

Abstract
Over the past few decades, major strides have advanced the techniques for early detection and treatment of cancer. However, metastatic tumor growth still accounts for the majority of cancer-related deaths worldwide. In fact, breast cancers are notorious for relapsing years or decades after the initial clinical treatment, and this relapse can vary according to the type of breast cancer. In estrogen receptor-positive breast cancers, late tumor relapses frequently occur whereas relapses in estrogen receptor-negative cancers or triple negative tumors arise early resulting in a higher mortality risk. One of the main causes of metastasis is tumor dormancy in which cancer cells remain concealed, asymptomatic, and untraceable over a prolonged period of time. Under certain conditions, dormant cells can re-enter into the cell cycle and resume proliferation leading to recurrence. However, the molecular and cellular regulators underlying this transition remain poorly understood. To date, three mechanisms have been identified to trigger tumor dormancy including cellular, angiogenic, and immunologic dormancies. In addition, recent studies have suggested that DNA repair mechanisms may contribute to the survival of dormant cancer cells. In this article, we summarize the recent experimental and clinical evidence governing cancer dormancy. In addition, we will discuss the role of DNA repair mechanisms in promoting the survival of dormant cells. This information provides mechanistic insight to explain why recurrence occurs, and strategies that may enhance therapeutic approaches to prevent disease recurrence.
Keywords: Quiescence, Homologous recombination, Non-homologous end joining, Tumor dormancy, DNA repair
Core tip: One of the main causes of metastasis is tumor dormancy in which cancer cells remain concealed, asymptomatic, and untraceable over a prolonged period of time. Recent studies have suggested that DNA repair mechanisms may contribute to the survival of dormant cancer cells. Under certain conditions, dormant cells can re-enter into the cell cycle and resume proliferation leading to recurrence. Understanding the molecular and cellular regulators underlying the transition from tumor dormancy to metastatic disease may provide insight into how recurrence occurs and also discover strategies that may enhance therapeutic approaches to prevent metastatic cancer.
INTRODUCTION
Metastatic tumor growth can account for the majority of cancer-related deaths worldwide[1]. In fact, nearly 30% of breast cancers will relapse years or decades after the initial treatment[2-4]. Different subtypes of breast cancer display different recurrence behaviors. For examples, late tumor relapses frequently occur in estrogen receptor-positive (ER+) breast cancers whereas relapses in estrogen receptor-negative breast cancers or triple negative breast tumors arise early resulting in a higher mortality risk[2,5]. Tumor dormancy, one of the main causes of metastasis, occurs when disseminated tumor cells remain concealed, asymptomatic, and untraceable over a prolonged period of time. Cancer cells can become dormant at the onset of disease or after the initial therapeutic treatment, and can remain dormant for years or even decades after the first treatment[6]. Dormant cells can be characterized by exhibiting slow growth rates, having the ability to escape frontline treatment and the host’s immune system, and demonstrating the capability to self-renew. Multiple studies have shown that many cancers such as breast and prostate cancers, melanoma, B-cell lymphoma, leukemia, and carcinoma contain dormant cancer cells[7-15]. Therefore, it is important to understand the molecular mechanisms that govern the transition of dormant cells into metastatic disease.
To date, three mechanisms have been identified to trigger tumor dormancy including cellular, angiogenic, and immunologic dormancies (Figure 1)[16]. Cellular dormancy is characterized as a state in which cells are quiescent and halted in the G0 phase of the cell cycle (Figure 1). The microenvironment of tumors can prompt cancer cells to enter into cellular dormancy like hypoxic environments, which is associated with malignancies, and causes cancer cell proliferation to decrease[17]. Under certain circumstances such as the addition of growth factor, cytokines, nutrients or chemical agents, dormant cells can re-enter into the cell cycle and resume proliferation. Many cancer therapeutic treatments target the cell cycle which permits the cells to enter into quiescence. This allows the cancer cells to escape treatment subsequently leading to disease recurrence[16,18-20]. Once dormant cancer cells exit G0 arrest, a second mechanism termed angiogenic dormancy can limit the tumor size by preventing angiogenesis and therefore the tumor cannot obtain the nutrients required for continual growth. These cells can maintain a balance between proliferation and apoptosis resulting in the inability to detect the tumor[6,16] (Figure 1). The immune system can also contribute to cancer cell dormancy by maintaining a balance between clearance and proliferation[16] (Figure 1). During immunologic dormancy, DTCs can be eliminated or they can stay in an equilibrium state and, over time, environmental factors and genomic instability can cause the cells to exit the equilibrium state resulting in tumor growth and recurrence[21].
Figure 1.
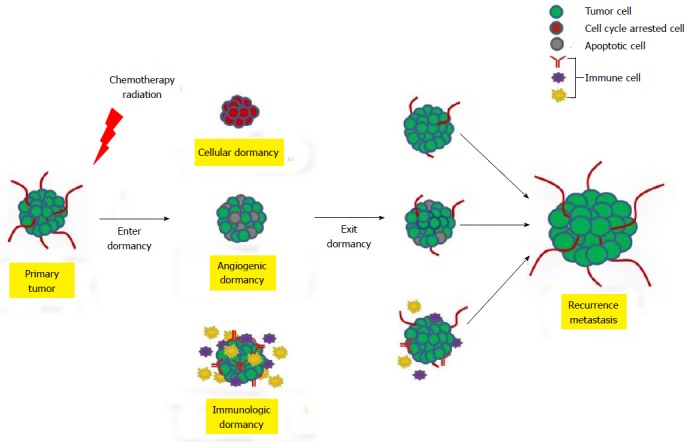
Mechanisms of human tumor dormancy. Schematic depicting three mechanisms that lead to tumor dormancy after the initial clinical treatment. Tumor dormancy can result from cell cycle arrest (cellular dormancy), tumor size limitation due to a lack of functional blood vessels (angiogenic dormancy), or immunosurveillance (immunologic dormancy). Figure adapted from Almog[16] (2010) and Wang and Lin[6] (2013).
The precise molecular mechanism in which cancer cells enter and exit dormancy remains to be elucidated. One mechanism that plays a major role in cancer growth is the DNA repair pathways, and recently, studies indicate that the DNA repair pathways can lead to tumor dormancy[15,22]. Therefore, it may be possible to target dormant cancer cells through these pathways. Below, we will discuss the current understanding of the three mechanism of tumor dormancy and the role of double-strand breaks (DSBs) DNA repair pathways in dormant cancer cells. This information may improve the development of relevant study models and enhance therapeutic approaches to prevent disease recurrence.
CELLULAR DORMANCY
Cellular dormancy or quiescence is a process that occurs naturally in normal adult stem cells such as hemopoietic and spermatogonial stem cells. These stem cells serve as a source for self-renewal and maintenance of tissues throughout a person’s lifetime. However, in a heterogeneous cancer cell population, dormancy can be disadvantageous because cancer cells can evade treatments leading to metastatic recurrence[16,18-20] (Figure 1).
Several studies have demonstrated that the expression of the cellular proliferation, Ki-67, and apoptotic markers are significantly diminished in patients with clinical dormancy[23-27]. In addition, positive Ki-67 expression was correlated with breast cancer recurrence and poor prognosis[28]. The stepwise progression of the cell cycle is regulated by cyclins and cyclin-dependent kinases (CDKs). In particular, cellular quiescence is controlled either directly or indirectly by these regulators. Within the microenvironment, the interactions between the CDK inhibitors, p27 (Kip1) and p21 (Cip1, Waf1), maintain a balance between proliferative and dormant hematopoietic stem cells[29]. Recently, Fitzgerald et al[30] (2015) demonstrated that treatment of head and neck squamous cell carcinoma patients with radiation resulted in cellular quiescence via the upregulation of p21. In addition, the DREAM complex which consist of a Retinoblastoma (Rb)-like pocket protein, E2F, and mutilvulval class B (MuvB) proteins, is a critical regulator of cell cycle arrest[31]. The MuvB protein is known to recruit, bind, and direct transcription regulators to the promoter of key cell cycle genes during various stages within the cell cycle[32]. During dormancy, MuvB binds to all of the components of the DREAM complex and represses the transcription of all cell cycle-dependent genes[32-34]. Disruption of various components of the DREAM complex results in the inability to repress the cell-cycle dependent genes and subsequently the cells re-enter the cell cycle[35,36]. Quiescence is also established by the dual specificity tyrosine phosphorylation-regulated kinase (DYRK). This protein activates the DREAM complex by phosphorylating a MuvB subunit, LIN52, which promotes the interaction of MutB with the other core components of the DREAM complex[31]. An isoform of DYRK, DYRK1B, can stabilize p27 (Kip1) which increases the turnover of cyclin D consequently inhibiting cell from entering into the cell cycle[37,38]. CDK4 and CDK6 inactivate the tumor suppressor, Rb, subsequently allowing cells to enter into the cell cycle. By pharmaceutically blocking these kinases, Rb-cells can exit the cell cycle and enter into a dormant state[39]. These results clearly demonstrate the need for balance between the DREAM and proliferative complexes in order to maintain cells in a quiescent state.
Mis-regulation of cell cycle proteins can result in tumor formation, dormancy, and recurrence. Prostate cancer, breast cancer, and renal cell carcinoma are linked to the loss of p27 (Kip1)[40-42]. In addition, reduction in p27 (Kip1) is used as a strong prognostic marker for recurrence and poor outcomes in renal cell carcinoma patients[42]. Loss of p53, the upstream regulator of p21, was correlated with drug resistance and recurrence in colorectal cancer[43]. Overexpression of cyclin D is associated with recurrence of multiple neoplasms including breast, lymphomas, prostate, and non-small cell lung cancers[44-46]. Overexpression of cyclin D1 can occur via a multitude of different mechanisms including genetic rearrangements, amplification of the gene locus, oncogenic signaling, and mutation in the gene that result in the inability to degrade the protein[44]. Recently, Kim et al[47] (2014) reported that overexpression of the cell cycle regulators CDK4, CDK6, pRB, and cyclin D1 was correlated with the recurrence of atypical meningioma. Furthermore, some evidence suggested that overexpression of CDK4 may be connected to nasopharyngeal carcinoma tumor aggression and serve as a diagnostic biomarker[48]. Clearly, these results demonstrate the importance in controlling the cell cycle and how aberrant regulation may lead to tumor recurrence and poor prognosis.
ANGIOGENIC DORMANCY
The majority of tumors require the recruitment of blood vessels to support continual growth. When tumors fail to establish a sufficient vasculature, then they enter into a state of avascular or angiogenic dormancy (Figure 1). Tumor dormancy via angiogenesis requires the interaction between the microenvironment and cell cycle regulators including p21, p27, Myc, urokinase receptor (u-PAR), extracellular regulated kinase (ERK), and p38[49]. Blockage of the metastasis-associated u-PAR, intergrins, focal adhesion kinase or epithelial growth factor receptor can result in tumor suppression and induction of tumor dormancy[49]. U-PAR can also regulate tumor dormancy by favoring p38 activation over ERK activation[50]. In addition, the activation of the PI3K/c-Myc pathway controls the level of thrombospondin (TSP), a vital factor of tumor dormancy[16]. Troyanovsky et al[51] (2001) also discovered that the expression of angiostatin can control tumor dormancy by suppressing tumor growth, and one mediator of angiostatin, angiomotin, was highly elevated in dormant cells.
The transition from avascular tumor to a highly vascularized tumor is termed the “angiogenic switch”[16,21]. Balancing the pro-angiogenic and anti-angiogenic factors is vital in regulating the angiogenic switch. Satchi-Fainaro et al[52] (2012) discovered that dormant glioblastoma cells express high levels of anti-angiogenic factors including TSP, angiomotin, and insulin-like growth factor binding protein 5, and low levels of pro-angiogenic proteins (endothelial cell-specific marker 1 and epithelial growth factor receptor). Furthermore, TSP-1 and endothelial-derived perlecan were found to maintain breast cancer cells in a dormant state therefore suppressing tumor growth[53,54]. Another key protein that plays a role in controlling the switch from dormancy to tumor growth is heat shock protein 27 (HSP27)[55]. Decreased expression of HSP27 in breast cancer cells resulted in reduced cell proliferation and migration caused by lower levels of secreted vascular endothelial growth factor (VEGF) and basic fibroblast growth factor, known pro-angiogenic factors[55]. Recently, the hypoxia inducible factor, HIF-2α, was shown to promote angiogenesis in hepatocellular carcinoma[56]. HIF-2α increased plasminogen activator inhibitor 1 which lowered active plasmin concentrations resulting in increased angiogenesis[56].
The formation of dormant cell niches can be controlled by the microenvironment. Several proteins such as latent transforming growth factor β (TGF-β) binding protein (LTBP), bone morphogenetic protein 7 (BMP7), and osteopontin (OPN) all influence the establishment of quiescent cell niches[57-59]. Overexpression of LTBP in nasopharyngeal carcinoma induced cancer cell dormancy and reduced VEGF expression thus inhibiting the migration and angiogenesis of tumor cells[57]. BMP7, a member of the TGF-β superfamily, signaling facilitates the balance between dormant prostate cancer cells and metastasis[58]. Administration of BMP7 in mice significantly reduced tumor growth whereas inhibition of BMP7, via the secreted antagonist COCO, resulted in metastasis[58,59]. Leukemic dormancy occurs within bone marrow niches and is influenced by the expression of OPN[14]. Acute lymphobaslic leukemia blasts express high levels of the OPN receptor, VLA-4, which permits the cells to adhere to stroma-derived OPN secreted by osteoblasts within the bone marrow niche[14]. This interaction drives leukemia blast into dormancy and this causes the cells to escape chemotherapy and/or radiation treatment[14]. In addition, antibody neutralization of OPN resulted in leukemia blast to exit dormancy and re-enter the cell cycle[14]. Taken together, these data support the notion that communication between cancer cells and cells associated with the tumor microenvironment is important for controlling the transition between dormancy and angiogenesis.
IMMUNOLOGIC DORMANCY
Tumor dormancy can be established by preserving equilibrium between immune response and tumor cells (Figure 1). The mechanism of how tumor cells enter and exit immunologic dormancy is not well understood. The immune system can control dormancy via three different methods including elimination, equilibrium, and escape. The innate and adaptive immune systems work together to detect and eliminate transformed cancer cell prior to the host becoming clinically symptomatic. If the tumor cells are not completely eliminated, then the host’s immunity can restrict tumor growth resulting in the continuance of cells within a dormant state. Over time, the tumor cells can adapt to the immune environment causing cells to exit dormancy leading to recurrence[60-62] and tumor metastasis (Figure 1). For example, DTC can reduce T-cell activation which weakens the cytotoxic T-lymphocyte response thus cells escape apoptosis[63]. Direct tumor immunosuppression can mediate the escape from dormancy by driving the overexpression of B7 homolog 1 (B7-H1) which inhibits T-cell activation and the cytotoxic T lymphocyte (CTL) response[63]. In addition, cancer cells can escape tumor dormancy by inhibiting antigen presentation and by methylating cytokine signaling 1 thus leading to resistance to CTL-induced apoptosis[63]. Furthermore, loss of CD4+ or CD8+ T-cells can result in tumor cell dormancy escape[64]. Several cell types within the immune system can indirectly regulate the escape from dormancy by secreting proteins that promote angiogenesis. Interleukin 23, produced by macrophages, suppresses anti-tumor effectors responses, whereas interleukin 12 represses tumor growth[65,66]. The glycoprotein, macrophage stimulating 1 (MS1) can bind to its receptor, MS1 receptor (MST1R), thus suppressing antitumor immune response and promoting cell proliferation, survival, and chemotaxis. The loss of MST1R increases antitumor CD8+ T-cell responses resulting in higher levels of secreted tumor necrosis factor α subsequently leading to the inability of micrometastatic cancer cells to generate macrometastases[67,68]. In addition, myeloid-derived suppressor cells, regulatory T-cells, and tumor-associated macrophages can also indirectly promote tumor cells to escape dormancy[63]. These cells can secrete mitogens and proangiogenic molecules which promote cell proliferation, angiogenesis and immunosupression causing the cells to exit dormancy[63]. These results demonstrate the importance in controlling the immune system to prevent tumor recurrence and metastasis.
Genomic instability may facilitate the escape of dormant cancer cells from immunological dormancy. Over time, if cancers cells do not have the capability to repair their DNA, they can accumulate mutations allowing the cells to evade anti-tumor immunity leading to recurrence. Therefore, understanding how DNA repair mechanism function in dormant cells may lead to new developments to detect and treat dormant cancer cells.
DNA REPAIR MECHANISMS
Many cancer drugs induce high levels of DNA lesions both single-stranded (SSB) and double-stranded, which results in the death of proliferating cells. Mechanism involved in SSB and DSBs break repair significantly affect the cancer cells ability to evade radiation and chemotherapy treatments. SSBs are repair through the base excision repair pathway. The damaged base is recognized and excised by DNA glycosylases which generates abasic sites. PARP1 and PARP2 proteins sense the SSB and recruit other factors such as XRCC1 to the damaged region[69]. Loss of heterozygosity of OGG1, a DNA glycosylase, is associated with papillary thyroid cancer[70].
DSBs are considered to be the most toxic form of DNA lesions[71-73]. When DNA lesions occur, cells can utilize DNA damage repair pathways to restore the DNA and maintain the genomic integrity of the cell. Two of the major DSBs repair pathways are homologous recombination (HR) and non-homologous end jointing (NHEJ). HR utilizes the DNA sequence from the homologous sister chromatid to repair the DSBs, and occurs predominately in the S and G2 phases of the cell cycle. HR is a major mechanism to ensure the high fidelity of genetic information and because this process uses the homologous sequence as a template, it is considered to be a more error-free repair pathway. Once the HR process is initiated, the DSB is resected to create a 3’ overhang that becomes coated with ssDNA-binding protein RPA. Once this filament is formed, RPA is replaced by RAD51 in an ATM/CHK2/BRCA1/BRCA2/PALB2-dependent manner[69]. RAD51 is a key HR repair protein with recombinase activity. One of the main functions of RAD51 is to invade the sister chromatid and identify the template sequence, and reduced RAD51 expression is associated with decreased HR activities[74].
In contrast to HR pathway, NHEJ takes place throughout the cell cycle and involves the direct ligation of broken ends without the need of homologous templates which results in more errors being incorporated within the DNA sequence[75]. Upon initiation of NHEJ, Ku70 and Ku80 form heterodimers that detect and bind the DNA ends. The Ku proteins will then recruit the catalytic subunit, DNA-Protein Kinase (DNA-PK). This step is required for XRCC4 and Lig4-mediated rejoining of the damaged DNA ends during NHEJ[69]. DNA-PK complex acts as a molecular sensor for NHEJ repair[76,77], and cells lacking DNA-PK function fail to show proper NHEJ[78-84]. Additionally, PARP1 may compete with Ku protein to bind the DSB ends resulting in an alternative NHEJ pathway.
Many cancers have abnormalities in the DNA repair pathways, therefore several therapeutics have been developed to exploit these defects. The NHEJ catalytic subunit, DNA-PK, is considered to be up-regulated in radiation-resistant glioblastoma and prostate cancers[85,86]. Recently, clinical trials have shown that inhibitors of DNA-PK have increased the sensitivity of cancer cells to DNA damaging agents however these drugs have been avoided due to the toxicity to normal cells[87]. Small molecular inhibitors of DNA ligase IV, which is involved in NHEJ, have also been used to decrease cell proliferation and increase the tumor inhibitory effect of chemotherapeutics that cause DSBs[88]. The mis-regulation of genes associated with HR, RAD51, BRCA1, ERCC1, APE1, and PARP1, are also observed in various cancers and are associated with resistance to chemotherapies[87]. Specifically, mutations in BRCA1, BRCA2, ATM, CHEK2, and RAD50 have been identified in several cancers including lung, ovarian, pancreatic, and leukemia[69]. Besides drugs that target RAD51, currently there are very little therapeutics that target other proteins involved in HR[87]. Alternatively, targeting the alternative NHEK pathway via PARP1 inhibitors have been used to treat BRCA1 or BRCA2-defected cancers[69].
DNA repair pathways have been shown to play a vital role in the survival of dormant cancer cells after the initial therapeutic treatments. In hepatocellular carcinoma, the stem cell population switches from actively dividing to dormant after the first round of chemotherapy, which allows for the survival of malignant cells[89,90]. The dormant cells contain less DSBs after chemotherapy treatment, and Nishikawa et al[15,22] (2012) demonstrated that these cells activated the NHEJ pathway to repair the DNA damage[15,22]. Furthermore, our unpublished data indicates that the NHEJ pathway is important in facilitating DSBs repair in ER+ dormant breast cancer cells after exposure to chemotherapy or radiation. In addition, we discovered that when these cells were treated with chemotherapeutics and exited dormancy, genomic instability increased leading to more aggressive phenotypes and chemotherapy resistance (Lin, unpublished data).
HR may also be involved in DNA repair of dormant cancer cells. The human Fanconi anemia monoubiquitination pathway has been implicated in promoting DNA repair via HR[91]. Recently, defects in this pathway resulted in the accumulation of DNA damage causing hematopoietic stem cells to exit their dormant state. The repeated activation of the hematopoietic stem cells out of their quiescent state can lead to the complete collapse of the hematopoietic system triggering diseases such as Fanconi anemia and leukemia[92].
CONCLUSION
One of the most difficult clinical challenges that we face today is the effective treatment of malignant diseases due to the inability to detect dormant cancer cells[93]. Recently, Kim et al[94] (2012) established a dormancy gene signature in ER+ breast cancer cells. When two of these genes, BHLHE41 and NR2F1, are knocked-down in the breast cancer cells, in vivo cell growth increased[94]. While these data are promising in identifying dormant cells, it has yet to be used diagnostically. Therefore, it is important to continue investigating the mechanism that control cancer dormancy. Targeting pathways involved in cellular, angiogeneic or immunologic dormancy may provide a way to detect dormant cells as well as treating metastatic cancer.
A possible mechanism to target dormant cancer cells is through the DNA repair pathways, and recent studies have suggested that DNA repair mechanisms may contribute to the survival of dormant cancer cells. In particular, the NHEJ pathway may cause a high frequency of spontaneous mutagenesis subsequently resulting in genomic instability and tumor progression[75]. However, more studies need to be performed to determine if other DNA repair mechanism facilitate the maintenance and survival of dormant cells. In addition, these pathways are not intrinsic to dormant cancer cells. Therefore, understanding the mechanisms of how dormancy is involved in recurrence is urgent for the prevention of secondary tumors. Several advancements have been made to characterized dormant cancer cells, however, to date, there is a lack of suitable model systems to detect and maintain cells in a dormant state. Development of in vivo and in vitro model systems are imperative to identify key molecular determinants of dormancy, which may lead to strategies for detecting and eliminating dormant cancer cells thus preventing recurrence and reducing cancer mortality.
ACKNOWLEDGMENTS
We would like to thank Jeane Govan for critical reading of the manuscript.
Footnotes
Supported by The DOD Innovator and Scholar Concept Award, No. W81XWH-12-1-0372.
Conflict-of-interest statement: The authors declare no conflicts of interest regarding this manuscript.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 29, 2015
First decision: June 4, 2015
Article in press: July 27, 2015
P- Reviewer: Muntane J, Panda C S- Editor: Ji FF L- Editor: A E- Editor: Jiao XK
References
- 1.Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448–456. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- 2.Karrison TG, Ferguson DJ, Meier P. Dormancy of mammary carcinoma after mastectomy. J Natl Cancer Inst. 1999;91:80–85. doi: 10.1093/jnci/91.1.80. [DOI] [PubMed] [Google Scholar]
- 3.Pfitzenmaier J, Ellis WJ, Arfman EW, Hawley S, McLaughlin PO, Lange PH, Vessella RL. Telomerase activity in disseminated prostate cancer cells. BJU Int. 2006;97:1309–1313. doi: 10.1111/j.1464-410X.2006.06194.x. [DOI] [PubMed] [Google Scholar]
- 4.Weckermann D, Müller P, Wawroschek F, Harzmann R, Riethmüller G, Schlimok G. Disseminated cytokeratin positive tumor cells in the bone marrow of patients with prostate cancer: detection and prognostic value. J Urol. 2001;166:699–703. [PubMed] [Google Scholar]
- 5.Blows FM, Driver KE, Schmidt MK, Broeks A, van Leeuwen FE, Wesseling J, Cheang MC, Gelmon K, Nielsen TO, Blomqvist C, et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010;7:e1000279. doi: 10.1371/journal.pmed.1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang SH, Lin SY. Tumor dormancy: potential therapeutic target in tumor recurrence and metastasis prevention. Exp Hematol Oncol. 2013;2:29. doi: 10.1186/2162-3619-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, Beitsch PD, Leitch M, Hoover S, Euhus D, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10:8152–8162. doi: 10.1158/1078-0432.CCR-04-1110. [DOI] [PubMed] [Google Scholar]
- 8.Pound CR, Partin AW, Eisenberger MA, Chan DW, Pearson JD, Walsh PC. Natural history of progression after PSA elevation following radical prostatectomy. JAMA. 1999;281:1591–1597. doi: 10.1001/jama.281.17.1591. [DOI] [PubMed] [Google Scholar]
- 9.Amling CL, Blute ML, Bergstralh EJ, Seay TM, Slezak J, Zincke H. Long-term hazard of progression after radical prostatectomy for clinically localized prostate cancer: continued risk of biochemical failure after 5 years. J Urol. 2000;164:101–105. [PubMed] [Google Scholar]
- 10.Faries MB, Steen S, Ye X, Sim M, Morton DL. Late recurrence in melanoma: clinical implications of lost dormancy. J Am Coll Surg. 2013;217:27–34; discussion 34-36. doi: 10.1016/j.jamcollsurg.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Callaway MP, Briggs JC. The incidence of late recurrence (greater than 10 years); an analysis of 536 consecutive cases of cutaneous melanoma. Br J Plast Surg. 1989;42:46–49. doi: 10.1016/s0007-1226(89)90111-2. [DOI] [PubMed] [Google Scholar]
- 12.Davis TA, Maloney DG, Czerwinski DK, Liles TM, Levy R. Anti-idiotype antibodies can induce long-term complete remissions in non-Hodgkin’s lymphoma without eradicating the malignant clone. Blood. 1998;92:1184–1190. [PubMed] [Google Scholar]
- 13.Press OW, Leonard JP, Coiffier B, Levy R, Timmerman J. Immunotherapy of Non-Hodgkin’s lymphomas. Hematology Am Soc Hematol Educ Program. 2001:221–240. doi: 10.1182/asheducation-2001.1.221. [DOI] [PubMed] [Google Scholar]
- 14.Boyerinas B, Zafrir M, Yesilkanal AE, Price TT, Hyjek EM, Sipkins DA. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood. 2013;121:4821–4831. doi: 10.1182/blood-2012-12-475483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishikawa S, Ishii H, Haraguchi N, Kano Y, Fukusumi T, Ohta K, Ozaki M, Sakai D, Satoh T, Nagano H, et al. Genotoxic therapy stimulates error-prone DNA repair in dormant hepatocellular cancer stem cells. Exp Ther Med. 2012;3:959–962. doi: 10.3892/etm.2012.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Almog N. Molecular mechanisms underlying tumor dormancy. Cancer Lett. 2010;294:139–146. doi: 10.1016/j.canlet.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 17.Okuyama H, Inoue M. [Hypoxic microenvironment and cancer dormancy] Gan To Kagaku Ryoho. 2011;38:1559–1564. [PubMed] [Google Scholar]
- 18.Patrikidou A, Chabaud S, Ray-Coquard I, Bui BN, Adenis A, Rios M, Bertucci F, Duffaud F, Chevreau C, Cupissol D, et al. Influence of imatinib interruption and rechallenge on the residual disease in patients with advanced GIST: results of the BFR14 prospective French Sarcoma Group randomised, phase III trial. Ann Oncol. 2013;24:1087–1093. doi: 10.1093/annonc/mds587. [DOI] [PubMed] [Google Scholar]
- 19.Boichuk S, Parry JA, Makielski KR, Litovchick L, Baron JL, Zewe JP, Wozniak A, Mehalek KR, Korzeniewski N, Seneviratne DS, et al. The DREAM complex mediates GIST cell quiescence and is a novel therapeutic target to enhance imatinib-induced apoptosis. Cancer Res. 2013;73:5120–5129. doi: 10.1158/0008-5472.CAN-13-0579. [DOI] [PubMed] [Google Scholar]
- 20.Mellor HR, Ferguson DJ, Callaghan R. A model of quiescent tumour microregions for evaluating multicellular resistance to chemotherapeutic drugs. Br J Cancer. 2005;93:302–309. doi: 10.1038/sj.bjc.6602710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enderling H, Nava Almog, Lynn Hlatky. Systems biology of tumor dormany. In: Cohen IR, A , editors. Lajtha, J.D. Lambris, R. Paoletti Advances in experimental medicine and biology. New York: Springer Science; 2013. p. 290. [Google Scholar]
- 22.Nishikawa S, Dewi DL, Ishii H, Konno M, Haraguchi N, Kano Y, Fukusumi T, Ohta K, Noguchi Y, Ozaki M, et al. Transcriptomic study of dormant gastrointestinal cancer stem cells. Int J Oncol. 2012;41:979–984. doi: 10.3892/ijo.2012.1531. [DOI] [PubMed] [Google Scholar]
- 23.Rameshwar P. Breast cancer cell dormancy in bone marrow: potential therapeutic targets within the marrow microenvironment. Expert Rev Anticancer Ther. 2010;10:129–132. doi: 10.1586/era.10.3. [DOI] [PubMed] [Google Scholar]
- 24.Hou JM, Krebs MG, Lancashire L, Sloane R, Backen A, Swain RK, Priest LJ, Greystoke A, Zhou C, Morris K, et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J Clin Oncol. 2012;30:525–532. doi: 10.1200/JCO.2010.33.3716. [DOI] [PubMed] [Google Scholar]
- 25.Fehm T, Müller V, Alix-Panabières C, Pantel K. Micrometastatic spread in breast cancer: detection, molecular characterization and clinical relevance. Breast Cancer Res. 2008;10 Suppl 1:S1. doi: 10.1186/bcr1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pantel K, Brakenhoff RH, Brandt B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer. 2008;8:329–340. doi: 10.1038/nrc2375. [DOI] [PubMed] [Google Scholar]
- 27.Panteleakou Z, Lembessis P, Sourla A, Pissimissis N, Polyzos A, Deliveliotis C, Koutsilieris M. Detection of circulating tumor cells in prostate cancer patients: methodological pitfalls and clinical relevance. Mol Med. 2009;15:101–114. doi: 10.2119/molmed.2008.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan J, Liu XL, Han LZ, Xiao G, Li NL, Deng YN, Yin LC, Ling LJ, Yu XY, Tan CL, et al. Relation between Ki-67, ER, PR, Her2/neu, p21, EGFR, and TOP II-α expression in invasive ductal breast cancer patients and correlations with prognosis. Asian Pac J Cancer Prev. 2015;16:823–829. doi: 10.7314/apjcp.2015.16.2.823. [DOI] [PubMed] [Google Scholar]
- 29.Zou P, Yoshihara H, Hosokawa K, Tai I, Shinmyozu K, Tsukahara F, Maru Y, Nakayama K, Nakayama KI, Suda T. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 2011;9:247–261. doi: 10.1016/j.stem.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 30.Fitzgerald AL, Osman AA, Xie TX, Patel A, Skinner H, Sandulache V, Myers JN. Reactive oxygen species and p21Waf1/Cip1 are both essential for p53-mediated senescence of head and neck cancer cells. Cell Death Dis. 2015;6:e1678. doi: 10.1038/cddis.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011;25:801–813. doi: 10.1101/gad.2034211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sadasivam S, DeCaprio JA. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer. 2013;13:585–595. doi: 10.1038/nrc3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sadasivam S, Duan S, DeCaprio JA. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012;26:474–489. doi: 10.1101/gad.181933.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmit F, Korenjak M, Mannefeld M, Schmitt K, Franke C, von Eyss B, Gagrica S, Hänel F, Brehm A, Gaubatz S. LINC, a human complex that is related to pRB-containing complexes in invertebrates regulates the expression of G2/M genes. Cell Cycle. 2007;6:1903–1913. doi: 10.4161/cc.6.15.4512. [DOI] [PubMed] [Google Scholar]
- 35.Hurford RK, Cobrinik D, Lee MH, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 1997;11:1447–1463. doi: 10.1101/gad.11.11.1447. [DOI] [PubMed] [Google Scholar]
- 36.Gaubatz S, Lindeman GJ, Ishida S, Jakoi L, Nevins JR, Livingston DM, Rempel RE. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol Cell. 2000;6:729–735. doi: 10.1016/s1097-2765(00)00071-x. [DOI] [PubMed] [Google Scholar]
- 37.Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 2006;20:47–64. doi: 10.1101/gad.1384406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng X, Ewton DZ, Friedman E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res. 2009;69:3317–3324. doi: 10.1158/0008-5472.CAN-08-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baughn LB, Di Liberto M, Wu K, Toogood PL, Louie T, Gottschalk R, Niesvizky R, Cho H, Ely S, Moore MA, et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 2006;66:7661–7667. doi: 10.1158/0008-5472.CAN-06-1098. [DOI] [PubMed] [Google Scholar]
- 40.Sirma H, Broemel M, Stumm L, Tsourlakis T, Steurer S, Tennstedt P, Salomon G, Michl U, Haese A, Simon R, et al. Loss of CDKN1B/p27Kip1 expression is associated with ERG fusion-negative prostate cancer, but is unrelated to patient prognosis. Oncol Lett. 2013;6:1245–1252. doi: 10.3892/ol.2013.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stendahl M, Nilsson S, Wigerup C, Jirström K, Jönsson PE, Stål O, Landberg G. p27Kip1 is a predictive factor for tamoxifen treatment response but not a prognostic marker in premenopausal breast cancer patients. Int J Cancer. 2010;127:2851–2858. doi: 10.1002/ijc.25297. [DOI] [PubMed] [Google Scholar]
- 42.Sgambato A, Camerini A, Genovese G, De Luca F, Viacava P, Migaldi M, Boninsegna A, Cecchi M, Sepich CA, Rossi G, et al. Loss of nuclear p27(kip1) and α-dystroglycan is a frequent event and is a strong predictor of poor outcome in renal cell carcinoma. Cancer Sci. 2010;101:2080–2086. doi: 10.1111/j.1349-7006.2010.01644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Geng L, Talmon G, Wang J. MicroRNA-520g confers drug resistance by regulating p21 expression in colorectal cancer. J Biol Chem. 2015;290:6215–6225. doi: 10.1074/jbc.M114.620252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Casimiro MC, Velasco-Velázquez M, Aguirre-Alvarado C, Pestell RG. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: past and present. Expert Opin Investig Drugs. 2014;23:295–304. doi: 10.1517/13543784.2014.867017. [DOI] [PubMed] [Google Scholar]
- 45.Velasco-Velázquez MA, Li Z, Casimiro M, Loro E, Homsi N, Pestell RG. Examining the role of cyclin D1 in breast cancer. Future Oncol. 2011;7:753–765. doi: 10.2217/fon.11.56. [DOI] [PubMed] [Google Scholar]
- 46.Gautschi O, Ratschiller D, Gugger M, Betticher DC, Heighway J. Cyclin D1 in non-small cell lung cancer: a key driver of malignant transformation. Lung Cancer. 2007;55:1–14. doi: 10.1016/j.lungcan.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 47.Kim MS, Kim KH, Lee EH, Lee YM, Lee SH, Kim HD, Kim YZ. Results of immunohistochemical staining for cell cycle regulators predict the recurrence of atypical meningiomas. J Neurosurg. 2014;121:1189–1200. doi: 10.3171/2014.7.JNS132661. [DOI] [PubMed] [Google Scholar]
- 48.Chen TJ, Lee SW, Lin LC, Lin CY, Chang KY, Li CF. Cyclin-dependent kinase 4 overexpression is mostly independent of gene amplification and constitutes an independent prognosticator for nasopharyngeal carcinoma. Tumour Biol. 2014;35:7209–7216. doi: 10.1007/s13277-014-1884-2. [DOI] [PubMed] [Google Scholar]
- 49.Favaro E, Amadori A, Indraccolo S. Cellular interactions in the vascular niche: implications in the regulation of tumor dormancy. APMIS. 2008;116:648–659. doi: 10.1111/j.1600-0463.2008.01025.x. [DOI] [PubMed] [Google Scholar]
- 50.Ranganathan AC, Adam AP, Aguirre-Ghiso JA. Opposing roles of mitogenic and stress signaling pathways in the induction of cancer dormancy. Cell Cycle. 2006;5:1799–1807. doi: 10.4161/cc.5.16.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Troyanovsky B, Levchenko T, Månsson G, Matvijenko O, Holmgren L. Angiomotin: an angiostatin binding protein that regulates endothelial cell migration and tube formation. J Cell Biol. 2001;152:1247–1254. doi: 10.1083/jcb.152.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Satchi-Fainaro R, Ferber S, Segal E, Ma L, Dixit N, Ijaz A, Hlatky L, Abdollahi A, Almog N. Prospective identification of glioblastoma cells generating dormant tumors. PLoS One. 2012;7:e44395. doi: 10.1371/journal.pone.0044395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, Stainier DY, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013;15:807–817. doi: 10.1038/ncb2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franses JW, Baker AB, Chitalia VC, Edelman ER. Stromal endothelial cells directly influence cancer progression. Sci Transl Med. 2011;3:66ra5. doi: 10.1126/scitranslmed.3001542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Straume O, Shimamura T, Lampa MJ, Carretero J, Øyan AM, Jia D, Borgman CL, Soucheray M, Downing SR, Short SM, et al. Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer. Proc Natl Acad Sci USA. 2012;109:8699–8704. doi: 10.1073/pnas.1017909109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Geis T, Döring C, Popp R, Grossmann N, Fleming I, Hansmann ML, Dehne N, Brüne B. HIF-2alpha-dependent PAI-1 induction contributes to angiogenesis in hepatocellular carcinoma. Exp Cell Res. 2015;331:46–57. doi: 10.1016/j.yexcr.2014.11.018. [DOI] [PubMed] [Google Scholar]
- 57.Chen H, Ko JM, Wong VC, Hyytiainen M, Keski-Oja J, Chua D, Nicholls JM, Cheung FM, Lee AW, Kwong DL, et al. LTBP-2 confers pleiotropic suppression and promotes dormancy in a growth factor permissive microenvironment in nasopharyngeal carcinoma. Cancer Lett. 2012;325:89–98. doi: 10.1016/j.canlet.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 58.Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A, Shen R, Brogi E, Brivanlou AH, Giancotti FG. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell. 2012;150:764–779. doi: 10.1016/j.cell.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, Pai SK, Liu W, Fukuda K, Chambers C, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med. 2011;208:2641–2655. doi: 10.1084/jem.20110840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vesely MD, Schreiber RD. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Ann N Y Acad Sci. 2013;1284:1–5. doi: 10.1111/nyas.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quesnel B. Dormant tumor cells as a therapeutic target? Cancer Lett. 2008;267:10–17. doi: 10.1016/j.canlet.2008.02.055. [DOI] [PubMed] [Google Scholar]
- 62.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 63.Hensel JA, Flaig TW, Theodorescu D. Clinical opportunities and challenges in targeting tumour dormancy. Nat Rev Clin Oncol. 2013;10:41–51. doi: 10.1038/nrclinonc.2012.207. [DOI] [PubMed] [Google Scholar]
- 64.Teng MW, Swann JB, Koebel CM, Schreiber RD, Smyth MJ. Immune-mediated dormancy: an equilibrium with cancer. J Leukoc Biol. 2008;84:988–993. doi: 10.1189/jlb.1107774. [DOI] [PubMed] [Google Scholar]
- 65.Teng MW, Vesely MD, Duret H, McLaughlin N, Towne JE, Schreiber RD, Smyth MJ. Opposing roles for IL-23 and IL-12 in maintaining occult cancer in an equilibrium state. Cancer Res. 2012;72:3987–3996. doi: 10.1158/0008-5472.CAN-12-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, Basham B, McClanahan T, Kastelein RA, Oft M. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- 67.Eyob H, Ekiz HA, Welm AL. RON promotes the metastatic spread of breast carcinomas by subverting antitumor immune responses. Oncoimmunology. 2013;2:e25670. doi: 10.4161/onci.25670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eyob H, Ekiz HA, Derose YS, Waltz SE, Williams MA, Welm AL. Inhibition of ron kinase blocks conversion of micrometastases to overt metastases by boosting antitumor immunity. Cancer Discov. 2013;3:751–760. doi: 10.1158/2159-8290.CD-12-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dietlein F, Thelen L, Reinhardt HC. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014;30:326–339. doi: 10.1016/j.tig.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 70.Royer MC, Zhang H, Fan CY, Kokoska MS. Genetic alterations in papillary thyroid carcinoma and hashimoto thyroiditis: An analysis of hOGG1 loss of heterozygosity. Arch Otolaryngol Head Neck Surg. 2010;136:240–242. doi: 10.1001/archoto.2010.20. [DOI] [PubMed] [Google Scholar]
- 71.Hsiang YH, Lihou MG, Liu LF. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989;49:5077–5082. [PubMed] [Google Scholar]
- 72.Markovits J, Pommier Y, Kerrigan D, Covey JM, Tilchen EJ, Kohn KW. Topoisomerase II-mediated DNA breaks and cytotoxicity in relation to cell proliferation and the cell cycle in NIH 3T3 fibroblasts and L1210 leukemia cells. Cancer Res. 1987;47:2050–2055. [PubMed] [Google Scholar]
- 73.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 74.Bindra RS, Schaffer PJ, Meng A, Woo J, Måseide K, Roth ME, Lizardi P, Hedley DW, Bristow RG, Glazer PM. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol. 2004;24:8504–8518. doi: 10.1128/MCB.24.19.8504-8518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 76.Kienker LJ, Shin EK, Meek K. Both V(D)J recombination and radioresistance require DNA-PK kinase activity, though minimal levels suffice for V(D)J recombination. Nucleic Acids Res. 2000;28:2752–2761. doi: 10.1093/nar/28.14.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kurimasa A, Kumano S, Boubnov NV, Story MD, Tung CS, Peterson SR, Chen DJ. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol Cell Biol. 1999;19:3877–3884. doi: 10.1128/mcb.19.5.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen S, Inamdar KV, Pfeiffer P, Feldmann E, Hannah MF, Yu Y, Lee JW, Zhou T, Lees-Miller SP, Povirk LF. Accurate in vitro end joining of a DNA double strand break with partially cohesive 3’-overhangs and 3’-phosphoglycolate termini: effect of Ku on repair fidelity. J Biol Chem. 2001;276:24323–24330. doi: 10.1074/jbc.M010544200. [DOI] [PubMed] [Google Scholar]
- 79.Baumann P, West SC. DNA end-joining catalyzed by human cell-free extracts. Proc Natl Acad Sci USA. 1998;95:14066–14070. doi: 10.1073/pnas.95.24.14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rosenzweig KE, Youmell MB, Palayoor ST, Price BD. Radiosensitization of human tumor cells by the phosphatidylinositol3-kinase inhibitors wortmannin and LY294002 correlates with inhibition of DNA-dependent protein kinase and prolonged G2-M delay. Clin Cancer Res. 1997;3:1149–1156. [PubMed] [Google Scholar]
- 81.Roth DB, Lindahl T, Gellert M. Repair and recombination. How to make ends meet. Curr Biol. 1995;5:496–499. doi: 10.1016/s0960-9822(95)00101-1. [DOI] [PubMed] [Google Scholar]
- 82.Jackson SP, Jeggo PA. DNA double-strand break repair and V(D)J recombination: involvement of DNA-PK. Trends Biochem Sci. 1995;20:412–415. doi: 10.1016/s0968-0004(00)89090-8. [DOI] [PubMed] [Google Scholar]
- 83.Hartley KO, Gell D, Smith GC, Zhang H, Divecha N, Connelly MA, Admon A, Lees-Miller SP, Anderson CW, Jackson SP. DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell. 1995;82:849–856. doi: 10.1016/0092-8674(95)90482-4. [DOI] [PubMed] [Google Scholar]
- 84.Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 85.Bouchaert P, Guerif S, Debiais C, Irani J, Fromont G. DNA-PKcs expression predicts response to radiotherapy in prostate cancer. Int J Radiat Oncol Biol Phys. 2012;84:1179–1185. doi: 10.1016/j.ijrobp.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 86.Kase M, Vardja M, Lipping A, Asser T, Jaal J. Impact of PARP-1 and DNA-PK expression on survival in patients with glioblastoma multiforme. Radiother Oncol. 2011;101:127–131. doi: 10.1016/j.radonc.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 87.Hosoya N, Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014;105:370–388. doi: 10.1111/cas.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Srivastava M, Nambiar M, Sharma S, Karki SS, Goldsmith G, Hegde M, Kumar S, Pandey M, Singh RK, Ray P, et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell. 2012;151:1474–1487. doi: 10.1016/j.cell.2012.11.054. [DOI] [PubMed] [Google Scholar]
- 89.Haraguchi N, Ishii H, Mimori K, Tanaka F, Ohkuma M, Kim HM, Akita H, Takiuchi D, Hatano H, Nagano H, et al. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. 2010;120:3326–3339. doi: 10.1172/JCI42550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haraguchi N, Ishii H, Nagano H, Doki Y, Mori M. The future prospects and subject of the liver cancer stem cells study for the clinical application. Gastroenterology. 2011 doi: 10.1053/j.gastro.2011.02.035. [DOI] [PubMed] [Google Scholar]
- 91.Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D’Andrea AD, Wang ZQ, Jasin M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci USA. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle B, Brocks D, Bayindir I, Kaschutnig P, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520:549–552. doi: 10.1038/nature14131. [DOI] [PubMed] [Google Scholar]
- 93.Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14:611–622. doi: 10.1038/nrc3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim RS, Avivar-Valderas A, Estrada Y, Bragado P, Sosa MS, Aguirre-Ghiso JA, Segall JE. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS One. 2012;7:e35569. doi: 10.1371/journal.pone.0035569. [DOI] [PMC free article] [PubMed] [Google Scholar]