

Abstract
Colorectal cancer represents the third most common and the second deadliest type of cancer for both men and women in the United States claiming over 50000 lives in 2014. The 5-year survival rate for patients diagnosed with metastatic colon and rectal cancer is < 15%. Early detection and more effective treatments are urgently needed to reduce morbidity and mortality of patients afflicted with this disease. Here we will review the risk factors and current treatment paradigms for colorectal cancer, with an emphasis on the role of chemoprevention as they relate to epidermal growth factor receptor (EGFR) blockade. We will discuss how various EGFR ligands are upregulated in the presence of Western diets high in saturated and N-6 polyunsaturated fats. We will also outline the various mechanisms of EGFR inhibition that are induced by naturally occurring chemopreventative agents such as ginseng, green tea, and curcumin. Finally, we will discuss the current role of targeted chemotherapy in colon cancer and outline the limitations of our current treatment options, describing mechanisms of resistance and escape.
Keywords: Chemoprevention, Colon cancer, Epidermal growth factor receptor, Western diet, Curcumin, Green tea, Ginseng
Core tip: This review article will summarize the risk factors and current treatment paradigms for colorectal cancer, with an emphasis on the role of targeted chemotherapy and chemoprevention as they relate to epidermal growth factor receptor (EGFR) blockade. It will include an overview of the structure and function of EGFR as well as intracellular pathways regulated by its activity. It will discuss how various EGFR ligands are upregulated in the presence of Western diets that are high in saturated and N-6 unsaturated fat, and will outline the various mechanisms of EGFR inhibition observed with several naturally occurring chemopreventative agents including ginseng, green tea, and curcumin.
INTRODUCTION
A total of 1665540 new cancer cases and 585720 cancer deaths were projected to occur in the United States in 2014. Of these, colon and rectal cancer (CRC) will account for 8% of new cases, representing the third most common and the second deadliest type of cancer for both men and women[1], claiming over 50000 lives in 2014[1,2]. The 5-year survival rate for patients diagnosed with metastatic CRC is < 15%[1]. Early detection and treatment is crucial for the improvement in morbidity and mortality of patients afflicted with this disease.
Overexpression of epidermal growth factor receptor (EGFR) is common in many tumors. Specifically in CRC, EGFR is estimated to be overexpressed in 60%-80% of tumors, and is associated with a poor prognosis[2]. For these reasons EGFR has been targeted as a locus for treatment with small molecule inhibitors and monoclonal antibodies, with the latter playing a role in the treatment of metastatic disease. This review article will discuss risk factors and current treatment modalities for colorectal cancer and examine the roles of chemotherapy and chemoprevention.
RISK FACTORS FOR COLORECTAL CANCER
Many factors have been identified contributing to the risk of colon cancer. These risk factors are believed to increase the rate at which genetic mutations occur in various oncogenes and tumor suppressor genes, and/or result in growth-promoting epigenetic modifications. Generally, these factors can be classified into the following categories: germline genetic mutations, environmental exposures, personal or family history of CRC, associated diseases, and demographic considerations.
There are several germline genetic mutations that greatly increase the incidence of colon cancer through distinct molecular mechanisms. The two syndromes that account for most of the hereditary diseases are Lynch syndrome, and familial adenomatous polyposis (FAP) syndrome. Recent estimates indicate that Lynch syndrome accounts for approximately 3% of CRC cases, while FAP syndrome contributes an additional 0.01%[3,4]. Lynch syndrome is caused by mutations in one or more of the DNA mismatch repair genes MLH1, MSH2, MSH6, PMS2, and EPCAM. The two most common forms of FAP syndrome are a result of a germline mutation in the APC gene. Other germline - inherited colorectal cancer syndromes include MUTYH-associated polyposis, Cowden syndrome, Peutz-Jeghers syndrome, and juvenile polyposis syndrome.
Environmental exposures associated with an increased risk of CRC include a history of abdominal radiation, smoking, alcohol use, and diet[5-8]. Of particular interest with respect to the EGFR receptor is the role of a high fat Western diet, which has been shown to promote the development of experimental colon cancer via an EGFR-mediated mechanism. The role of this pathway will be discussed in detail later.
Personal history of CRC or large adenomatous polyps (> 1 cm) or polyps with villous features increase the risk of colorectal cancer[9]. Family history of colon cancer or adenomatous polyps confers an increased risk of disease, even if these histories do not meet the criteria for the syndromes discussed above. US guidelines reflect this increased risk, with the ACG recommending earlier screening if a single first-degree relative was diagnosed with CRC or had an advanced adenoma diagnosed at age < 60 years or if two first-degree relatives were diagnosed with CRC or advanced adenomas[10].
Disease states associated with an increased incidence of colon cancer include IBD (both ulcerative colitis and Crohn’s disease), diabetes, and obesity. As with many cancers, risk for CRC increases with age. CRC incidence is approximately equal in males and females, although there is an increased incidence and higher mortality rate among African Americans and an increased mortality among men. Recent studies suggest that testosterone effects in males rather the protective effects of estrogens in females account for increased male risk[11].
APPROACH TO CRC MANAGEMENT
The management of CRC includes screening, staging, and treatment with surgery, chemotherapy, and/or radiation. As more than 20% of patients with CRC will present with metastatic disease with a 5 year survival rate < 15%[1], prevention is critical in colorectal cancer. Colorectal cancer prevention is primarily based on screening methods, which include stool tests, radiographic imaging, and colonoscopy to identify adenomatous polyps, a precursor lesion for colon cancer. Colonic polyps may be identified through these screening methods and then may be removed during colonoscopy. Colorectal cancer, once diagnosed, is defined as either colon or rectal cancer based on the anatomical location of the lesion, with the rectum being defined as the region extending from the transitional mucosa of the anal dentate line to the sigmoid colon at the peritoneal reflection. Recent studies of CRC suggest that tumors arising in the proximal and distal colon have different molecular phenotypes with different prognostic outcomes. Interestingly, rectal cancers and tumors in the distal colon share many molecular features[12].
Upon diagnosis of CRC, staging is primarily accomplished through CT (with certain situations calling for additional PET-CT) of the chest, abdomen, and pelvis, using the TMN system, with the goal of identifying tumors appropriate for resection. If amenable to resection, the tumor is removed. Pathological staging and subsequent assessment of high-risk features for systemic recurrence are performed to help guide the utility of adjuvant chemotherapy with 5-FU based chemotherapies. In this regard, determining the presence of nodal disease is of particular importance. For metastatic disease, assessment of RAS gene status (KRAS/NRAS) and BRAF status (if KRAS is WT) determines whether or not the tumor is likely to respond to anti-EGFR monoclonal antibodies such as panitumumab and cetuximab. The rationale for this treatment paradigm and the specific pathways involved will be discussed later. In addition to genetic testing for individuals with CRC at younger ages or with CRC positive family history, search for metastatic lesions must be pursued to determine if patients are likely to benefit from resection of isolated metastasis. The timing of colectomy with resection of metastasis, and the use of various 5-FU based chemotherapeutics as neoadjuvant forms of chemotherapy such as FOLFOX, FOLFIRI, and CapeOX, along with bevacizumab, panitumumab, or cetuximab, depend on the individual patient and tumor characteristics. If resection of metastatic disease is impossible, neoadjuvant chemotherapy should be administered first if there is no imminent risk of obstruction or significant bleeding. In addition, the patient should undergo periodic re-assessment regarding the resectability of metastatic lesions[13].
For rectal cancer, endorectal ultrasound is important to assess the presence of LN involvement. In clinical T1-T2 node negative rectal cancer, surgical management should be pursued with a pathological assessment of TMN stage. High grade T lesions or node positive disease should be treated with adjuvant chemotherapy and radiation. In advanced clinical stage disease (T3 or higher or any node positive disease), neoadjuvant chemoradiation should be offered with adjuvant chemotherapy. The chemotherapeutics recommended in rectal cancer include the 5-FU based agents with oxaliplatin. In metastatic disease, there is a role for panitumumab and cetuximab if the tumors are KRAS/NRAS WT. As with colon cancer, the goal in metastatic rectal cancer is to periodically reassess the potential for resection of metastases. Treatment regimens for rectal vs colon cancer share many similarities, with the major difference being the use of radiation therapy for rectal cancer as outlined above[13]. There is, however, some data suggesting a benefit for adjuvant RT in colon cancer in select patients with high-risk features for local recurrence[14].
EGFR PATHWAYS IN COLORECTAL CANCER
EGFR was one of the first targets to be exploited in cancer treatment. The receptor also known as HER (human EGF receptor) or c-erbB1, is a 170-kDa transmembrane protein with intrinsic protein tyrosine kinase activity. EGFR is one of four members of the c-erb subfamily of receptor protein tyrosine kinases. Two cysteine-rich domains comprise the ligand-binding region on the extracellular aspect of the cell. A single alpha-helical transmembrane domain connects the ligand-binding region to the intracellular receptor, which is comprised of three domains. One domain serves as a site for feedback attenuation by PKC and erk MAP kinases, another is a tyrosine kinase domain, and the third is a carboxy-terminal tail. EGFR is present on all epithelial and stromal cells, and is expressed on many glial and smooth muscle cells as well. It is a multifunctional receptor that plays a key role in cell division and apoptosis, cell differentiation and dedifferentiation, migration, and organogenesis[15]. EGFR executes these functions by activation of multiple signaling pathways including PLC-gamma-1, RAS-RAF-MEK-MAPKs, phosphatidylinositol-3 kinase and Akt, Src, the stress-activated protein kinases, PAK-JNKK-JNK, and the signal transducers and activators of transcription. Binding of a diverse array of ligands (EGF, TGF, amphiregulin, heparin-binding EGF, betacellulin, or epiregulin) induces receptor homodimerization or heterdimerization with other ErBb2 members (Figure 1).
Figure 1.
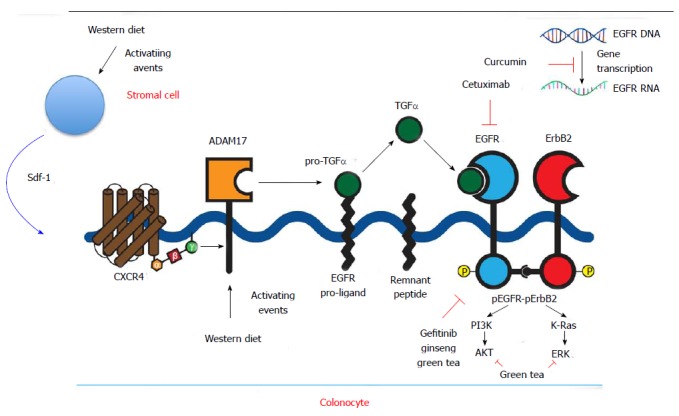
Epidermal growth factor receptor pathways, western diet, chemoprevention, synthetic inhibitors. EGFR: Epidermal growth factor receptor; CXCR4: C-X-C chemokine receptor type 4; TGF: Transforming growth factor; PI3K: Phosphatidylinositol-3 kinase; ERK: Extracellular regulated protein kinases.
EGFR ligands are released from membrane bound proligand forms by membrane bound metalloprotease enzymes of the ADAM family. ADAM17 is a key enzyme regulating release of EGFR ligands: EGF, amphiregulin, and heparin-binding EGF[16].
When liganded, the EGFR undergoes autophosphorylation in trans in the cytoplasmic kinase domain. Phosphorylated tyrosine residues function as docking sites that are recognized by adapter or effector proteins that contain src homology 2 domains or protein tyrosine binding domains. EGFR signal responses are cell-type specific and modulated by the specific activating EGFR ligand, the particular homo or heterodimeric ErbB partners formed and the availability of downstream effector pathways[17].
EGFR is expressed in 60%-80% of CRCs[2]. The mechanisms by which EGFR promotes tumorigenesis are diverse and involve both cell cycle dysregulation and the promotion of factors that aid in tumor survival. Studies in other tumors have dissected some of the mechanisms involved. In breast cancer cells, increased levels of EGFR have been associated with increased proliferative and angiogenic activity. Increased proliferation and angiogenesis are thought to be induced TGF, which correlated with increased mitotic activity. EGFR ligands TGFα and EGF have also been shown to function as chemoattractants for endothelial cells, with TGFα additionally promoting the expression of VEGF[18-20]. EGFR overexpression blocks apoptosis through various mechanisms - in prostate cancer, the Ras/Raf/MEK cascade and the Rac/PAK1 signaling pathway have been implicated in the inactivation of the proapoptotic protein BAD that is inhibited by phosphorylation[21]. In breast cancer EGF and amphiregulin upregulated the expression of certain matrix metalloproteinases implicated in tumor progression and metastasis even in the presence of EGFR inhibition that blocked cell proliferation, suggesting that low levels of EGFR activation may promote MMP9 induction[22]. Finally, microRNAs have been shown to mediate EGFR effects on tumorigenesis. Specifically, miRNA-143 and -145 have been demonstrated to be downregulated when mice with wild type EGFR are fed a western diet high in fat, with increased expression of RAS and MYC implicated as some of the several important G1 regulators mediating this oncogenic effect. Colon cancers seen in EGFR mutant specimens demonstrated an increase in these same miRNAs without an increase in RAS and MYC activity, suggesting an alternate pathway of tumorigenesis in these tumors[23].
EGFR, DIET, AND CHEMOPREVENTION
There is a strong association between Western diet and the incidence of colorectal cancer. This association was initially observed in the late 1960s, in epidemiological studies of the incidence of colon cancer in Japanese-American emigrants over the course of two generations following their adoption of a Western style diet, high in animal fat and red meat[24]. This association has been investigated in the azoxymethane (AOM) model of colon cancer that mimics many of the clinical, histological and molecular features of sporadic human colon cancer. AOM causes O6 methylation of DNA guanine bases resulting in activating mutations in K-ras and CTNNB1 (which codes for β-catenin)[25]. In this model, EGFR is required for tumor promotion by Western diet[26,27]. To demonstrate EGFR requirement, mice with wild type Egfr and mice homozygous for loss-of-function Waved-2 Egfr mutations were fed standard vs high-fat diets and cancer was induced by treating with AOM, followed by tumor promoting dextran sulfate sodium. The Waved-2 Egfr lacks 90% of wild type receptor kinase activity. The Egfr wild type mice in the high-fat group had a significantly higher tumor incidence compared to mice on standard diet but this tumor promoting effect of high fat diet did not occur in mice with mutant Egfr[7]. The proto-oncogenes CTNNB1, MYC, CNND1, and PTGS2 and the EGFR ligand TGFα were also found to be expressed at significantly higher levels in tumors from Egfr wild type mice treated with the high fat diet compared to tumors from mice with mutant Egfr[7].
In more recent preliminary studies we showed that Western diet increases ADAM17 expression and up-regulates EGFR ligands TGF-α and amphiregulin[28]. Stroma-derived factor 1 alpha (Sdf1α) was also increased by WD. Sdf1α is a ligand for the G-protein coupled receptor CXCR4. In other preliminary colon cancer studies we showed that Sdf1α induces the activation of EGFR (EGFR transactivation) by stimulating ADAM17 (Figure 1). ADAM17 is increased in human colon cancer that likely contributes to increases in EGFR ligands and signals observed in these tumors[29]. This mechanism of ligand-driven EGFR signals contrasts with activating EGFR mutations or gene amplification seen in other cancers such as brain and lung cancer[30].
CTNNB1 codes for β-catenin which is an integral part of the cell cytoskeleton as well as an important transcription factor in colonic tumorigenesis, which regulates many key tumor-promoting genes including MYC, CCND1, and PTGS2[31-33]. EGFR is an upstream regulator of β-catenin causing deacetylation that blocks β-catenin degradation and leads to nuclear localization of this molecule[34]. Nuclear localization was increased in all tumors. MYC was also expressed in all tumors and was highest in the mice with wild type Egfr fed a Western diet. CCND1 codes for cyclin D1 that controls G1-> S cell cycle progression and its expression was greater in mice with wild type Egfr compared to those with mutant Egfr. PTGS2 codes for Cox-2 that is also linked to Egfr status, with Cox-2 being 7-8 fold higher in mice with wild type Egfr fed a Western diet compared to standard diet. This finding is of particular interest as prior studies have demonstrated that K-Ras and β-catenin are required to induce Cox-2 in colon cancer cells, underscoring the importance of the EGFR-Kras-Cox-2 signaling cascade. Finally, the expression of the EGFR ligand TGFα was shown to correlate with tumor burden in both genotypes, with a stronger association with the wild type Egfr noted[7].
In addition to EGFR other factors have been implicated in high-fat diet promoted tumorigenesis, including increases in colonic secondary bile acids, elevations of serum insulin, insulin-like growth factor which can also stimulate EGFR through various mechanisms, and diet-induced changes in the microbiome[35-38].
In the study showing EGFR was required for Western diet to promote tumorigenesis, mice fed a Western diet exhibited weight gain, increased visceral fat and insulin resistance, consistent with the development of a metabolic syndrome, which is also implicated in colon cancer causation[7].
Ginseng
The high morbidity and mortality rates of late stage presentation of colon cancer have prompted more investigation into preventative strategies. Ginseng as a chemopreventive agent has been shown to decrease the incidence of various forms of cancer in case control and prospective cohort studies[39-41]. Several studies have demonstrated the anti-tumor effects of ginseng extract, focusing on the diverse group of biologically active chemical structures called ginsenosides, glycosides with dammarane skeletons with varying sugar types, numbers, and linkage positions. Several have been isolated and administered to mice, resulting in statistically significant decreases in lung tumor incidence and reduced growth of colon tumor xenografts. Several mechanisms have been implicated in the anti-tumorigenic properties of ginseng including antioxidant, anti-proliferative, pro-apoptotic and anti-inflammatory actions of ginseng and more recently EGFR inhibitory effects have been identified[42-45]. Additionally, in a mouse model of colitis-associated colon cancer, American ginseng was shown to inhibit inflammation and suppress EGFR signaling, effects that are postulated to contribute to ginseng’s anti tumorigenic properties[46] (Figure 1).
In studies of mice treated with a combination of Western diet alone or WD plus ginseng, colonic mucosal EGFR signals were noted to be increased in the Western diet group and Ginseng inhibited these increases. Ginseng also appears to inhibit tumorigenesis through other mechanisms, including the induction of apoptosis. Ginseng’s anti tumor effects likely require ginseng metabolite activation by the colonic microbiome as several biologically active metabolites of ginsenosides are synthesized by gut microbes. One metabolite in particular, 20-O-b-(D-glucopyranosyl)-20(S)-protopanaxadiol or compound K, was shown to suppress growth of colon tumor xenografts[47].
Green tea
Several other naturally occurring products have been studied as potential chemopreventative agents and been shown to inhibit EGFR signals. A bioactive green tea polyphenol, epigallocatechin-3-gallate (EGCG), has been shown to selectively inhibit EGF-dependent signaling in cervical cancer cells, leading to growth cessation and cell apoptosis. The mechanism of this selective inhibition was shown to involve suppression of EGFR-induced ERK1/2 (aka MAPK1 and 3) and AKT activation as well as direct suppression of ERK and AKT[48] (Figure 1). These kinases have been implicated in cell cycle progression; ERK1/2 signals both activation of the intrinsic or extrinsic apoptotic pathway depending on the ligand and cell type, and AKT has been shown to regulate cell proliferation and survival, with constitutive up-regulation of activated AKT demonstrated in many types of human cancers[49-51]. The importance of these cellular pathways is underscored by the observation that only selective kinases downstream of EGFR were inhibited, but not others. Increasing concentrations of EGCG exerted both short term reversible effects on cell cycle progression and long term cellular changes with increased rates of apoptosis[49].
Curcumin
Another naturally occurring substance that has drawn the attention of the scientific community is curcumin, the yellow pigment of tumeric found in curry. It is produced by the rhizome of the plant tumeric and has been safely consumed and utilized for its medicinal properties for centuries. This substance has been shown to inhibit the growth of cancer cells by suppressing gene expression of cyclinD1 and EGFR[52]. Recent studies have demonstrated that curcumin inhibits binding of the transcription factor EGR-1 to the EGFR promoter as well as suppresing EGR-1 gene expression through the ERK signal pathway, thereby suppressing EGR-1 transactivation activity[53] (Figure 1). Of note, the concentrations required to achieve this growth suppression in vitro, are much higher than those normally achieved in blood and tissue in vivo following curcumin ingestion, but for colon cancer prevention colonic luminal concentrations may be more relevant. Recent developments of more stable curcumin analogues may also increase the efficacy of this compound[54].
EGFR AS A CHEMOTHERAPEUTIC TARGET
With the potential central role of EGFR in tumorigenesis, several groups have successfully developed neutralizing antibodies or kinase inhibitors. Of particular interest are the monoclonal antibodies cetuximab and panitumaumab, as well as the small molecule inhibitors gefitinib and erlotinib.
Cetuximab and panitumumab act by binding the extracellular domain of EGFR and thereby inhibiting ligand-dependent activation and receptor dimerization. Cetuximab also may induce an immune response by antibody-dependent cell-mediated cytotoxicity[55-58] (Figure 1). In colon cancer, cetuximab is currently FDA approved for EGFR-positive metastatic disease in patients who cannot tolerate irinotecan-based therapy, or in combination with oxaliplatin, irinotecan, and 5-FU. These recommendations are based on a 2009 study that examined the use of cetuximab as a first-line treatment with FOLFOX, with assessment of tumor response in KRAS wildtype vs KRAS mutant tumors. Tumors with KRAS mutations resulting in constitutively active GTP-binding protein were shown to be resistant to EGFR inhibitors[59,60]. This trial confirmed previous findings and demonstrated significant differences between tumor response and risk of disease progression in the KRAS mutant and KRAS WT groups with the addition of cetuximab, though a difference of progression-free survival was not detected[61]. Panitumumab is also used in metastatic CRC and also requires WT KRAS for efficacy[62]. More recently a study suggested that tumors with KRAS mutations in codon 13 may remain susceptible to Cetuximab, whereas those with KRAS codon 12 mutations did not[63].
Small molecule EGFR receptor tyrosine kinase inhibitors, gefitinib and erlotinib are not used in the treatment of CRC. Gefitinib was initially approved for third-line treatment of patients with non-small cell lung cancer (NSCLC) based on preliminary small clinical trials but later studies demonstrated conflicting results of its efficacy[64]. A phase II RCT of FOLFIRI vs gefitinib plus FOLFIRI did not show any benefit and demonstrated high toxicity[65]. There have since been studies looking at the efficacy of gefitinib in select groups of patients, initially based on demographic considerations such as non-smokers, Asians, and women, and later based on specific activating mutations[66,67], underscoring the importance of careful patient selection in maximizing the success of these targeted agents. Erlotinib is currently approved for second-line treatment of patients with locally advanced or metastatic NSCLC and first-line treatment for patients with locally advanced, unresectable, or metastatic pancreatic cancer in combination with gemcitabine[55]. Recent studies have looked at the combination of cetuximab and erlotinib in the treatment of chemotherapy-refractory metastatic CRC with promising results. These studies demonstrated improvement in response rates and progression free survival in patients with tumors having wild type EGFR compared to failures in the patients with tumors having mutant EGFR[68].
These studies point to the importance of assessing the mutation status of EGFR and KRAS when using EGFR targeted therapies. It should be noted that there are many other factors that determine a given patient’s initial and subsequent response to therapy. This is highlighted by the fact that KRAS mutations only account for approximately 30%-40% of nonresponsive patients[60,69]. Mutations in other downstream signaling molecules such as BRAF have been shown to correlate with unresponsiveness to cetuximab and panitumumab[70]. Raf proteins are principal downstream effectors of KRAS in the RAS-RAF-MEK-MAPKs signaling cascade. They are activated directly by KRAS and serve to phosphorylate and activate the downstream kinase MEK, which phosphorylates ERK leading to numerous Ras-induced cellular responses[71,72]. Specifically, BRAF has a higher affinity for MEK leading to stronger MEK stimulation than A-Raf or c-Raf, and plays a critical role in promoting cell survival by activating the MAPK pathway[73]. The prognostic significance of these mutations with respect to survival is less clear, with some data indicating that gender may a role how these mutations affect tumor virulence. In one prospective cohort study, BRAF mutations were associated with a reduced cancer specific survival in men, particularly in lymph node positive disease, when compared to women. Additionally in microsatellite stable tumors, BRAF was found to be an independent predictor of poor prognosis in men[74]. The exact mechanisms of how gender may interact with BRAF mutation status are not yet clear. However, even when adjusting for BRAF mutant tumors to assess nonresponders to cetuximab and panitumumab, approximately 41% of nonresponders are left unaccounted for, suggesting the presence of other unknown mechanisms of resistance[70].
Responses even in selected groups of patients with wild type EGFR, KRAS, and BRAF alleles is not uniform, and all patients will ultimately develop acquired resistance to targeted therapy with monoclonal antibodies. Increased ERBB2 signaling has been shown to be one such mediator in resistant clones of previously cetuximab-sensitive cell lines via the up-regulation of ERK1/2 signaling[75]. This can occur through the amplification of ERBB2 itself or through the overexpression of hereguglin, one of the ERBB3 ligands. Increased c-Met signaling may be another mechanism for EGFR antibody resistance[76,77]. Importantly, restoration of sensitivity to cetuximab has been demonstrated with the application of interfering RNA or small molecule inhibitors such as gefitinib, suggesting a potential valuable role of these small molecule kinase inhibitors in restoring efficacy of EGFR targeted therapies.
CONCLUSION
We have reviewed the risk factors and current treatment paradigm for colorectal cancer, with an emphasis on the role of targeted chemotherapy and chemoprevention as they relate to EGFR blockade. The complex interplay between other growth promoting pathways that cross talk with EGFR and downstream EGFR effectors that can be driven by activating mutations make strategies that target EGFR vulnerable to several escape mechanisms. The role of Western diet and the exciting field of chemoprevention offer opportunities to target EGFR signaling cascade which plays a critical role in tumor promotion and progression. Future development of anti-EGFR directed nanoparticles restricted to the gut that could inhibit over active EGFR signals might hold promise to safely reduce colorectal cancer risk.
Footnotes
Conflict-of-interest statement: None.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 5, 2015
First decision: June 3, 2015
Article in press: July 23, 2015
P- Reviewer: Li LW, Scaggiante B, Schiavina R, Surlin VM S- Editor: Tian YL L- Editor: A E- Editor: Jiao XK
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Cohen RB. Epidermal growth factor receptor as a therapeutic target in colorectal cancer. Clin Colorectal Cancer. 2003;2:246–251. doi: 10.3816/CCC.2003.n.006. [DOI] [PubMed] [Google Scholar]
- 3.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Clendenning M, Sotamaa K, Prior T, Westman JA, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26:5783–5788. doi: 10.1200/JCO.2008.17.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bülow S, Faurschou Nielsen T, Bülow C, Bisgaard ML, Karlsen L, Moesgaard F. The incidence rate of familial adenomatous polyposis. Results from the Danish Polyposis Register. Int J Colorectal Dis. 1996;11:88–91. doi: 10.1007/BF00342466. [DOI] [PubMed] [Google Scholar]
- 5.Fedirko V, Tramacere I, Bagnardi V, Rota M, Scotti L, Islami F, Negri E, Straif K, Romieu I, La Vecchia C, et al. Alcohol drinking and colorectal cancer risk: an overall and dose-response meta-analysis of published studies. Ann Oncol. 2011;22:1958–1972. doi: 10.1093/annonc/mdq653. [DOI] [PubMed] [Google Scholar]
- 6.Henderson TO, Oeffinger KC, Whitton J, Leisenring W, Neglia J, Meadows A, Crotty C, Rubin DT, Diller L, Inskip P, et al. Secondary gastrointestinal cancer in childhood cancer survivors: a cohort study. Ann Intern Med. 2012;156:757–766, W-260. doi: 10.1059/0003-4819-156-11-201206050-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dougherty U, Cerasi D, Taylor I, Kocherginsky M, Tekin U, Badal S, Aluri L, Sehdev A, Cerda S, Mustafi R, et al. Epidermal growth factor receptor is required for colonic tumor promotion by dietary fat in the azoxymethane/dextran sulfate sodium model: roles of transforming growth factor-{alpha} and PTGS2. Clin Cancer Res. 2009;15:6780–6789. doi: 10.1158/1078-0432.CCR-09-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Botteri E, Iodice S, Bagnardi V, Raimondi S, Lowenfels AB, Maisonneuve P. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300:2765–2778. doi: 10.1001/jama.2008.839. [DOI] [PubMed] [Google Scholar]
- 9.Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med. 1992;326:658–662. doi: 10.1056/NEJM199203053261002. [DOI] [PubMed] [Google Scholar]
- 10.Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected] Am J Gastroenterol. 2009;104:739–750. doi: 10.1038/ajg.2009.104. [DOI] [PubMed] [Google Scholar]
- 11.Amos-Landgraf JM, Heijmans J, Wielenga MC, Dunkin E, Krentz KJ, Clipson L, Ederveen AG, Groothuis PG, Mosselman S, Muncan V, et al. Sex disparity in colonic adenomagenesis involves promotion by male hormones, not protection by female hormones. Proc Natl Acad Sci USA. 2014;111:16514–16519. doi: 10.1073/pnas.1323064111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fensterheim R. Colon cancer. NCCN Guidelines. Available from: http://www.nccn.org/about/nhl.pdf.
- 14.Czito BG, Bendell J, Willett CG. Radiation therapy for resectable colon cancer. Is there a role in the modern chemotherapy era? Oncology (Williston Park) 2006;20:179–187; discussion 187-188, 192. [PubMed] [Google Scholar]
- 15.Wells A. EGF receptor. Int J Biochem Cell Biol. 1999;31:637–643. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- 16.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arteaga CL. The epidermal growth factor receptor: from mutant oncogene in nonhuman cancers to therapeutic target in human neoplasia. J Clin Oncol. 2001;19:32S–40S. [PubMed] [Google Scholar]
- 18.de Jong JS, van Diest PJ, van der Valk P, Baak JP. Expression of growth factors, growth-inhibiting factors, and their receptors in invasive breast cancer. II: Correlations with proliferation and angiogenesis. J Pathol. 1998;184:53–57. doi: 10.1002/(SICI)1096-9896(199801)184:1<53::AID-PATH6>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 19.Grotendorst GR, Soma Y, Takehara K, Charette M. EGF and TGF-alpha are potent chemoattractants for endothelial cells and EGF-like peptides are present at sites of tissue regeneration. J Cell Physiol. 1989;139:617–623. doi: 10.1002/jcp.1041390323. [DOI] [PubMed] [Google Scholar]
- 20.Gille J, Swerlick RA, Caughman SW. Transforming growth factor-alpha-induced transcriptional activation of the vascular permeability factor (VPF/VEGF) gene requires AP-2-dependent DNA binding and transactivation. EMBO J. 1997;16:750–759. doi: 10.1093/emboj/16.4.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sastry KS, Karpova Y, Kulik G. Epidermal growth factor protects prostate cancer cells from apoptosis by inducing BAD phosphorylation via redundant signaling pathways. J Biol Chem. 2006;281:27367–27377. doi: 10.1074/jbc.M511485200. [DOI] [PubMed] [Google Scholar]
- 22.Kondapaka SB, Fridman R, Reddy KB. Epidermal growth factor and amphiregulin up-regulate matrix metalloproteinase-9 (MMP-9) in human breast cancer cells. Int J Cancer. 1997;70:722–726. doi: 10.1002/(sici)1097-0215(19970317)70:6<722::aid-ijc15>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 23.Zhu H, Dougherty U, Robinson V, Mustafi R, Pekow J, Kupfer S, Li YC, Hart J, Goss K, Fichera A, et al. EGFR signals downregulate tumor suppressors miR-143 and miR-145 in Western diet-promoted murine colon cancer: role of G1 regulators. Mol Cancer Res. 2011;9:960–975. doi: 10.1158/1541-7786.MCR-10-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haenszel W, Kurihara M. Studies of Japanese migrants. I. Mortality from cancer and other diseases among Japanese in the United States. J Natl Cancer Inst. 1968;40:43–68. [PubMed] [Google Scholar]
- 25.Takahashi M, Wakabayashi K. Gene mutations and altered gene expression in azoxymethane-induced colon carcinogenesis in rodents. Cancer Sci. 2004;95:475–480. doi: 10.1111/j.1349-7006.2004.tb03235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fichera A, Little N, Jagadeeswaran S, Dougherty U, Sehdev A, Mustafi R, Cerda S, Yuan W, Khare S, Tretiakova M, et al. Epidermal growth factor receptor signaling is required for microadenoma formation in the mouse azoxymethane model of colonic carcinogenesis. Cancer Res. 2007;67:827–835. doi: 10.1158/0008-5472.CAN-05-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dougherty U, Sehdev A, Cerda S, Mustafi R, Little N, Yuan W, Jagadeeswaran S, Chumsangsri A, Delgado J, Tretiakova M, et al. Epidermal growth factor receptor controls flat dysplastic aberrant crypt foci development and colon cancer progression in the rat azoxymethane model. Clin Cancer Res. 2008;14:2253–2262. doi: 10.1158/1078-0432.CCR-07-4926. [DOI] [PubMed] [Google Scholar]
- 28.Dougherty U, Mustafi R, Valuckaite V, Konda VJ, Pekow J, Sadiq F, Haider HI, Adhikari S, Hart J, Joseph L, et al. Western diet up-regulates ADAM17, a key meditator of EGFR signaling via activation of colonic renin-angiotensin system and inhibition of miR-145,-148a and -152. Gastroenterology. 2015;148:S–99. [Google Scholar]
- 29.Blanchot-Jossic F, Jarry A, Masson D, Bach-Ngohou K, Paineau J, Denis MG, Laboisse CL, Mosnier JF. Up-regulated expression of ADAM17 in human colon carcinoma: co-expression with EGFR in neoplastic and endothelial cells. J Pathol. 2005;207:156–163. doi: 10.1002/path.1814. [DOI] [PubMed] [Google Scholar]
- 30.Barber TD, Vogelstein B, Kinzler KW, Velculescu VE. Somatic mutations of EGFR in colorectal cancers and glioblastomas. N Engl J Med. 2004;351:2883. doi: 10.1056/NEJM200412303512724. [DOI] [PubMed] [Google Scholar]
- 31.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 32.Araki Y, Okamura S, Hussain SP, Nagashima M, He P, Shiseki M, Miura K, Harris CC. Regulation of cyclooxygenase-2 expression by the Wnt and ras pathways. Cancer Res. 2003;63:728–734. [PubMed] [Google Scholar]
- 33.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Zhang X, Polakiewicz RD, Yao TP, Comb MJ. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J Biol Chem. 2008;283:12686–12690. doi: 10.1074/jbc.C700185200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGarr SE, Ridlon JM, Hylemon PB. Diet, anaerobic bacterial metabolism, and colon cancer: a review of the literature. J Clin Gastroenterol. 2005;39:98–109. [PubMed] [Google Scholar]
- 36.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 37.van der Veeken J, Oliveira S, Schiffelers RM, Storm G, van Bergen En Henegouwen PM, Roovers RC. Crosstalk between epidermal growth factor receptor- and insulin-like growth factor-1 receptor signaling: implications for cancer therapy. Curr Cancer Drug Targets. 2009;9:748–760. doi: 10.2174/156800909789271495. [DOI] [PubMed] [Google Scholar]
- 38.Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, Schloss PD. The gut microbiome modulates colon tumorigenesis. MBio. 2013;4:e00692–e00613. doi: 10.1128/mBio.00692-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yun TK, Choi SY. Preventive effect of ginseng intake against various human cancers: a case-control study on 1987 pairs. Cancer Epidemiol Biomarkers Prev. 1995;4:401–408. [PubMed] [Google Scholar]
- 40.Yun TK, Zheng S, Choi SY, Cai SR, Lee YS, Liu XY, Cho KJ, Park KY. Non-organ-specific preventive effect of long-term administration of Korean red ginseng extract on incidence of human cancers. J Med Food. 2010;13:489–494. doi: 10.1089/jmf.2009.1275. [DOI] [PubMed] [Google Scholar]
- 41.Yun TK, Choi SY. Non-organ specific cancer prevention of ginseng: a prospective study in Korea. Int J Epidemiol. 1998;27:359–364. doi: 10.1093/ije/27.3.359. [DOI] [PubMed] [Google Scholar]
- 42.Yun TK. Experimental and epidemiological evidence on non-organ specific cancer preventive effect of Korean ginseng and identification of active compounds. Mutat Res. 2003;523-524:63–74. doi: 10.1016/s0027-5107(02)00322-6. [DOI] [PubMed] [Google Scholar]
- 43.Qi LW, Wang CZ, Yuan CS. American ginseng: potential structure-function relationship in cancer chemoprevention. Biochem Pharmacol. 2010;80:947–954. doi: 10.1016/j.bcp.2010.06.023. [DOI] [PubMed] [Google Scholar]
- 44.Yun TK, Lee YS, Lee YH, Kim SI, Yun HY. Anticarcinogenic effect of Panax ginseng C.A. Meyer and identification of active compounds. J Korean Med Sci. 2001;16 Suppl:S6–18. doi: 10.3346/jkms.2001.16.S.S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han HJ, Yoon BC, Lee SH, Park SH, Park JY, Oh YJ, Lee YJ. Ginsenosides inhibit EGF-induced proliferation of renal proximal tubule cells via decrease of c-fos and c-jun gene expression in vitro. Planta Med. 2002;68:971–974. doi: 10.1055/s-2002-35659. [DOI] [PubMed] [Google Scholar]
- 46.Cui X, Jin Y, Poudyal D, Chumanevich AA, Davis T, Windust A, Hofseth A, Wu W, Habiger J, Pena E, et al. Mechanistic insight into the ability of American ginseng to suppress colon cancer associated with colitis. Carcinogenesis. 2010;31:1734–1741. doi: 10.1093/carcin/bgq163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dougherty U, Mustafi R, Wang Y, Musch MW, Wang CZ, Konda VJ, Kulkarni A, Hart J, Dawson G, Kim KE, et al. American ginseng suppresses Western diet-promoted tumorigenesis in model of inflammation-associated colon cancer: role of EGFR. BMC Complement Altern Med. 2011;11:111. doi: 10.1186/1472-6882-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sah JF, Balasubramanian S, Eckert RL, Rorke EA. Epigallocatechin-3-gallate inhibits epidermal growth factor receptor signaling pathway. Evidence for direct inhibition of ERK1/2 and AKT kinases. J Biol Chem. 2004;279:12755–12762. doi: 10.1074/jbc.M312333200. [DOI] [PubMed] [Google Scholar]
- 49.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–395. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 50.Cobb MH, Hepler JE, Cheng M, Robbins D. The mitogen-activated protein kinases, ERK1 and ERK2. Semin Cancer Biol. 1994;5:261–268. [PubMed] [Google Scholar]
- 51.Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J. 2010;277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 52.Chen A, Xu J. Activation of PPAR{gamma} by curcumin inhibits Moser cell growth and mediates suppression of gene expression of cyclin D1 and EGFR. Am J Physiol Gastrointest Liver Physiol. 2005;288:G447–G456. doi: 10.1152/ajpgi.00209.2004. [DOI] [PubMed] [Google Scholar]
- 53.Chen A, Xu J, Johnson AC. Curcumin inhibits human colon cancer cell growth by suppressing gene expression of epidermal growth factor receptor through reducing the activity of the transcription factor Egr-1. Oncogene. 2006;25:278–287. doi: 10.1038/sj.onc.1209019. [DOI] [PubMed] [Google Scholar]
- 54.Kanwar SS, Yu Y, Nautiyal J, Patel BB, Padhye S, Sarkar FH, Majumdar AP. Difluorinated-curcumin (CDF): a novel curcumin analog is a potent inhibitor of colon cancer stem-like cells. Pharm Res. 2011;28:827–838. doi: 10.1007/s11095-010-0336-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chabner B, Barnes J, Neal J, Olson E, Mujagic H, Sequist L, Wilson W, Longo D, Mitsiades C, Richardson P. Targeted Therapies: Tyrosine Kinase Inhibitors, Monoclonal Antibodies, and Cytokines. In: Goodman and Gilman’s The Pharmacological Basis of Therapeutics., editor. New York, NY: McGraw-Hill;; 2011. [Google Scholar]
- 56.Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin Cancer Res. 1995;1:1311–1318. [PubMed] [Google Scholar]
- 57.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 58.Kurai J, Chikumi H, Hashimoto K, Yamaguchi K, Yamasaki A, Sako T, Touge H, Makino H, Takata M, Miyata M, et al. Antibody-dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin Cancer Res. 2007;13:1552–1561. doi: 10.1158/1078-0432.CCR-06-1726. [DOI] [PubMed] [Google Scholar]
- 59.Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 60.De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, Biesmans B, Van Laethem JL, Peeters M, Humblet Y, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19:508–515. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 61.Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–671. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 62.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 63.Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol. 2012;30:3570–3577. doi: 10.1200/JCO.2012.42.2592. [DOI] [PubMed] [Google Scholar]
- 64.Maruyama R, Nishiwaki Y, Tamura T, Yamamoto N, Tsuboi M, Nakagawa K, Shinkai T, Negoro S, Imamura F, Eguchi K, et al. Phase III study, V-15-32, of gefitinib versus docetaxel in previously treated Japanese patients with non-small-cell lung cancer. J Clin Oncol. 2008;26:4244–4252. doi: 10.1200/JCO.2007.15.0185. [DOI] [PubMed] [Google Scholar]
- 65.Santoro A, Comandone A, Rimassa L, Granetti C, Lorusso V, Oliva C, Ronzoni M, Siena S, Zuradelli M, Mari E, et al. A phase II randomized multicenter trial of gefitinib plus FOLFIRI and FOLFIRI alone in patients with metastatic colorectal cancer. Ann Oncol. 2008;19:1888–1893. doi: 10.1093/annonc/mdn401. [DOI] [PubMed] [Google Scholar]
- 66.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 67.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 68.Weickhardt AJ, Price TJ, Chong G, Gebski V, Pavlakis N, Johns TG, Azad A, Skrinos E, Fluck K, Dobrovic A, et al. Dual targeting of the epidermal growth factor receptor using the combination of cetuximab and erlotinib: preclinical evaluation and results of the phase II DUX study in chemotherapy-refractory, advanced colorectal cancer. J Clin Oncol. 2012;30:1505–1512. doi: 10.1200/JCO.2011.38.6599. [DOI] [PubMed] [Google Scholar]
- 69.Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboüé R, Tuech JJ, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer. 2007;96:1166–1169. doi: 10.1038/sj.bjc.6603685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 71.Hill CS, Treisman R. Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell. 1995;80:199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- 72.Zhang BH, Guan KL. Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J. 2000;19:5429–5439. doi: 10.1093/emboj/19.20.5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Erhardt P, Schremser EJ, Cooper GM. B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol Cell Biol. 1999;19:5308–5315. doi: 10.1128/mcb.19.8.5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wangefjord S, Sundström M, Zendehrokh N, Lindquist KE, Nodin B, Jirström K, Eberhard J. Sex differences in the prognostic significance of KRAS codons 12 and 13, and BRAF mutations in colorectal cancer: a cohort study. Biol Sex Differ. 2013;4:17. doi: 10.1186/2042-6410-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra86. doi: 10.1126/scitranslmed.3002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Inno A, Di Salvatore M, Cenci T, Martini M, Orlandi A, Strippoli A, Ferrara AM, Bagalà C, Cassano A, Larocca LM, et al. Is there a role for IGF1R and c-MET pathways in resistance to cetuximab in metastatic colorectal cancer? Clin Colorectal Cancer. 2011;10:325–332. doi: 10.1016/j.clcc.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 77.Liu C, Park M, Tsao MS. Overexpression of c-met proto-oncogene but not epidermal growth factor receptor or c-erbB-2 in primary human colorectal carcinomas. Oncogene. 1992;7:181–185. [PubMed] [Google Scholar]