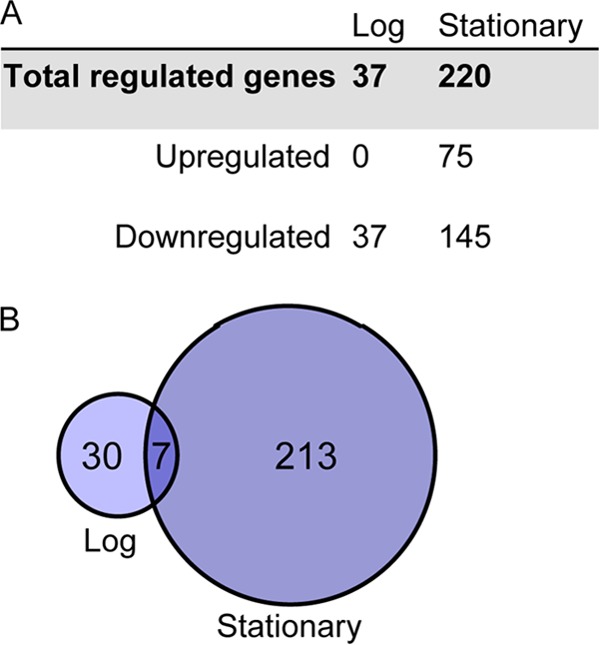
Histone-regulated gene expression is growth phase dependent. (A) The number of genes differentially expressed in the Δura3 ΔhpyA strain versus the Δura3 parent strain is dependent on the phase of growth. (B) Of the histone-regulated genes, only 7 are affected by histone deletion in both logarithmic and stationary phases. Circles in the Venn diagram are scaled by gene number.
Histone regulates genes in many processes, including archaeal cell wall (S-layer) and membrane synthesis.
No significant functional enrichment for histone-regulated genes in either gene ontology terms or archaeal clusters of orthologous genes (GO and arCOGs, respectively [33]) (data not shown) was detected, suggesting that histone regulates genes in a wide range of cellular processes. However, out of 220 genes differentially regulated in stationary phase, many (3%) are putatively involved in cell wall or membrane biogenesis. Those with experimentally characterized cell wall functions in haloarchaea are repressed in the Δura3 ΔhpyA strain relative to the parent strain (Table 2), suggesting that these genes require HpyA for activation. Further analysis of proteins encoded by these genes revealed strong homology to enzymes required to glycosylate the cell surface S-layer and maintain cell shape in the related halophile Haloferax volcanii (34) (Table 2). Importantly, the csg product, the major protein component of the S-layer, is also downregulated in stationary-phase histone deletion mutants. However, we were unable to detect significant differences in membrane integrity or S-layer glycosylation between the histone deletion mutant and the Δura3 parent strain (see Text S2 and Fig. S1 in the supplemental material). Together, these results suggest that while histone is involved in regulation of genes in many different processes, it also regulates the expression of key components of the cell wall synthesis machinery.
TABLE 2 .
Histone-regulated genes associated with S-layer synthesis and glycosylation
| Gene ID | Annotationa | GE ratiob | Homologc | E value |
|---|---|---|---|---|
| VNG0001H | S-layer domain | −1.17 | NA | NA |
| VNG0062G | Glycosyltransferase | −1.59 | NA | NA |
| VNG1048G | Predicted UDP-glucose 6-dehydrogenase | −1.64 | aglM, HVO1531 | 2.00E−108 |
| VNG1055G | dTDP-glucose pyrophosphorylase | −1.02 | aglF, HVO1527 | 4.00E−108 |
| VNG1062G | Glycosyltransferase | −1.23 | aglI, HVO1528 | 3.00E−46 |
| VNG2679G | Cell surface protein | −1.05 | csg, HVO2072 | 2.00E−155 |
Annotations from arCOG (33). Details of arCOG annotations for each differentially expressed gene are listed in Table S2 in the supplemental material.
GE ratio, log2 gene expression ratio of ΔhpyA::Δura3 during stationary phase.
Homolog, experimentally characterized homolog with greatest amino acid similarity to encoded proteins. HVO numbers correspond to gene identifiers in the Haloferax volcanii genome (68). NA, no experimentally characterized homolog.
Proteomics analysis of fractionated cells is consistent with open chromatin.
In order to identify the principal protein components of chromatin in H. salinarum, chromatin was fractionated via sucrose gradient ultracentrifugation. Two major peaks of nucleic acid were detected in sucrose fractions by absorbance at 260 nm. Analysis by nuclease digestion in parallel samples revealed that lower-density sucrose fractions 1 to 6 consist of RNA (see Fig. S3 in the supplemental material), whereas mid-density fractions 8 to 14 contain most of the DNA content and include an additional population of heavier RNA. Protein from the DNA fractions, as well as control protein without DNA from lighter fractions, was isolated from mid-logarithmic and stationary-phase samples (Fig. 5). A total of 1,256 unique proteins were identified in all samples using liquid chromatography-tandem mass spectrometry (LC-MS/MS) (~53% total coverage of the H. salinarum proteome [see Table S3]). This is a highly sensitive detection level relative to other proteomics experiments conducted thus far for H. salinarum (35). Surprisingly, the majority of proteins detected exclusively in the DNA fractions throughout growth were putatively membrane associated (43.5%) rather than DNA associated as expected (3.8% transcription factors and putative NAPs [see Table S3]). Furthermore, the five putative NAPs with strong homology to those important for chromatin function in bacteria and other archaea (HpyA, MC1, Sph1, Sph2, and DpsA) were present in low abundance in both DNA and control fractions across both log and stationary growth phases: less than 0.41% of total observed peptides was detected for each (Table 3). Histone protein (HpyA) accounted for only 0.06% of the observed peptides in any given sample. A linear model was used to determine which proteins common to both DNA and control populations were significantly enriched over expected frequency in the DNA fractions (see Materials and Methods). Of those proteins enriched in the DNA fractions, 69% of proteins enriched in log phase and 68% of proteins enriched in stationary phase are annotated as ribosome or proteasome subunits (see Table S4). This is consistent with the behavior of rapidly sedimenting large protein complexes. Therefore, previous reports of rapidly sedimenting DNA in H. salinarum lysate (26) as well as the observations made here are unlikely to be primarily due to the presence of DNA-binding proteins. Taken together, these proteomics results are consistent with open, transcriptionally active chromatin throughout growth, rather than chromatin compacted through the binding action of NAP or histone.
FIG 5 .
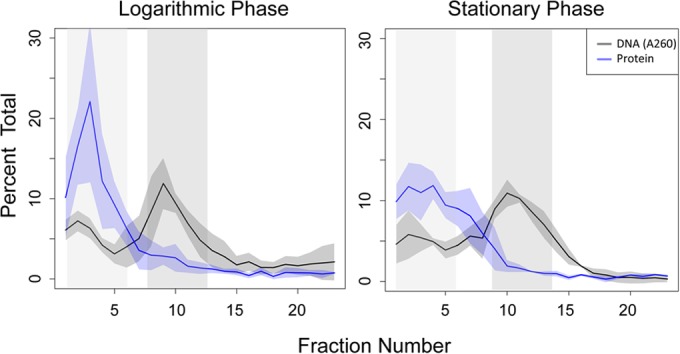
Sucrose gradient profiles of DNA and protein from each phase of growth. Proteins present in low-density fractions containing RNA (light-gray-shaded region) and higher-density fractions containing DNA (dark-gray-shaded region) were identified by tandem mass spectrometry. Nucleic acid profiles (black line; A260) and protein profiles (blue line; Bradford assay) represent the means from 3 to 4 biological replicate samples; the shaded area around each line indicates the standard deviation from the mean. Fraction 25, corresponding to unsheared lysate (26), is not shown for clarity.
TABLE 3 .
Proportion of total peptides detected for each putative nucleoid-associated protein in DNA and control sucrose fractions
| Protein name | Description | Molecular mass (kDa) | % of peptides observed at phase: |
|||
|---|---|---|---|---|---|---|
| Log |
Stationary |
|||||
| Control | DNA | Control | DNA | |||
| HpyA | Archaeal histone | 16 | 0.06 | 0.02 | 0.03 | 0.02 |
| DpsA | DNA protection during starvation protein | 20 | 0.13 | 0.12 | 0.09 | 0.15 |
| MC1 | Archaeal NAP | 12 | 0.06 | 0.02 | 0.07 | 0.02 |
| Sph1 | Smc-like protein | 71 | 0.13 | 0.41 | 0.20 | 0.24 |
| Sph2 | Smc-like protein | 76 | 0.21 | 0.28 | 0.05 | 0.07 |
Expression of putative haloarchaeal NAPs suggests a dynamic DNA landscape.
Gene expression and shape changes in histone mutants are growth phase dependent (Fig. 3 and 4), but NAPs and histone proteins were detected in both growth phases (Table 3; see also Table S3 in the supplemental material). We reasoned that, because the proteomics experiment was semiquantitative, this difference might be due to dynamic changes in expression of NAPs throughout growth. To test this, we reanalyzed wild-type and Δura3 strain data from previously published experiments in which gene and protein expression was monitored at several time points throughout growth in batch culture (36) and during oxygen and growth rate perturbations in a turbidostat (37), respectively. Clear differences in expression of the five putative NAPs discussed above (Table 3) were observed between log growth and stationary phase (Fig. 6A). Expression of hpyA mRNA was higher during rapid growth than slow growth. In contrast, dpsA and mc1 expression dynamics were anticorrelated with those of hpyA (Fig. 6A). Expression of sph1 and sph2 was modestly affected by growth. Similar patterns were observed for the proteins encoded by these genes in quantitative proteomics data (Fig. 6B). In the histone mutant background, mc1 expression is slightly repressed during stationary phase (P < 0.05; see Table S2), suggesting a regulatory interrelationship between these genes. These results are consistent with the hypothesis that, although detected at levels too low to compact chromatin, a dynamic, diverse NAP population may be responsible for growth-phase-dependent differential gene expression and phenotypic consequences as observed for the Δura3 ΔhpyA strain (Fig. 3 and 4).
FIG 6 .
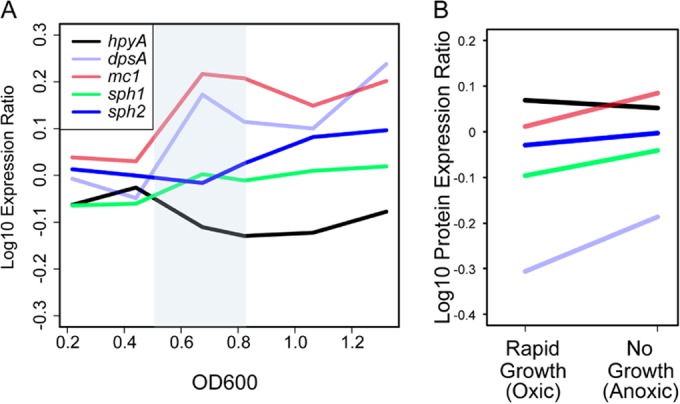
Gene and protein expression of putative NAPs as a function of growth. (A) Gene expression profiles as a function of growth phase. Normalized expression ratios for hpyA (VNG0134G), dpsA (VNG2443G), mc1 (VNG2508C), sph1 (VNG6320C), and sph2 (VNG6173C) from wild-type NRC-1 and Δura3 strain growth data from reference 36 were binned according to optical density, and average values within each bin for each gene (Materials and Methods) were plotted. The blue-shaded region highlights the period of transition from log to stationary phase. Line colors for each gene are explained in the legend. (B) Protein expression ratios of NAPs during rapid growth (oxic conditions) versus slow or no growth (anoxic conditions). Each protein profile corresponds to the mean expression under each condition. Line colors are as in panel A. Original data were obtained from reference 37.
DISCUSSION
Unexpected phenotypic consequences of histone gene dosage alteration suggest a unique function for haloarchaeal histones.
Genetics experiments conducted here suggest that histone is not essential for growth but is required for maintenance of cell shape in H. salinarum. These observations are in contrast to those from histone mutants generated in other archaea and the eukaryotes, which require at least one histone fold domain for viability (16, 28, 29, 38) and survival under stress conditions similar to those tested here (17). In eukaryotes, histone protein overexpression also leads to cell elongation; however, this is likely related to disruption of cell division dynamics, as growth is severely perturbed in yeast (39). Loss of natural transformation competence due to deletion of the histone subunit HTkA in the hyperthermophilic archaeon Thermococcus kodakarensis suggests that membrane or cell wall integrity may be compromised (16), although the morphology of these mutants has not been tested to our knowledge. Granted that H. salinarum is not naturally competent, the Δura3 ΔhpyA strain was still readily transformed (Fig. 3C and D) despite gross morphological differences from the parent strain (Fig. 3). Deletion mutants also maintained membrane and S-layer integrity (see Fig. S1 in the supplemental material). Together, these phenotypic observations point to a novel role for histone as a regulator of cell shape in haloarchaea.
Gene expression data presented here suggest potential pathways linking histone, gene expression, and cell shape. Functional analysis of genes perturbed by histone deletion suggests that HpyA is required for wild-type expression of several genes whose products are putatively associated with the membrane and the cell wall. These include transporters, S-layer (archaeal cell wall) glycosylation enzymes, and a putative phospholipase D (34) (Table 2; see also Table S2 in the supplemental material). Some transport proteins are required for maintenance of wild-type cell morphology in Escherichia coli, albeit indirectly (40). Further, S-layer glycosylation is required to maintain cell shape in H. salinarum (41, 42). Although the overall integrity of the S-layer of Δura3 ΔhpyA mutants remained grossly intact (see Fig. S1), more sensitive detection methods such as mass spectrometry might be required to detect subtle differences in glycosylation (43). Nevertheless, future investigation into these interesting possibilities could determine mechanisms by which histone affects morphology in H. salinarum.
Histone as a regulator of gene expression in open chromatin.
In H. salinarum, histone and other putative NAPs appear to be present in low abundance in both the cytosolic control and the DNA fractions, according to the semiquantitative metric of number of peptides observed per protein (Table 3). This is in sharp contrast to eukaryotes, where histone proteins compact the entirety of the genome and are one of the most abundant proteins in the cell. For example, in humans histones comprise ~60% of all proteins associated with chromosomes by mass (44). In the euryarchaea T. kodakarensis and Methanothermobacter thermautotrophicus, nucleosomes have also been shown to occupy nucleosome-positioning motifs occurring frequently throughout the genome. Histones may therefore also be highly abundant in cells of these species (45, 46). In E. coli, despite the large proportion of protein-free DNA, NAPs make up a significant fraction of all cellular protein. For example, Fis proteins make up approximately 12% of all protein molecules in exponential phase (47). “Beads-on-a-string” and rod-like DNA morphology have been observed in naked DNA in vitro under dehydrated and high-ionic-strength conditions (48), pointing to a possibility for nonenzymatic compaction of DNA in the high-salt cytosol (nearly saturated K+) of H. salinarum. Therefore, HpyA and NAPs in H. salinarum are unlikely to play a major role in the sedimentation rate observed here or in “beads-on-a-string” structures and transcription start site phasing observed previously (25–27).
Our data suggest that HpyA is required for modulating global gene expression, acting as a repressor or an activator depending on the growth phase (Fig. 4; see also Table S2 in the supplemental material). Similarly, many different DNA architectural proteins also regulate gene expression in the eukaryotic and bacterial domains of life. In eukaryotes, nucleosomes inhibit transcription nonspecifically by blocking transcription factors and the general transcriptional machinery from accessing DNA. In vitro studies suggest that archaeal histones from hyperthermophilic methanogens conform to this paradigm. However, understanding of general trends in archaeal histone function awaits evidence from other classes of archaeal organisms (13–15). The number of genes affected by histone deletion in H. salinarum is similar to that observed in T. kodakarensis single histone mutants (16) and Escherichia coli NAP mutants (5). The directions of gene expression changes are also growth phase dependent in E. coli NAP mutants (5, 6). In bacteria, growth-phase-dependent effects are commonly attributed to the dynamic protein composition of the genomic landscape (5, 49), consistent with the phase-dependent dynamics of putative H. salinarum NAP expression observed here (Fig. 6).
However, the function of H. salinarum HpyA differs from bacterial and archaeal NAPs and eukaryotic histones in important ways. The polar distribution of charges on the surface of HpyA is more reminiscent of halophilic transcription factors (24) than canonical eukaryotic histones (Fig. 1C). Histone fold domain-containing proteins, a structural motif conserved from archaea to humans (18), have been shown to function as sequence-specific transcription factors (50) regulating certain functions such as development (51) in eukaryotes. As described above, our sucrose fractionation proteomics data (Table 3) and the high-salt cytosol suggest that HpyA is unlikely to compact DNA. Taken together, our results are consistent with the hypothesis that histone in H. salinarum does not play a significant role in genome compaction but is required for the maintenance of cell morphology and retains the ability to affect gene expression, possibly as a transcription factor.
MATERIALS AND METHODS
Strains and growth conditions.
All strains described are originally derived from Halobacterium salinarum NRC-1 (ATCC 700922). All other strains used in this study are listed in Table 1. Cultures were routinely incubated at 42°C with agitation in complex medium (CM; 250 g ⋅ liter−1 NaCl, 20 g ⋅ liter−1 MgSO4⋅7H2O, 3 g ⋅ liter−1 sodium citrate, 2 g ⋅ liter−1 KCl, 10 g ⋅ liter−1 peptone). For strains generated in the Δura3 background, uracil was added to a final concentration of 50 µg ⋅ ml−1. For strains containing the overexpression vector pMTFCHA and its derivatives (Table 1), mevinolin was added to a final concentration of 1 µg ⋅ ml−1 to maintain the plasmid.
Strain construction.
Plasmid pKAD01 for in-frame deletion of hpyA was constructed by Gateway cloning (Invitrogen, Carlsbad, CA) using PCR splicing by overhang extension (SOEing) and destination vector pRSK01 as described in reference 52. Knockout clones were transformed and selected in the Δura3 uracil auxotroph parent strain as described in reference 53, and the knockout genotype was confirmed with PCR, Sanger sequencing, and Southern blotting (Fig. 2). Overexpression vector pKAD02, constructed as described previously (54), contains the hpyA (VNG0134G) coding sequence driven by the strong constitutive promoter of VNG2293G in backbone vector pMTFCHA (52). Complementation plasmid pKAD03 was synthesized by cloning the native promoter for the rpa-hpyA-aup operon (Prpa200 [see Text S2 in the supplemental material]) upstream of the hpyA coding sequence in pMTFCHA using Gibson assembly (55). In order to cure the Δura3/pKAD02 strain of the plasmid, triplicate cultures were grown in nonselective medium for 5 days and subcultured prior to phenotyping. All plasmid constructs and plasmid-bearing strains were screened using PCR and confirmed by Sanger sequencing. All primers used for strain construction are described in detail in Table S5 in the supplemental material.
Southern blotting and detection.
Genomic DNA was purified and digested with the restriction enzymes BamHI and KpnI. DNA samples were run on an 0.8% agarose gel prior to blotting on a BioRad Zeta-probe membrane. An alkali-labile digoxigenin II (DIG-II)-dUTP probe of 86 bp internal to hpyA was synthesized using PCR primers K51 and K52 (see Table S5 in the supplemental material), gel purified, and prepared according to the manufacturer’s instructions. Blots were hybridized overnight at 50°C, and chemiluminescent probe detection was performed as described in reference 56.
Sequence and structural analyses.
All amino acid sequence alignments were performed using Clustal Omega (57). Histone sequences from the haloarchaea (21) were retrieved from NCBI and manually curated to remove duplicate or spurious entries. The isoelectric points (pIs) of the remaining 69 histone protein sequences were calculated using the sequence manipulation suite (58). Prior to pI analysis, N- and C-terminal histone domains of haloarchaeal histone sequences were separated and linker sequences were removed. Significant differences in pIs between all N-terminal domains and all C-terminal domains were determined by t test. The histone protein consensus sequence logo for haloarchaea (Fig. 1B) was generated using WebLogo3.3 (59). For structural modeling, histones H3 and H4 from S. cerevisiae were trimmed of N-terminal tails and fused in silico using the HpyA linker region (see Text S1 in the supplemental material). Modeling of yeast H3 to H4 and H. salinarum HpyA onto the HMk scaffold from Methanopyrus kandleri (PDB identifier 1f1eA [10]) and calculation of the surface charge distribution were performed using SWISS-MODEL (60). Structural models were visualized with DeepView/PDB-Viewer (61). Charge as shown in Fig. 1C was calculated for both HpyA and yeast histones at the default dielectric constant of 80.00. Adjusting the constant to 48.4 to account for a high-salt environment (24) slightly affected the magnitude of observed charges but had no detectable effect on charge distribution.
Growth rate measurements.
Growth experiments were performed in a high-throughput multiplate reader as follows: at least three biological replicates, in technical triplicate, for each of Δura3 parent, Δura3 ΔhpyA, Δura3 empty vector control, and hpyA overexpression strains (Fig. 2C; Table 1) were grown to mid-exponential phase and subcultured to an optical density at 600 nm (OD600) of 0.05 in CM with uracil. Subcultures were grown in a multiwell plate at 42°C for 48 h under continuous shaking (~225 rpm) in a Bioscreen C microbial growth analyzer (Growth Curves USA, Piscataway, NJ). OD600 was automatically measured every 30 min. Growth rate was calculated from the linear regression of the log2 optical density during early exponential growth for each replicate. Growth rates from technical replicate cultures were averaged; the mean and standard deviation were calculated for growth rates of biological replicate cultures.
Light microscopy.
Cells were visualized at mid-logarithmic (OD600 of 0.3 to 0.5) and stationary (OD600 of ≥1.3) phases of growth with a Zeiss Axio Scope A1 microscope under 100× phase contrast. Images were captured with a Pixelink PL-E421M camera. Cell counting and morphological quantitation were performed with ImageJ and R statistical software. Ellipses were fitted to each cell, and measurements of the area, length, and width were collected. t tests were performed on equivalent numbers of cells that had been randomly sampled from images of biological triplicate cultures. Resultant P values are listed in Table S1 in the supplemental material.
Gene expression microarrays and analysis.
For Δura3 and Δura3 ΔhpyA strains, three biological replicate cultures were grown to mid-log phase in CM with uracil and then subcultured to an OD600 of 0.05 for further growth. Four-milliliter aliquots were harvested from 50-ml cultures at mid-logarithmic (OD600 of 0.5) and stationary (OD600 of 1.5) phases. Culture harvest, RNA extraction, labeling, and hybridization to custom Agilent ORF arrays (6 probes per gene) were performed as described in reference 62. Each sample was hybridized against H. salinarum NRC-1 wild type grown under standard conditions (CM to OD600 of 0.4 at 37°C with 225-rpm shaking [63]). Dye swaps were performed for each biological replicate. A total of 36 replicate data points were collected per gene in each sample. Spot ratios were determined using Agilent feature extraction, and all further analysis was performed in the R statistical computing environment. Ratios were normalized within and between arrays using the R package Limma (64) in a pipeline adapted from reference 62. Resultant normalized gene expression data were subjected to Student’s t test using the TM4 multiple experiment viewer (65). A cutoff of P < 0.05 between biological replicates of parent strain versus histone mutant was used to designate those genes that showed significant changes in gene expression in response to histone deletion. Ratios for genes with significant expression changes by t test were calculated by subtracting the mean log2 ratios of the mutant biological replicates from those of the wild type. Resultant ratios for genes meeting the P < 0.05 significance cutoff that were also differentially expressed 2-fold or more in the ΔhpyA background over the Δura3 parent yielded the final list of 250 genes (Fig. 4). As evidenced by comparing resultant microarray ratios to those obtained by RT-qPCR (see Fig. S2 and Table S2 in the supplemental material), the dynamic range of array ratios was narrow, a known intrinsic feature of microarrays. Therefore, prior to setting a 2-fold threshold for differential expression, a correction factor derived from the linear relationship between the RT-qPCR and microarray data sets was applied to scale the microarray data (corrected log2 array value = 3.5763 × array ratio − 0.3758).
Protein identification and analysis.
Wild-type H. salinarum cultures were harvested in log (OD600 of 0.5) or stationary (OD600 of 1.5) phase and fractionated over a sucrose density gradient as described in reference 26, except that cultures were fixed for 30 min in 1% formaldehyde, lysates were sheared with a needle, and ultracentrifugation was performed at 160,000 relative centrifugal force (RCF) for 5 h at 4°C. Nucleic acid concentration in each fraction was determined by absorbance at 260 nm, and protein concentration was determined by the Bradford assay (66). From each phase of growth, six samples were prepared for protein identification: two biological replicates of DNA (fractions 8 to 13 in log phase and 9 to 14 in stationary phase [Fig. 5]) and one of cytosolic components, including free RNA (fractions 1 to 6) as a control. Fractions for either DNA or control samples were pooled, and protein was precipitated with 10% trichloroacetic acid. Protein precipitates were separated via SDS-PAGE. All proteins less than 100 kDa were excised to avoid the abundant S-layer protein and subsequently digested with trypsin. Peptide samples were analyzed via LC-MS/MS using a Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Scientific). MS/MS peptide and protein assignments were performed using Mascot (Matrix Science, London, United Kingdom; version 2.5.0). Details of assignment are described in Text S2 in the supplemental material. The numbers of peptides detected per protein from biological replicate samples in each of the DNA and control fractions were added together prior to analysis in order to avoid the effect of missing values. To obtain the values reported in Table 3, we calculated the number of peptides detected for each protein relative to the total number of peptides detected under each condition (e.g., log phase, DNA fractions, etc.). This quantity is here referred to as “peptide proportion.” Peptide proportions for each protein were normalized to predicted protein mass (Table 3). Significant enrichment of peptide proportion in control versus DNA fractions was calculated by determining the linear relationship for peptide proportion of each protein between fractions using the R package robustbase (R package version 0.92-5; http://CRAN.R-project.org/package=robustbase). Proteins with residuals for peptide proportion at least 2 standard deviations from the mean were considered enriched in a given sample and are reported in the text.
Comparison of NAP gene expression and quantitative proteomics data during growth.
Gene expression profiles throughout a high-resolution time course of the H. salinarum growth curve (Fig. 6) (36) were retrieved from the GEO database (GSE14976). Normalized data points from the NRC-1 and Δura3 strains were binned into equal intervals from OD600s of 0.1 to 1.4, where each interval contains at least 2 measurements. Averages of optical densities and expression values from each bin were plotted. For analysis of NAP expression profiles, available quantitative iTRAQ proteomics data were analyzed from H. salinarum cultures exposed to slow-growth conditions (low oxygen) versus rapid-growth conditions (high oxygen) in a turbidostat over time (37). Protein level values from high-oxygen conditions were averaged to yield rapid-growth value, whereas values from low-oxygen conditions were averaged to yield slow-growth values (Fig. 6B). We focused on five genes encoding putative NAPs within these data sets: mc1 (VNG2508C), the bacterial-type NAP dpsA (VNG2443G), histone (hypA), and two genes encoding homologs to the universally conserved DNA cohesion and condensation protein Smc1, sph1 (VNG6320C) and sph2 (VNG6173C).
Microarray data accession number.
All raw and normalized microarray data have been deposited in NCBI’s Gene Expression Omnibus (67) and are accessible through GEO Series accession number GSE54599.
SUPPLEMENTAL MATERIAL
Amino acid sequences of histones from haloarchaeal genomes currently curated in the NCBI database used to produce Fig. 1A and C. Download
Supplemental text, including supplemental methods and results corresponding to supplemental figures and tables and references for this material. Download
ΔhpyA strain morphological phenotypes are not due to membrane/cell wall integrity defects or stress sensitivity. Download
RT-qPCR verification of strains and microarray gene expression data. Download
Detection of nucleic acids in sucrose gradient fractions. Download
Results of t tests from quantitative analysis of mutant morphology.
Genes differentially expressed in ΔhpyA strain versus Δura3 parent strain.
All proteins identified by mass spectrometry on sucrose gradient-fractionated H. salinarum lysates.
Enrichment of proteins in DNA versus control sucrose gradient fractions.
Primers used in this study.
ACKNOWLEDGMENTS
We thank Claudia Ofori-Marfoh and Callan Corcoran for technical assistance, Marc Facciotti for the generous gift of plasmids pRSK01 and pMTFCHA, and the Duke University School of Medicine Proteomics and Metabolomics Shared Resource for the protein identification service.
This research was supported by grants to A.K.S. from the National Science Foundation (NSF-MCB-10-52290 and NSF-MCB-1417750) and the Duke Arts and Sciences Council Committee on Faculty Research.
Footnotes
Citation Dulmage KA, Todor H, Schmid AK. 2015. Growth-phase-specific modulation of cell morphology and gene expression by an archaeal histone protein. mBio 6(5):e00649-15. doi:10.1128/mBio.00649-15.
REFERENCES
- 1.Clark DJ. 2010. Nucleosome positioning, nucleosome spacing and the nucleosome code. J Biomol Struct Dyn 27:781–793. doi: 10.1080/073911010010524945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kornberg RD. 2007. The molecular basis of eukaryotic transcription. Proc Natl Acad Sci U S A 104:12955–12961. doi: 10.1073/pnas.0704138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. 2011. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luijsterburg MS, Noom MC, Wuite GJ, Dame RT. 2006. The architectural role of nucleoid-associated proteins in the organization of bacterial chromatin: a molecular perspective. J Struct Biol 156:262–272. doi: 10.1016/j.jsb.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Prieto AI, Kahramanoglou C, Ali RM, Fraser GM, Seshasayee AS, Luscombe NM. 2012. Genomic analysis of DNA binding and gene regulation by homologous nucleoid-associated proteins IHF and HU in Escherichia coli K12. Nucleic Acids Res 40:3524–3537. doi: 10.1093/nar/gkr1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kahramanoglou C, Seshasayee AS, Prieto AI, Ibberson D, Schmidt S, Zimmermann J, Benes V, Fraser GM, Luscombe NM. 2011. Direct and indirect effects of H-NS and Fis on global gene expression control in Escherichia coli. Nucleic Acids Res 39:2073–2091. doi: 10.1093/nar/gkq934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandman K, Reeve JN. 2006. Archaeal histones and the origin of the histone fold. Curr Opin Microbiol 9:520–525. doi: 10.1016/j.mib.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z, Guo L, Huang L. 2012. Archaeal chromatin proteins. Sci China Life Sci 55:377–385. doi: 10.1007/s11427-012-4322-y. [DOI] [PubMed] [Google Scholar]
- 9.Decanniere K, Babu AM, Sandman K, Reeve JN, Heinemann U. 2000. Crystal structures of recombinant histones HMfA and HMfB from the hyperthermophilic archaeon Methanothermus fervidus. J Mol Biol 303:35–47. doi: 10.1006/jmbi.2000.4104. [DOI] [PubMed] [Google Scholar]
- 10.Fahrner RL, Cascio D, Lake JA, Slesarev A. 2001. An ancestral nuclear protein assembly: crystal structure of the Methanopyrus kandleri histone. Protein Sci 10:2002–2007. doi: 10.1110/ps.10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pavlov NA, Cherny DI, Jovin TM, Slesarev AI. 2002. Nucleosome-like complex of the histone from the hyperthermophile Methanopyrus kandleri (MkaH) with linear DNA. J Biomol Struct Dyn 20:207–214. doi: 10.1080/07391102.2002.10506836. [DOI] [PubMed] [Google Scholar]
- 12.Tomschik M, Karymov MA, Zlatanova J, Leuba SH. 2001. The archaeal histone-fold protein HMf organizes DNA into bona fide chromatin fibers. Structure 9:1201–1211. doi: 10.1016/S0969-2126(01)00682-7. [DOI] [PubMed] [Google Scholar]
- 13.Soares D, Dahlke I, Li WT, Sandman K, Hethke C, Thomm M, Reeve JN. 1998. Archaeal histone stability, DNA binding, and transcription inhibition above 90 degrees C. Extremophiles 2:75–81. doi: 10.1007/s007920050045. [DOI] [PubMed] [Google Scholar]
- 14.Thomm M, Sandman K, Frey G, Koller G, Reeve JN. 1992. Transcription in vivo and in vitro of the histone-encoding gene hmfB from the hyperthermophilic archaeon Methanothermus fervidus. J Bacteriol 174:3508–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilkinson SP, Ouhammouch M, Geiduschek EP. 2010. Transcriptional activation in the context of repression mediated by archaeal histones. Proc Natl Acad Sci U S A 107:6777–6781. doi: 10.1073/pnas.1002360107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cubonova L, Katano M, Kanai T, Atomi H, Reeve JN, Santangelo TJ. 2012. An archaeal histone is required for transformation of Thermococcus kodakarensis. J Bacteriol 194:6864–6874. doi: 10.1128/JB.01523-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weidenbach K, Glöer J, Ehlers C, Sandman K, Reeve JN, Schmitz RA. 2008. Deletion of the archaeal histone in Methanosarcina mazei Go1 results in reduced growth and genomic transcription. Mol Microbiol 67:662–671. doi: 10.1111/j.1365-2958.2007.06076.x. [DOI] [PubMed] [Google Scholar]
- 18.Arents G, Moudrianakis EN. 1995. The histone fold: a ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc Natl Acad Sci U S A 92:11170–11174. doi: 10.1073/pnas.92.24.11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chartier F, Laine B, Sautiere P. 1988. Characterization of the chromosomal protein MC1 from the thermophilic archaebacterium Methanosarcina sp. CHTI 55 and its effect on the thermal stability of DNA. Biochim Biophys Acta 951:149–156. doi: 10.1016/0167-4781(88)90035-8. [DOI] [PubMed] [Google Scholar]
- 20.Laine B, Chartier F, Imbert M, Lewis R, Sautiere P. 1986. Primary structure of the chromosomal protein HMb from the archaebacteria Methanosarcina barkeri. Eur J Biochem 161:681–687. doi: 10.1111/j.1432-1033.1986.tb10493.x. [DOI] [PubMed] [Google Scholar]
- 21.Becker EA, Seitzer PM, Tritt A, Larsen D, Krusor M, Yao AI, Wu D, Madern D, Eisen JA, Darling AE, Facciotti MT. 2014. Phylogenetically driven sequencing of extremely halophilic Archaea reveals strategies for static and dynamic osmo-response. PLoS Genet 10:e1004784. doi: 10.1371/journal.pgen.1004784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng WV, et al. 2000. Genome sequence of Halobacterium species NRC-1. Proc Natl Acad Sci U S A 97(22):12176–12181. doi: 10.1073/pnas.190337797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malik HS, Henikoff S. 2003. Phylogenomics of the nucleosome. Nat Struct Biol 10:882–891. doi: 10.1038/nsb996. [DOI] [PubMed] [Google Scholar]
- 24.Kennedy SP, Ng WV, Salzberg SL, Hood L, DasSarma S. 2001. Understanding the adaptation of Halobacterium species NRC-1 to its extreme environment through computational analysis of its genome sequence. Genome Res 11:1641–1650. doi: 10.1101/gr.190201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shioda M, Sugimori K, Shiroya T, Takayanagi S. 1989. Nucleosomelike structures associated with chromosomes of the archaebacterium Halobacterium salinarium. J Bacteriol 171:4514–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takayanagi S, Morimura S, Kusaoke H, Yokoyama Y, Kano K, Shioda M. 1992. Chromosomal structure of the halophilic archaebacterium Halobacterium salinarium. J Bacteriol 174:7207–7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ammar R, Torti D, Tsui K, Gebbia M, Durbic T, Bader GD, Giaever G, Nislow C. 2012. Chromatin is an ancient innovation conserved between Archaea and Eukarya. Elife 1:e00078. doi: 10.7554/eLife.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsubara K, Sano N, Umehara T, Horikoshi M. 2007. Global analysis of functional surfaces of core histones with comprehensive point mutants. Genes Cells 12:13–33. doi: 10.1111/j.1365-2443.2007.01031.x. [DOI] [PubMed] [Google Scholar]
- 29.Nakanishi S, Sanderson BW, Delventhal KM, Bradford WD, Staehling-Hampton K, Shilatifard A. 2008. A comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat Struct Mol Biol 15:881–888. doi: 10.1038/nsmb.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soares DJ, Sandman K, Reeve JN. 2000. Mutational analysis of archaeal histone-DNA interactions. J Mol Biol 297:39–47. doi: 10.1006/jmbi.2000.3546. [DOI] [PubMed] [Google Scholar]
- 31.Zerulla K, Soppa J. 2014. Polyploidy in haloarchaea: advantages for growth and survival. Front Microbiol 5:274. doi: 10.3389/fmicb.2014.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coker JA, DasSarma P, Capes M, Wallace T, McGarrity K, Gessler R, Liu J, Xiang H, Tatusov R, Berquist BR, DasSarma S. 2009. Multiple replication origins of Halobacterium sp. strain NRC-1: properties of the conserved orc7-dependent oriC1. J Bacteriol 191:5253–5261. doi: 10.1128/JB.00210-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolf YI, Makarova KS, Yutin N, Koonin EV. 2012. Updated clusters of orthologous genes for Archaea: a complex ancestor of the Archaea and the byways of horizontal gene transfer. Biol Direct 7:46. doi: 10.1186/1745-6150-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guan Z, Naparstek S, Kaminski L, Konrad Z, Eichler J. 2010. Distinct glycan-charged phosphodolichol carriers are required for the assembly of the pentasaccharide N-linked to the Haloferax volcanii S-layer glycoprotein. Mol Microbiol 78:1294–1303. doi: 10.1111/j.1365-2958.2010.07405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van PT, Schmid AK, King NL, Kaur A, Pan M, Whitehead K, Koide T, Facciotti MT, Goo YA, Deutsch EW, Reiss DJ, Mallick P, Baliga NS. 2008. Halobacterium salinarum NRC-1 PeptideAtlas: toward strategies for targeted proteomics and improved proteome coverage. J Proteome Res 7:3755–3764. doi: 10.1021/pr800031f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Facciotti MT, Pang WL, Lo FY, Whitehead K, Koide T, Masumura K, Pan M, Kaur A, Larsen DJ, Reiss DJ, Hoang L, Kalisiak E, Northen T, Trauger SA, Siuzdak G, Baliga NS. 2010. Large scale physiological readjustment during growth enables rapid, comprehensive and inexpensive systems analysis. BMC Syst Biol 4:64. doi: 10.1186/1752-0509-4-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmid AK, Reiss DJ, Kaur A, Pan M, King N, Van PT, Hohmann L, Martin DB, Baliga NS. 2007. The anatomy of microbial cell state transitions in response to oxygen. Genome Res 17:1399–1413. doi: 10.1101/gr.6728007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M'Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 39.Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, Snyder M, Oliver SG, Cyert M, Hughes TR, Boone C, Andrews B. 2006. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell 21:319–330. doi: 10.1016/j.molcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 40.Vega DE, Young KD. 2014. Accumulation of periplasmic enterobactin impairs the growth and morphology of Escherichia coli tolC mutants. Mol Microbiol 91:508–521. doi: 10.1111/mmi.12473. [DOI] [PubMed] [Google Scholar]
- 41.Mescher MF, Strominger JL. 1976. Structural (shape-maintaining) role of the cell surface glycoprotein of Halobacterium salinarium. Proc Natl Acad Sci U S A 73:2687–2691. doi: 10.1073/pnas.73.8.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Todor H, Dulmage K, Gillum N, Bain JR, Muehlbauer MJ, Schmid AK. 2014. A transcription factor links growth rate and metabolism in the hypersaline adapted archaeon Halobacterium salinarum. Mol Microbiol 93:1172–1182. doi: 10.1111/mmi.12726. [DOI] [PubMed] [Google Scholar]
- 43.Cohen-Rosenzweig C, Yurist-Doutsch S, Eichler J. 2012. AglS, a novel component of the Haloferax volcanii N-glycosylation pathway, is a dolichol phosphate-mannose mannosyltransferase. J Bacteriol 194:6909–6916. doi: 10.1128/JB.01716-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uchiyama S, Kobayashi S, Takata H, Ishihara T, Hori N, Higashi T, Hayashihara K, Sone T, Higo D, Nirasawa T, Takao T, Matsunaga S, Fukui K. 2005. Proteome analysis of human metaphase chromosomes. J Biol Chem 280:16994–17004. doi: 10.1074/jbc.M412774200. [DOI] [PubMed] [Google Scholar]
- 45.Maruyama H, Harwood JC, Moore KM, Paszkiewicz K, Durley SC, Fukushima H, Atomi H, Takeyasu K, Kent NA. 2013. An alternative beads-on-a-string chromatin architecture in Thermococcus kodakarensis. EMBO Rep 14:711–717. doi: 10.1038/embor.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nalabothula N, Xi L, Bhattacharyya S, Widom J, Wang JP, Reeve JN, Santangelo TJ, Fondufe-Mittendorf YN. 2013. Archaeal nucleosome positioning in vivo and in vitro is directed by primary sequence motifs. BMC Genomics 14:391. doi: 10.1186/1471-2164-14-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ali Azam T, Iwata A, Nishimura A, Ueda S, Ishihama A. 1999. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J Bacteriol 181:6361–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eickbush TH, Moudrianakis EN. 1978. The compaction of DNA helices into either continuous supercoils or folded-fiber rods and toroids. Cell 13:295–306. doi: 10.1016/0092-8674(78)90198-8. [DOI] [PubMed] [Google Scholar]
- 49.Rimsky S, Travers A. 2011. Pervasive regulation of nucleoid structure and function by nucleoid-associated proteins. Curr Opin Microbiol 14:136–141. doi: 10.1016/j.mib.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 50.Baxevanis AD, Arents G, Moudrianakis EN, Landsman D. 1995. A variety of DNA-binding and multimeric proteins contain the histone fold motif. Nucleic Acids Res 23:2685–2691. doi: 10.1093/nar/23.14.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen X, Hiller M, Sancak Y, Fuller MT. 2005. Tissue-specific TAFs counteract Polycomb to turn on terminal differentiation. Science 310:869–872. doi: 10.1126/science.1118101. [DOI] [PubMed] [Google Scholar]
- 52.Wilbanks EG, Larsen DJ, Neches RY, Yao AI, Wu CY, Kjolby RA, Facciotti MT. 2012. A workflow for genome-wide mapping of archaeal transcription factors with ChIP-seq. Nucleic Acids Res 40:e74. doi: 10.1093/nar/gks063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peck RF, DasSarma S, Krebs MP. 2000. Homologous gene knockout in the archaeon Halobacterium salinarum with ura3 as a counterselectable marker. Mol Microbiol 35:667–676. doi: 10.1046/j.1365-2958.2000.01739.x. [DOI] [PubMed] [Google Scholar]
- 54.Schmid AK, Pan M, Sharma K, Baliga NS. 2011. Two transcription factors are necessary for iron homeostasis in a salt-dwelling archaeon. Nucleic Acids Res 39:2519–2533. doi: 10.1093/nar/gkq1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gibson DG. 2011. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498:349–361. doi: 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Selmecki A, Bergmann S, Berman J. 2005. Comparative genome hybridization reveals widespread aneuploidy in Candida albicans laboratory strains. Mol Microbiol 55:1553–1565. doi: 10.1111/j.1365-2958.2005.04492.x. [DOI] [PubMed] [Google Scholar]
- 57.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stothard P. 2000. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28:1102–1104. [DOI] [PubMed] [Google Scholar]
- 59.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guex N, Peitsch MC. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 62.Sharma K, Gillum N, Boyd JL, Schmid A. 2012. The RosR transcription factor is required for gene expression dynamics in response to extreme oxidative stress in a hypersaline-adapted archaeon. BMC Genomics 13:351. doi: 10.1186/1471-2164-13-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baliga NS, Bjork SJ, Bonneau R, Pan M, Iloanusi C, Kottemann MC, Hood L, DiRuggiero J. 2004. Systems level insights into the stress response to UV radiation in the halophilic archaeon Halobacterium NRC-1. Genome Res 14:1025–1035. doi: 10.1101/gr.1993504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smyth GK, Michaud J, Scott HS. 2005. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21:2067–2075. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- 65.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. 2003. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34:374–378. [DOI] [PubMed] [Google Scholar]
- 66.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 67.Edgar R, Domrachev M, Lash AE. 2002. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hartman AL, Norais C, Badger JH, Delmas S, Haldenby S, Madupu R, Robinson J, Khouri H, Ren Q, Lowe TM, Maupin-Furlow J, Pohlschroder M, Daniels C, Pfeiffer F, Allers T, Eisen JA. 2010. The complete genome sequence of Haloferax volcanii DS2, a model archaeon. PLoS One 5:e9605. doi: 10.1371/journal.pone.0009605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Amino acid sequences of histones from haloarchaeal genomes currently curated in the NCBI database used to produce Fig. 1A and C. Download
Supplemental text, including supplemental methods and results corresponding to supplemental figures and tables and references for this material. Download
ΔhpyA strain morphological phenotypes are not due to membrane/cell wall integrity defects or stress sensitivity. Download
RT-qPCR verification of strains and microarray gene expression data. Download
Detection of nucleic acids in sucrose gradient fractions. Download
Results of t tests from quantitative analysis of mutant morphology.
Genes differentially expressed in ΔhpyA strain versus Δura3 parent strain.
All proteins identified by mass spectrometry on sucrose gradient-fractionated H. salinarum lysates.
Enrichment of proteins in DNA versus control sucrose gradient fractions.
Primers used in this study.