

Abstract
Background
Modern horses represent heterogeneous populations specifically selected for appearance and performance. Genomic regions under high selective pressure show characteristic runs of homozygosity (ROH) which represent a low genetic diversity. This study aims at detecting the number and functional distribution of ROHs in different horse populations using next generation sequencing data.
Methods
Next generation sequencing was performed for two Sorraia, one Dülmen Horse, one Arabian, one Saxon-Thuringian Heavy Warmblood, one Thoroughbred and four Hanoverian. After quality control reads were mapped to the reference genome EquCab2.70. ROH detection was performed using PLINK, version 1.07 for a trimmed dataset with 11,325,777 SNPs and a mean read depth of 12. Stretches with homozygous genotypes of >40 kb as well as >400 kb were defined as ROHs. SNPs within consensus ROHs were tested for neutrality. Functional classification was done for genes annotated within ROHs using PANTHER gene list analysis and functional variants were tested for their distribution among breed or non-breed groups.
Results
ROH detection was performed using whole genome sequences of ten horses of six populations representing various breed types and non-breed horses. In total, an average number of 3492 ROHs were detected in windows of a minimum of 50 consecutive homozygous SNPs and an average number of 292 ROHs in windows of 500 consecutive homozygous SNPs. Functional analyses of private ROHs in each horse revealed a high frequency of genes affecting cellular, metabolic, developmental, immune system and reproduction processes. In non-breed horses, 198 ROHs in 50-SNP windows and seven ROHs in 500-SNP windows showed an enrichment of genes involved in reproduction, embryonic development, energy metabolism, muscle and cardiac development whereas all seven breed horses revealed only three common ROHs in 50-SNP windows harboring the fertility-related gene YES1. In the Hanoverian, a total of 18 private ROHs could be shown to be located in the region of genes potentially involved in neurologic control, signaling, glycogen balance and reproduction. Comparative analysis of homozygous stretches common in all ten horses displayed three ROHs which were all located in the region of KITLG, the ligand of KIT known to be involved in melanogenesis, haematopoiesis and gametogenesis.
Conclusions
The results of this study give a comprehensive insight into the frequency and number of ROHs in various horses and their potential influence on population diversity and selection pressures. Comparisons of breed and non-breed horses suggest a significant artificial as well as natural selection pressure on reproduction performance in all types of horse populations.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1977-3) contains supplementary material, which is available to authorized users.
Keywords: Runs of homozygosity, Horse population, Selection signature, Reproduction, KITLG
Background
The modern horse population represents a particularly heterogenous group influenced over the time by various selective pressures [1]. However, in studies on genetic diversity a contrasting homogeneity within breeds or non-breeds has been observed [2–4]. In particular breeds like the Arabian, Hanoverian and Saxon-Thuringian Heavy Warmblood have been shaped by intense human selection for specific abilities and characteristics that meet with requirements for optimal performance whereas environmental conditions have particularly influenced non-breed horses. The Dülmen Horse and the Sorraia can be characterized as non-breeds with a robust constitution for primitive living conditions not subjected to human selection criteria for specific breeding aims but to natural selection [5–7]. Strong selective pressures result in a reduction of genetic diversity which is characterized by long stretches of consecutive homozygous genotypes in the genome known as runs of homozygosity (ROH) [8–11]. Size and frequency of ROHs give evidence for relatedness within and in-between populations.
In horses, signals of selection have been investigated in 744 horses of 33 breeds using whole-genome single nucleotide polymorphism (SNP) array data [1]. Potential genomic targets of selection were observed within breeds by FST-based statistics in 500-kb windows and revealed common haplotypes in the region of coat color genes, size and performance traits. The highest signature of selection was found in the Paint and Quarter Horse on ECA18 in the region of the myostatin gene (MSTN). Positively selected loci for performance have also been detected in a Thoroughbred population study based on microsatellite markers [12]. Candidate regions for exercise adaption including fatty acid oxidation, increased insulin sensitivity and muscle strength have been suggested as potential selection targets.
Signals of selection have also been investigated in other mammals including cattle, dog, pig and human, frequently scanning ROHs as diversity indices [1, 9–11, 13]. In human, ROHs were considered valuable for population demographic analyses and allowed reliable differentiation of human indigenous populations from distinct continents [14]. Shorter homozygous stretches helped to characterize population specific properties whereas ROH longer than 0.5 Mb could be shown to be frequent in all populations [15, 16]. It was proposed that ROHs longer than 1 Mb were quite more common in outbred individuals. In addition to population genetic applications, ROH detection was suggested to be valuable for mapping of causative mutations for recessive diseases [17, 18]. A study for schizophrenia identified nine risk ROHs which were significantly more frequent in affected patients and harbored disease-associated genes [19].
In domestic animals, especially performance related phenotypes and breed specific characteristics were mainly in focus of ROH analyses [9, 20]. A whole-genome comparative detection of ROHs in a sliding window approach was applied for pigs in wild and domesticated populations [9]. Two overlapping ROHs were identified in the European breeds harboring genes involved in cell differentiation. In Large White and Landrace pigs an exclusive ROH could be shown to be located in the region of the growth related pleiomorphic adenoma gene 1 (PLAG1). Further breed specific ROH analyses for Chinese and Western pigs revealed loci under selection important for high-altitude adaption in Tibetan pigs as well as a coat color locus in the region of endothelin receptor type B (EDNRB) in Chinese belted pigs [21].
Signatures of selection affecting coat color and body size traits could also be observed in genome-wide ROH scans for dogs [22]. It was suggested that ancestral genetic variations were transformed into specific characteristics of different dog breeds [13, 22]. Next generation sequencing (NGS) data from dogs and wolves revealed regions of potential selection in domesticated dogs which affect metabolism and thus suggest a potential adaption to starch digestion [13, 23]. In the Lundehund, fifteen regions with long-range haplotypes indicated potential signatures of positive selection for polydactyly, body size and male fertility [24]. In cattle, a large number of ROHs have been shown to be widely distributed among various breeds and demonstrated its utility for prediction of inbreeding coefficients and relatedness [10, 25, 26]. Haplotype-frequency based approaches revealed signatures of selection in the region of genes affecting reproduction and muscle formation [27]. A genome-wide scan in Holstein cattle identified milk yield, composition, reproduction and behavioral traits in potentially selected regions [28]. Similar observations were made in an U.S. Holstein cattle study which investigated the distribution of ROHs in different milk production groups [29]. Forty genomic regions in potential signatures of selection were identified in SNP array data harboring loci for milk, fat and protein yield. However, the use of SNP arrays for ROH detection was suggested to be limited mainly for low SNP density reasons [27, 28, 30]. Higher resolution genomic analyses on basis of whole-genome data enabled the use of 15 million SNPs from 43 Fleckvieh cattle for powerful detection of selected traits [20]. Candidate regions for coat color, neurobehavioral functioning and sensory perception were found in ROH regions suggesting domestication-related signatures of selection. The accuracy of ROH detection in NGS data was shown to be high if corrected for bias by hidden errors in genotyping data [31].
In this study, whole-genome sequences of ten horses were used for analysis of ROHs in a sliding window approach. 50-SNP and 500-SNP windows were chosen for reliable detection of ROHs of different sizes. ROHs exclusively found in individual horses or breeds were further investigated for their gene content potentially affected by targeted selection for specific appearance and function.
Results
Sequencing and variant detection
Whole-genome sequences of a Dülmen Horse, two Sorraia, an Arabian, a Saxon-Thuringian Heavy Warmblood descendent from the Old-Oldenburg breed, a Thoroughbred and four Hanoverian were obtained using NGS. Mapping to the reference genome EquCab2.70 resulted in a mean coverage of 19.90X for the Dülmen Horse, 17.34X and 17.55X for the two Sorraia, 19.14X for the Arabian, 20.06X for the Saxon-Thuringian Heavy Warmblood, 5.92X for the Thoroughbred and 15.66X-35.18X for the four Hanoverian (Additional file 1). Raw data of variant detection revealed 3,865,613-7,147,081 SNPs and 698,724-992,338 insertions/deletions (INDELs) in each of the ten horses. After stringent quality control, a total of 11,325,777 SNPs were filtered out and used for ROH analysis. The mean heterozygosity for these SNPs per site was 0.28 in the Dülmen Horse and Arabian, 0.24 and 0.25 in the Sorraia horse, 0.29 in the Saxon-Thuringian Heavy Warmblood, 0.29 in the four Hanoverian and 0.21 in the Thoroughbred.
Sequence error estimation
We estimated sequence errors on the basis of SNP50 BeadChip data in five horses. The results of BeadChip analysis were assumed to be error free. The rate of false-negative SNPs was calculated based on heterozygous SNPs in BeadChip data which were homozygous in NGS data. We detected false-negative rates of 0.24–0.26 in the four Hanoverian and 0.22 in the Arabian (Table 1). The false-positive rate was at 3.8 x 10−4 to 9.8 × 10−4using all SNP positions of the BeadChip data. More stringent error estimations in long SNPChip ROH regions of >10 Mb compared to filtered NGS sequence revealed even lower error rates at 3.1 × 10−4 to 5.9 × 10−5.
Table 1.
Sequence error estimation
| Number of SNPs | Horse 4 | Horse 5 | Horse 6 | Horse 7 | Horse 9 |
|---|---|---|---|---|---|
| Hanoverian | Hanoverian | Hanoverian | Hanoverian | Arabian | |
| Heterozygous in NGS data | 891,514 | 815,810 | 833,149 | 832,549 | 812,952 |
| Heterozygous in BeadChip data | 16,426 | 15,469 | 15,873 | 15,987 | 14,008 |
| Homozygous in BeadChip data | 33,403 | 34,360 | 33,956 | 33,842 | 35,821 |
| Heterozygous in NGS and BeadChip data | 3,555 (7.14 %) | 3,239 (6.50 %) | 3,306 (6.64 %) | 3,396 (6.82 %) | 2,960 (5.94 %) |
| Heterozygous in BeadChip and homozygous in NGS data | 12,867 (25.83 %) | 12,223 (24.54 %) | 12,565 (25.22 %) | 12,589 (25.27 %) | 11,047 (22.17 %) |
| (False-negative rate) | |||||
| Homozygous in BeadChip and heterozygous in NGS data | 19 (3.8 × 10−4of all 49,813 positions validated in BeadChip) | 29 (5.8 × 10−4of all 49,813 positions validated in BeadChip) | 38 (7.6 × 10−4of all 49,815 positions validated in BeadChip) | 49 (9.8 × 10−4of all 49,823 positions validated in BeadChip) | 28 (5.6 × 10−4of all 49,824 positions validated in BeadChip) |
| (False-positive rate) | |||||
| Homozygous in >10 Mb BeadChip ROH regions and heterozygous in NGS data | 5.9 × 10−5 | 3.8 × 10−5 | 2.2 × 10−5 | 3.1 × 10−4 | 2.6 × 10−5 |
| (Robust false-positive rate) |
Number of detected Next Generation sequencing (NGS) -SNPs and their sequence error rates in comparison to Illumina SNP50 BeadChip data. The BeadChip data are assumed to be error free
ROH detection
An average number of 3492 ROHs was detected for the ten horses in windows of minimum amount of 50 homozygous SNPs and an average number of 292 ROHs in windows of 500 homozygous SNPs (Table 2). The number of smaller ROHs of 40–59 kb was almost equally distributed in all ten horses, whereas ROHs >59 kb were comparatively high in the two Sorraia horses and in the thoroughbred (Fig. 1). ROH detection in larger windows of >400 kb revealed even a more distinct distribution, showing ROHs particularly frequent in the Sorraia and Thoroughbred but also in the Arabian. As indicated by the number and size of ROH, the total length of ROHs in sliding windows of at least 50 SNPs was notably high in the Thoroughbred (953 Mb) and the two Sorraia (867 and 730 Mb) and comparatively high in the Arabian (566 Mb, Additional file 2). The FROH estimated for 50-SNP windows were 0.43 in the Thoroughbred, 0.39 and 0.33 in the Sorraia horses, 0.25 in the Arabian. The four Hanoverian as well as the Saxon-Thuringian Heavy Warmblood and the Dülmen Horse showed FROHranging from 0.18 to 0.22. Similar distributions of FROH could be observed for 500-SNP windows, showing the highest values of 0.18 in the Thoroughbred as well as 0.16 and 0.12 in the Sorraia horses.
Table 2.
Summary of runs of homozygosity (ROHs) detected in whole genome sequencing data of ten horses
| Horse 1 | Horse 2 | Horse 3 | Horse 4 | Horse 5 | Horse 6 | Horse 7 | Horse 8 | Horse 9 | Horse 10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Dülmen | Sorraia | Sorraia | Hanoverian | Hanoverian | Hanoverian | Hanoverian | Saxon-Thuringian Heavy Warmblood | Arabian | Thoroughbred (SRR1055837) | |
| Number of ROHs (50) | 2804 | 4395 | 3955 | 2965 | 3214 | 3104 | 3167 | 3138 | 3581 | 4595 |
| Number of private ROHs (50) | 722 | 1578 | 1349 | 538 | 752 | 662 | 709 | 739 | 934 | 1940 |
| Number of ROHs (500) | 188 | 541 | 422 | 145 | 202 | 179 | 175 | 211 | 259 | 599 |
| Number of private ROHs (500) | 152 | 352 | 285 | 80 | 150 | 84 | 109 | 123 | 153 | 459 |
The number of ROHs in windows of a minimum amount of 50 homozygous SNPs and 500 homozygous SNPs are shown. All ROHs were compared in-between horses and filtered for private ROH regions that could only be found in one horse
50 = A minimum amount of 50 homozygous SNPs
500 = A minimum amount of 500 homozygous SNPs
Fig. 1.
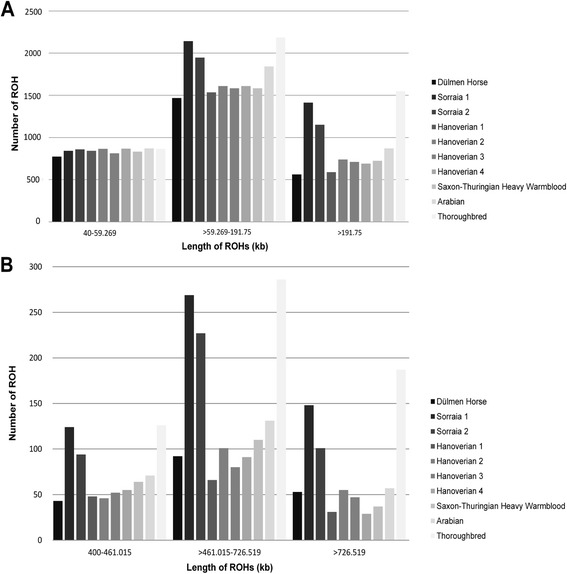
Number and size of runs of homozygosity (ROH) detected in ten horses. The length of ROHs was categorized into small, medium and large ROH regions. The results of plink analysis with windows of minimum amount of 50 homozygous SNPs (a) and 500 homozygous SNPs (b) are shown
Private ROHs and functional annotation
Functional annotation of genes located in private horse specific ROH regions, which could not be detected in one of the other horses under analysis, was performed in order to get an insight into biological processes affected by genes in horse specific homozygous segments. PANTHER gene list analysis for 50-SNP as well as 500-SNP windows revealed a high percentage of genes involved in cellular processes (GO:0009987), metabolic processes (GO:0008152) as well as biological regulations (GO:0065007), localization (GO:0051179) and developmental processes (GO:0032502) in all private ROHs of the analyzed ten horses (Table 3, Additional file 3). Further rates of gene hits affecting responses to stimulus (GO:0050896), cellular processes (GO:0071840; GO:0032501), immune system processes (GO:0002376), apoptotic processes (GO:0006915), biological adhesion (GO:0022610) and reproduction (GO:0000003) could also be observed in annotation results.
Table 3.
Functional annotations in private runs of homozygosity (ROH) of 50-SNP windows
| PANTHER gene ontology terms | Horse 1 | Horse 2 | Horse 3 | Horse 4 | Horse 5 | Horse 6 | Horse 7 | Horse 8 | Horse | Horse 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Dülmen | Sorraia (%) | Sorraia (%) | Hanoverian (%) | Hanoverian (%) | Hanoverian (%) | Hanoverian (%) | Saxon-Thuringian Heavy Warmblood (%) | Arabian (%) | Thoroughbred (SRR1055837) (%) | |
| Horse (%) | ||||||||||
| Cellular component organization or biogenesis (GO:0071840) | 3.6 | 3.1 | 2.8 | 3.2 | 4.0 | 4.5 | 5.3 | 4.4 | 3.5 | 4.0 |
| Cellular process (GO:0009987) | 18.7 | 19.9 | 18.2 | 18.8 | 21.5 | 20.2 | 20.3 | 20.8 | 19.3 | 18.6 |
| Localization (GO:0051179) | 8.3 | 9.3 | 10.2 | 10.3 | 7.5 | 11.3 | 9.9 | 11.2 | 10.6 | 8.6 |
| Apoptotic process (GO:0006915) | 2.2 | 1.5 | 2.8 | 2.1 | 1.7 | 1.9 | 1.8 | 1.4 | 2.7 | 2.6 |
| Reproduction (GO:0000003) | 1.9 | 1.8 | 3.7 | 2.1 | 1.3 | 2.0 | 0.7 | 1.2 | 0.4 | 1.8 |
| Biological regulation (GO:0065007) | 10.2 | 9.3 | 9.2 | 9.8 | 8.1 | 9.7 | 11.3 | 9.1 | 9.7 | 9.6 |
| Response to stimulus (GO:0050896) | 5.2 | 5.6 | 6.3 | 5 | 4.5 | 4.6 | 3.7 | 4.7 | 5.2 | 6.3 |
| Developmental process (GO:0032502) | 8.9 | 6.8 | 8.2 | 10.1 | 11.1 | 9.7 | 8.5 | 6.8 | 8.9 | 8.6 |
| Multicellular organismal process (GO:0032501) | 5.6 | 6.1 | 5.4 | 7.4 | 5.5 | 6.0 | 6.2 | 5.9 | 5.6 | 5.4 |
| Locomotion (GO:0040011) | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.4 | 0.0 |
| Biological adhesion (GO:0022610) | 2.8 | 4.2 | 2.4 | 3.7 | 6.8 | 2.8 | 3.2 | 3.5 | 2.7 | 3.1 |
| Metabolic process (GO:0008152) | 27 | 26.8 | 24.2 | 21.8 | 23.4 | 22.7 | 24.7 | 25.3 | 26.3 | 23.4 |
| Growth (GO:0040007) | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Immune system process (GO:0002376) | 5.4 | 5.5 | 6.6 | 5.6 | 4.5 | 4.5 | 4.2 | 5.6 | 4.8 | 8.2 |
PANTHER gene list analysis (http://www.pantherdb.org/) was performed for genes in private ROH regions which could be exclusively found in one specific horse. The percent of gene hits against total number of process hits involved in specific biological processes are shown
Analysis of shared private ROHs in specific breed horses revealed 18 ROHs common in all four Hanoverian but not in the other analyzed horses in 50-SNP windows and no shared ROHs in 500-SNP windows (Table 4). The 18 ROHs contained four novel genes and six genes known as dyslexia susceptibility 1 candidate 1 (DYX1C1), protein phosphatase 1, regulatory (inhibitor) subunit 14C (PPP1R14C), cilia and flagella associated protein 61 (CFAP61/C20orf26), cysteine sulfinic acid decarboxylase (CSAD), TBC1 domain family, member 30 (TBC1D30) and ALX homeobox 4 (ALX4), which were shown to be related by direct genetic interactions or co-expression (Fig. 2). A dense network of genetic interactions could also be found in-between genes located in private ROHs exclusively found in the non-breed horses Dülmen Horse and Sorraia (Fig. 3 and 4). In total, 198 ROHs could be detected in 50-SNP windows covering 139 genes (Additional file 4). The largest ROHs for non-breed horses of 324,707-163,116 base pairs were located in the region of the developmental and signaling genes secreted frizzled-related protein 2 (SFRP2), fraser extracellular matrix complex subunit 1 (FRAS1), interleukin-1 receptor-associated kinase 1 binding protein 1 (IRAK1BP1), pleckstrin homology domain interacting protein (PHIP), acyl-CoA synthetase short-chain family member 3 (ACSS3), protein tyrosine phosphatase, receptor type, f polypeptide, interacting protein (liprin), alpha 2 (PPFIA2) and did also cover a gene-rich region which included spermatogenesis associated 25 (SPATA25), acyl-CoA thioesterase 8 (ACOT8) and troponin C type 2 fast (TNNC2). ROH detection in 500-SNP windows revealed seven common ROH regions for non-breed horses. They were located on horse chromosomes 22 and 28 in or near by ROHs already found in 50-SNP windows. The largest region showed a size of 576,454 base pairs. In contrast to non-breeds, the whole group of breed horses (Hanoverian, Arabian, Saxon-Thuringian Heavy Warmblood and Thoroughbred) revealed only three common ROHs in 50-SNP windows and no common ROHs in 500-SNP windows (Additional file 5). The largest private ROH with 54,740 base pairs shared by all eight breed horses revealed a Tajima’s D of −1.0 and could be shown to harbor the V-Yes-1 Yamaguchi Sarcoma Viral Oncogene Homolog 1 (YES1).
Table 4.
Shared private runs of homozygosity (ROH) in 50-SNP windows
| ECA | Position | Number of SNPs in shared parts of ROH regions | Size of shared ROH (bp) | Gene ID | Human ortholog | Gene name/function |
|---|---|---|---|---|---|---|
| 1 | 135530600-135573693 | 192 | 43094 | ENSECAG00000010951, ENSECAG00000001112, ENSECAG00000004438, ENSECAG00000001272 | DYX1C1, novel gene, novel gene, novel gene | dyslexia susceptibility 1 candidate 1 |
| 4 | 13434735-13434933 | 17 | 199 | - | - | - |
| 4 | 16813531-16813643 | 4 | 113 | - | - | - |
| 4 | 48740665-48745235 | 7 | 4571 | - | - | - |
| 6 | 70162066-70181092 | 35 | 19027 | ENSECAG00000006878 | CSAD | cysteine sulfinic acid decarboxylase |
| 6 | 80540783-80586091 | 227 | 45309 | ENSECAG00000024798 | TBC1D30 | TBC1 domain family, member 30 |
| 6 | 81785611-81786700 | 28 | 1090 | - | - | - |
| 8 | 50498336-50582995 | 392 | 84660 | - | - | - |
| 12 | 9146140-9204195 | 70 | 58056 | ENSECAG00000019892 | ALX4 | ALX homeobox 4 |
| 14 | 30412967-30412967 | 1 | 1 | - | - | - |
| 14 | 85610289-85611904 | 69 | 1616 | - | - | - |
| 15 | 5973552-5973559 | 3 | 8 | - | - | - |
| 15 | 79802636-79806388 | 119 | 3753 | - | - | - |
| 16 | 23766626-23821105 | 311 | 54480 | ENSECAG00000002081 | novel gene | |
| 22 | 4841367-4841408 | 3 | 42 | ENSECAG00000004364 | CFAP61 | cilia and flagella associated protein 61 |
| 22 | 16310734-16310750 | 3 | 17 | - | - | - |
| 28 | 39200676-39218496 | 29 | 17821 | - | - | - |
| 31 | 16634998-16711978 | 347 | 76981 | ENSECAG00000023857 | PPP1R14C | protein phosphatase 1, regulatory (inhibitor) subunit 14C |
The consensus ROH regions and genes in private ROH regions shared in four Hanoverian are shown. The number of SNPs and size of shared ROH indicate the overlap of homozygous variants
Fig. 2.
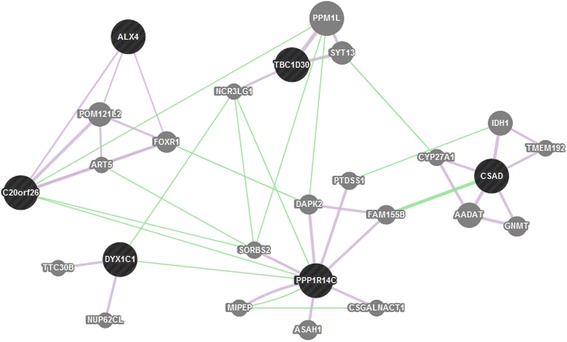
GeneMANIA network of six genes in ROH regions shared by the Hanoverian. The genes of interest are represented as black circles, related genes as grey circles. Genetic interactions are displayed as green lines and co-expressions as violet lines. All six genes are interrelated with each other
Fig. 3.
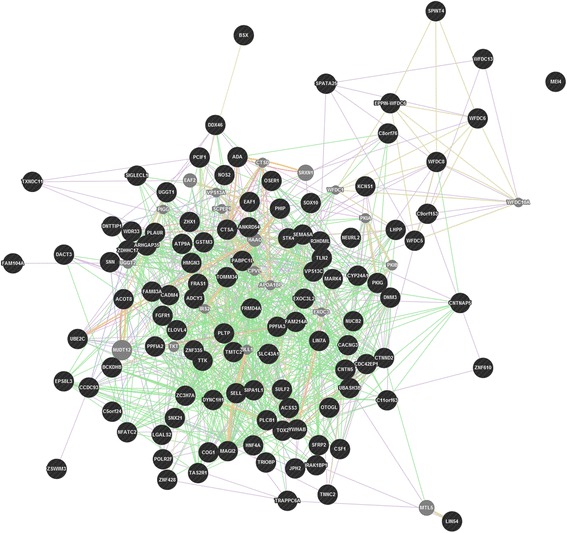
GeneMANIA network of 139 genes in 50-SNP window ROH regions shared by non-breed horses. The genes of interest are represented as black circles, related genes as grey circles. Genetic interactions are displayed as light green lines, predicted related genes as orange lines, physical interactions as red lines, co-localization as blue lines, shared protein domains as dark green lines and co-expressions as violet lines
Fig. 4.
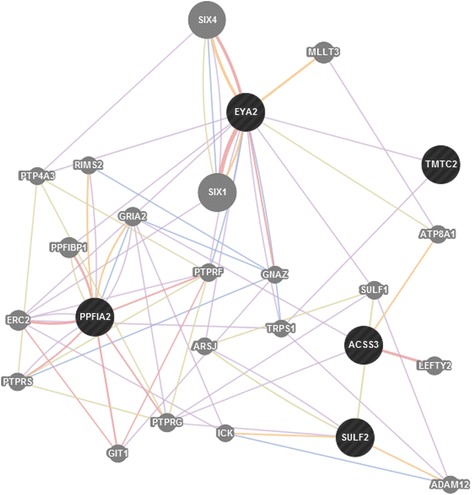
GeneMANIA network of 7 genes in 500-SNP window ROH regions shared by non-breed horses. The genes of interest are represented as black circles, related genes as grey circles. Predicted related genes are displayed as orange lines, physical interactions as red lines, co-localization as blue lines, shared protein domains as dark green lines and co-expressions as violet lines
Evaluations of consensus ROHs for all ten horses revealed three ROHs which were all located on chromosome 28 at 14,656,676–14,778,472 Mb in the region of KIT ligand (KITLG, Table 5). No common ROHs could be found in 500-SNP windows in all ten horses. Tajima’s D test statistics confirmed a deviation from neutrality in this region showing values below −1.2 in windows covering 14.65–14.78 Mb (Fig. 5, Additional file 6).
Table 5.
Shared runs of homozygosity (ROH) in 50-SNP windows
| ECA | Position | Number of SNPs in shared parts of ROH regions | Size of shared ROH (bp) | Gene ID | Human ortholog | Gene name/function |
|---|---|---|---|---|---|---|
| 28 | 14656676-14778472 | 310 | 121797 | ENSECAT00000000221 | KITLG | KIT ligand |
| 28 | 14781594-14843608 | 123 | 62015 | |||
| 28 | 14844608-14951801 | 175 | 107194 |
The consensus ROH regions and genes in all ten horses are shown. The number of SNPs and size of shared ROH indicate the overlap of homozygous variants
Fig. 5.
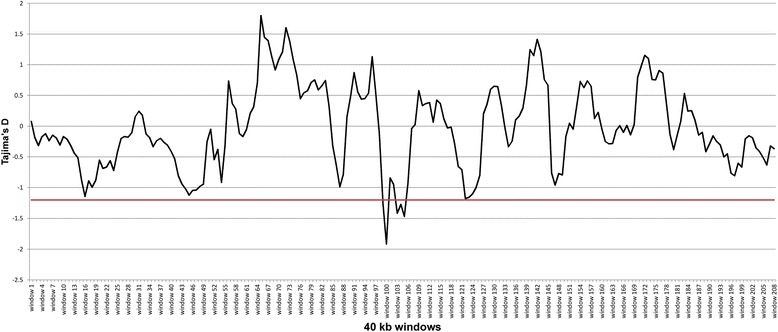
Tajima’s D estimate on equine chromosome 28 in the region of 13.68–15.75 Mb for all ten horses. Decreased Tajima’s D values below −1.2 can be observed in the consensus ROH extending over 14.65–14.78 Mb and harboring KITLG
Functional variations in ROH regions
Private ROHs were further investigated for variants which might have a functional impact on horse group specific traits. In non-breed horses, 166 mutations with predicted high or moderate effects within ROHs of 50-SNP windows and 5 mutations within ROHs of 500-SNP windows could be filtered out (Additional file 7 and 8). Three SNPs located on chromosome 10 at 19,334,666 (p.Val667Leu), 34,179,092 (p.Asp5Asn) and 34,221,357 Mb (p.Val208Ile) and one SNP on chromosome 28 at 8,441,975 Mb (p.Met260Thr) were found homozygous for the mutated allele in the Dülmen Horse and the two Sorraia horses but heterozygous or homozygous wild type in all breed horses. The Val667Leu variant in exon 10 of Histidine Rich Calcium Binding Protein (HRC) was predicted to be deleterious (SIFT score 0.01) whereas the other three variants located in Elongation Of Very Long Chain Fatty Acids Protein 4 (ELOVL4), Phosphotyrosine Picked Threonine-Protein Kinase (TTK) and ACSS3 were proposed to be tolerated (SIFT score 0.32, 0.34, 0.12). In contrast to non-breeds, breed horses harbored no variants with high or moderate effects in private ROHs. Nevertheless, the four Hanoverians could be shown to harbor four SNPs in their private ROH regions in the genes DYX1C1, CSAD and in the novel gene ENSECAG00000004438 (Additional file 9). These missense mutations showed no specific genotypes which could be exclusively found in the four Hanoverians. Furthermore, a closer examination of the consensus ROHs of all ten horses revealed a total of seven SNPs in the intronic region of KITLG but no variants with high or moderate effects.
Discussion
The detection of ROHs in ten horses of six different populations allowed us to estimate the genetic diversity in breeds or non-breeds and their signatures of potential selection. Smaller ROHs could be found in all horses to a very high number whereas ROHs of a larger size >59 kb and also longer stretches of consecutive homozygous genotypes >400 kb showed quite distinct distribution among different horse populations. Long homozygous stretches and consequently high inbreeding coefficients characterized the Sorraia and Thoroughbred, which were shown to be closed populations, as well as the Arabian derived from a relatively narrow genetic base [32–34]. Especially in the Thoroughbred the low genetic diversity was supposed to be a result of high selective pressures for specific traits of racing performance [12].
In contrast, the four Hanoverian sport horses in our study showed a low number of ROHs and relatively low values for FROH indicative for inbreeding. Nevertheless, they shared 18 ROHs which harbored six genes potentially important for appearance and performance in sport horses. One of these genes, the homeodomain transcription factor coding gene ALX4 was proposed to play an essential role in the skeletal mineralization and epidermal development in human and mice [35, 36]. Neurologic activity could be shown to be affected by CSAD, regulating intracellular calcium levels in neurons by its influence on taurine biosynthesis, and DYX1C1 involved in neuronal migration [37–39]. The candidate gene TBC1D30 has been characterized as a signal transducing peptide [40]. Comparative analyses of indicine and taurine cattle revealed signatures of selection and copy number variations in the region of TBC1D30 [41]. In KEPI (PPP1R14C)-knockout mice, a reduce response to repeated morphine injections suggested an important role of KEPI in the regulation of analgesic tolerance [42]. KEPI was shown to be expressed in brain regions of drug reward, locomotor control and nociception [42, 43]. Furthermore, it was supposed to play an important role for the regulation of glycogen synthase by its inhibitive effect on protein phosphatase 1 (PP1) [44]. A significant impact on fertility could be observed in association with CFAP61 which was shown to affect cilia and flagella motility [45, 46]. It can be assumed that these functional effects on neurologic control, signaling pathways, glycogen balance and reproduction might represent important targets of selection for the Hanoverian, which has become a specifically shaped breed into a modern sport horse type. In comparison to ROH analysis of all breed horses, the number of ROHs in the Hanoverian was relatively high probably as a result of breed specific similarities. However, despite significant differences in-between breeds, the whole group of breed horses revealed a region of potential selection harboring a fertility-related gene. YES1 could be shown to be an essential protein tyrosine kinase for self-defensive mechanisms in spermatocytes [47]. During testicular heat stress a significantly upregulated expression of YES1 was supposed to antagonize apoptotic processes to maintain spermatogenic differentiation and male fertility. In addition to that, it was even more intriguing that ROH analyses in non-breed horses also suggested a high positive selection for reproduction in mainly naturally selected horses. One of the largest ROHs could be shown to harbor the fertility related gene SPATA25 which is known to be mainly expressed in testis in human. Studies of obstructive azoospermia revealed a significantly reduced expression level in affected patients in comparison to fertile persons [48]. Other candidate genes were proposed to be involved in embryonic development. Analyses of FRAS1 deficient mice revealed phenotypic defects affecting embryonic epithelial basement membranes and internal organs [49]. Furthermore a number of genes involved in energy metabolism (Acyl-coenzyme A synthetase 3, ACSS3; thioesterase 8, ACOT8) [50, 51] and muscle development (NEURL2) [52, 53] could be found in large non-breed specific ROHs. The differentiation and survival of cardiomyocytes was supposed to be affected by SFRP2 [54]. It was shown that SFRP2 plays an important role in myocardial survival and is involved in ischemic injury repair of cardiomyocytes. The assumption of a potential non-breed specific effect on myocardial regulation for greater endurance in free range conditions was supported through the detection of a functional variant with a possibly deleterious impact on HRC. It was suggested that different expression levels of HRC can affect CA2+ homeostasis and contractile function of the heart [55, 56]. In human and mice affected with heart failures, the HRC expression levels could be shown to be significantly decreased. In conclusion, we propose that non-breed horses underlie a selection mainly driven by nature which affects reproduction, embryonic development, energy metabolism and cardiac development traits. These results confirm the suggestion that metabolic processes and morphogenesis play an important role for survival and maintenance in non-breeds [57].
Despite the specific genetic features in non-breeds as well as breeds and the general differences in the number and length of ROHs in various horse breeds, a functional enrichment of genes affecting cellular, metabolic and developmental as well as immune system and reproduction processes could be shown in ROHs in all ten horses.
These results suggest that despite the low number of individuals in some breeds or non-breeds these ten horses presumably represent a general phenomenon in horse populations. We assume that regions of genes involved in fundamental processes essential for development and sustainment of individuals and populations underlie high selective pressures and accordingly limited variations. A main focus which could be found in all breeds, specific breeds (Hanoverian) and also in non-breeds was a potential selection for traits of reproduction. Essential genes for processes affecting fertility, embryonic development and birth varied among different horse populations but could be assumed to play a key role in artificial or natural selection as well. Reproduction performance has been shown to be of high economic importance in breeds and of vital importance for non-breeds to ensure survival in the wild [5, 58]. Various studies in livestock came to the same conclusion and identified reproduction traits are essential targets of selection [9, 27, 28].
This assumption is supported by our detection of three consensus ROHs in all ten horses which harbor only one annotated gene, the KITLG, also known as Mast Cell Growth Factor, Stem Cell Factor or steel factor [59, 60]. Scans for signatures of diversifying selection in pigs proposed the KITLG locus to be a breed-specific signature in the Berkshire [61]. Due to its complex functional capacity, KITLG has fundamental impact on various essential processes affecting melanogenesis, haematopoiesis and gametogenesis [59, 62, 63]. Mutations in KITLG and its receptor KIT were shown to affect multiple cell formation stages parallelly during embryonic development and in fully-grown mice [63, 64]. The Steel Panda mutation at KITLG locus resulted in anemic black-eyed mice of white color with pigmented ears and scrotum and caused sterility in females. In human, a significant association for male infertility could be detected in KITLG affecting sperm count in patients [65]. In horses, the receptor of KITLG (KIT) was suggested to encode the dominant white (W) locus and to initiate severe disorders in haematopoietic system which might be responsible for the lethal consequences of homozygous W/W-genotype [66]. It was proposed that the dominant white phenotype is restricted in some breed registries due to the lethal effect of the homozygous dominant white mutation and also due to the risk of greater susceptibility to skin diseases. Therefore, we assume that the number of negative effects of KITLG mutations particularly affecting traits of reproduction and development have led to a strong positive selection of this region in horses that resulted in long ROHs.
The results of our study suggest that despite significant differences in-between breed and non-breed horses with regard to functional traits, all horse populations show strong signatures of selection in the region of genes affecting traits of reproduction.
Methods
Ethics statement
All animal work has been conducted according to the national and international guidelines for animal welfare. The EDTA-blood sampling was approved by the Institutional Animal Care and Use Committee (IACUC), the Lower Saxony state veterinary office at the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany (registration number 11A 160/7221.3-2.1-015/11, 8.84-02.05.20.12.066).
Samples and sequencing
Sequencing analysis was based on data from two Sorraia, one Dülmen Horse, one Arabian, one Saxon-Thuringian Heavy Warmblood, one Thoroughbred and four Hanoverian. Among these horses, six whole-genome sequences from two Hanoverian (SRX389480/SRX389477), one Arabian (SRX389472), one Sorraia (SRX389475), one Dülmen Horse (SRX384479) and one Thoroughbred (SRR1055837) were obtained from the Sequence Read Archive (NCBI). The remaining samples of a Sorraia mare, a Saxon-Thuringian Heavy Warmblood and two Hanoverian stallions were prepared for whole-genome sequencing. DNA was extracted from white blood cells derived from EDTA-blood sampling using Invisorb Spin Blood Mini kit according to the manufacturers’ protocol (Stratec Biomedical, Birkenfeld, Germany). Paired-end libraries of the two Hanoverian were prepared using the Illumina DNA sample preparation kit (Illumina, San Diego, CA). DNA-samples were sheared on the Covaris (Covaris, Woburn, Massachusetts) and purified with Agencourt AMPure XP beads (Beckman Coulter, Krefeld, Germany). The remaining two samples (Sorraia and Saxon-Thuringian Heavy Warmblood) were prepared using the Illumina Nextera DNA Sample Prep Kit according to the manufacturers’ protocol and purified with Agencourt AMPure XP beads as well. The whole genome of both Hanoverian was sequenced using an Illumina HiSeq2000 (Illumina) in paired-end mode (2 × 101 bp reads), whereas the Sorraia and Saxon-Thuringian Heavy Warmblood were run on an Illumina MiSeq with v2 Reagent Kits (2 x 250 bp reads) four times paired-end on a single lane flowcell to reach an adequate coverage for whole genome sequencing.
Quality control of FASTQ-files was done using fastqc 0.11.3 [67]. Reads were mapped to the reference genome EquCab2.70 using BWA 0.7.12 [68] and converted into binary format using SAMtools 1.2 [69]. PCR duplicated were marked using Picard tools (http://picard.sourceforge.net, version 1.130). Local realignment around INDELs, quality score recalibration and SNP calling was performed using GATK [70]. In order to get reliable data for variant detection we removed variants with a read depth <2 and >1000 and quality values <20 (qual). Variant annotation and effect prediction was done using SNPEff version 4.1 B (2015-02-13) [71]. The VCF file was adapted to PLINK 1.07 format using SAS/Genetics 9.4 (Statistical Analysis System, Cary, NC) and VCFtools 0.1.12b [72].
The data file for all ten horses re-sequenced is available at www.animalgenome.org (10horses.recode.vcf.gz). Raw data can be downloaded at the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra), BioProject ID PRJNA291776 (Submission ID: SUB1048258).
Runs of homozygosity
ROHs were detected using a trimmed dataset of 11,325,777 SNPs with a minimum read depth of 3, a maximum read depth of 60 and a minimum mean read depth of 12 for all ten samples. The X chromosome was omitted for this analysis. We defined ROHs as homozygous regions in sliding windows of 50 SNPs in a first run and 500 SNPs in a second approach using PLINK, version 1.07 (http://pngu.mgh.harvard.edu/purcell/plink/, [73]). Homozygous genotypes of >40 kb as well as >400 kb were defined as ROHs. The minimum distance of SNPs was estimated 0.8. This distance estimation was determined dividing the size of the genome covered with SNPs by the number of SNPs. No more than three SNPs with missing genotypes and three heterozygous SNPs were allowed in each window. The detected ROHs were categorized into small, medium and large ROHs and filtered for individual ROH regions for specific horses or breeds using SAS/Genetics, version 9.4. Private ROHs were determined by filtering out homozygous variants in ROHs in the horse of interest which could not be found in ROHs of other horses. Thus whole individual ROHs or individual parts of ROHs were detected as private ROHs for specific horses as well as for breeds or non-breeds. Consensus ROH regions were derived from intersections of homozygous variants in all ten horses. Furthermore inbreeding coefficients (FROH) were estimated for each horse dividing the size of ROHs in bp by the length of the genome (2,242,879,462 bp) covered with SNPs.
In addition to that, theta estimations and neutrality test statistics Tajima’s D, Fu&Li F’s, Fu&Li’s D, Fay’s H, Zeng’s E were obtained using ANGSD version 0.902 [74]. Analyses were performed for all detected private ROHs in breed, non-breed and Hanoverian horses and for the consensus ROHs as well. Run parameters were adjusted to control for sequencing errors using a minimum quality value of 20 (−minQ 20) and filtering for a read depth of 3 to 60 (−geno_minDepth 3, −geno_maxDepth 60). Sliding windows of 40 kb as well as 400 kb were chosen for analysis.
Sequence error detection by SNP50 BeadChip
In addition to whole-genome sequencing, two horses (Hanoverian) of a previous study [57] and three horses (two Hanoverian and one Arabian) of the current study were genotyped on the Illumina SNP50 BeadChip. Sequence errors were estimated in comparison with BeadChip data identifying heterozygous SNPs in BeadChip data which were homozygous in NGS data as false-negative and homozygous SNPs in BeadChip data which were heterozygous in NGS data as false-positive. For a more robust estimation of average false-positive error rates, long ROHs >1 Mb in sliding windows of 20 SNPs and a minimum distance of 50 were detected in BeadChip data using PLINK. No heterozygous SNPs and two missing called were admitted. These long ROH were assumed to hold error free homozygous genotypes and therefore ensure more precise error estimation in comparison with NGS-SNPs. The false-positive error rates were taken into account in the ROH detection admitting three heterozygous SNPs in each sliding window.
Functional annotation
Gene lists of horse specific ROH regions were obtained using SAS/Genetics for filtering PLINK summary files and Galaxy intersection tool (https://usegalaxy.org/) [75–77] for gene allocation to genomic regions. The chromosomal positions of ROHs were aligned with the refseq gene table from UCSC (Ensembl genes) in order to obtain all genes located in ROHs. To improve functional analysis, we converted these gene lists to human orthologous genes using g:Profiler [78, 79]. PANTHER gene list analysis [80] was performed for functional classification of biological processes affected by genes in private ROH regions. In addition to these horse specific evaluations, further analyses for consensus ROHs in all ten horses and shared private ROHs in breed horses (Hanoverian, Arabian, Saxon-Thuringian Heavy Warmblood and Thoroughbred), non-breed horses (Dülmen Horse, Sorraia) and in the Hanoverian were performed. Gene names and its human orthologues were obtained using the Galaxy intersect function and g:Profiler as well. Genetic relations in-between genes were obtained using GeneMANIA [81].
Functional variant detection
Functional variants with high or moderate effects were evaluated using SAS/Genetics for filtering SNPEff predictions categorized into high, moderate and low variant impacts. We determined the distribution of genotypes in relation to breed or non-breed groups and detected SIFT [82] prediction scores for functional effects using the Variant Effect Predictor [83].
Acknowledgements
The authors thank the Hanoverian state stud Celle, the Hanoverian Breeding Association, the Arabian Horse Society (Verband der Züchter und Freunde des Arabischen Pferdes e.V.) and all horse owners for donation of data and samples. We also thank J. Wrede for his help in data analysis.
Abbreviations
- ROH
Runs of homozygosity
- SNP
Single nucleotide polymorphism
- NGS
Next generation sequencing
- MSTN
Myostatin gene
- PLAG1
Pleiomorphic adenoma gene 1
- EDNRB
Endothelin receptor type B, DYX1C1, dyslexia susceptibility 1 candidate 1
- PPP1R14C
Protein phosphatase 1, regulatory (inhibitor) subunit 14C
- CFAP61/C20orf26
Cilia and flagella associated protein 61
- CSAD
Cysteine sulfinic acid decarboxylase
- TBC1D30
TBC1 domain family, member 30
- ALX4
ALX homeobox 4
- KITLG
KIT Ligand
Additional files
Summary of mapping metrics, sequence coverage and number of detected variants in ten horses. (DOCX 174 kb)
Inbreeding coefficients (F ROH ) based on runs of homozygosity (ROH). FROH was estimated dividing the total length of ROHs by the length of the genome covered by SNPs (2,242,879,462 bp). (DOCX 15 kb)
Functional annotations in private runs of homozygosity (ROH) of 500-SNP windows. PANTHER gene list analysis (http://www.pantherdb.org/) was performed for genes in private ROH regions which could be exclusively found in one specific horse. The percent of gene hits against total number of process hits involved in specific biological processes are shown. (DOCX 18 kb)
Shared runs of homozygosity (ROHs) in non-breed horses. The consensus private ROH regions and genes of the Dülmen Horse and two Sorraia in 50-SNP and 500-SNP windows are shown. The number of SNPs and size of shared ROH indicate the overlap of homozygous variants. (DOCX 78 kb)
Shared runs of homozygosity (ROHs) in breed horses. The consensus private ROH regions and genes of Hanoverian, Arabian, Saxon-Thuringian Heavy Warmblood and Thoroughbred horses in 50-SNP windows are shown. The number of SNPs and size of shared ROH indicate the overlap of homozygous variants. (DOCX 14 kb)
Theta estimations and neutrality test statistics for consensus and private ROHs. All private ROHs detected in the groups non-breed, breed and Hanoverian as well as in the region of KITLG were analyzed for Tajima’s D, Fu&Li F’s, Fu&Li’s D, Fay’s H, Zeng’s E using the software ANGSD (http://popgen.dk/angsd). (XLSX 18 kb)
Mutations with high or moderate effects in ROHs (50-SNP windows) of non-breed horses. The ROH position and size (EquCab2.70), the position of SNPs, their mutant allele, potential impact and type are shown. Impact estimations are derived from SNPEff predictions. (DOCX 65 kb)
Mutations with high or moderate effects in ROHs (500-SNP windows) of non-breed horses. The ROH position and size (EquCab2.70), the position of SNPs, their mutant allele, potential impact and type are shown. Impact estimations are derived from SNPEff predictions. (DOCX 16 kb)
Mutations with high or moderate effects in ROHs (50-SNP windows) of the four Hanoverians. The ROH position and size (EquCab2.70), the position of SNPs, their mutant allele, potential impact and type are shown. Impact estimations are derived from SNPEff predictions. (DOCX 15 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JM, MK and OD designed the study. JM and OD carried out the experiments and data analysis, drafted and finalized the manuscript. RT, SB, LA, MG and IG performed HiSeq next generation sequencing, performed part of raw data analysis and helped to finalize the manuscript. All authors read and approved the final manuscript.
Contributor Information
Julia Metzger, Email: julia.metzger@tiho-hannover.de.
Matthias Karwath, Email: Matthias.Karwath@smul.sachsen.de.
Raul Tonda, Email: rtonda@pcb.ub.cat.
Sergi Beltran, Email: sbeltrana@pcb.ub.cat.
Lídia Águeda, Email: lagueda@pcb.ub.cat.
Marta Gut, Email: mgut@pcb.ub.cat.
Ivo Glynne Gut, Email: igut@pcb.ub.es.
Ottmar Distl, Email: ottmar.distl@tiho-hannover.de.
References
- 1.Petersen JL, Mickelson JR, Rendahl AK, Valberg SJ, Andersson LS, Axelsson J, et al. Genome-wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet. 2013;9(1):e1003211. doi: 10.1371/journal.pgen.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Makvandi-Nejad S, Hoffman GE, Allen JJ, Chu E, Gu E, Chandler AM, Loredo AI, Bellone RR, Mezey JG, Brooks SA, Sutter NB. Four Loci explain 83 % of size variation in the horse. PLoS ONE. 2012;7(7):e39929. doi: 10.1371/journal.pone.0039929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orlando L, Ginolhac A, Zhang G, Froese D, Albrechtsen A, Stiller M, Schubert M, Cappellini E, Petersen B, Moltke I, Johnson PL, Fumagalli M, Vilstrup JT, Raghavan M, Korneliussen T, Malaspinas AS, Vogt J, Szklarczyk D, Kelstrup CD, Vinther J, Dolocan A, Stenderup J, Velazquez AM, Cahill J, Rasmussen M, Wang X, Min J, Zazula GD, Seguin-Orlando A, Mortensen C, et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature. 2013;499(7456):74–78. doi: 10.1038/nature12323. [DOI] [PubMed] [Google Scholar]
- 4.Brooks SA, Makvandi-Nejad S, Chu E, Allen JJ, Streeter C, Gu E, McCleery B, Murphy BA, Bellone R, Sutter NB. Morphological variation in the horse: defining complex traits of body size and shape. Anim Genet. 2010;41(Suppl 2):159–165. doi: 10.1111/j.1365-2052.2010.02127.x. [DOI] [PubMed] [Google Scholar]
- 5.Beckmann S. Roentgenologische Untersuchung zur Osteochondrosis dissecans an Fessel-, Sprung-und Kniegelenken bei 85 Duelmener Wildpferden. Berlin: Freie Universitaet Berlin, Diss., 2011; 2011. [Google Scholar]
- 6.Pinheiro M, Kjöllerström HJ, Oom MM. Genetic diversity and demographic structure of the endangered Sorraia horse breed assessed through pedigree analysis. Livest Sci. 2013;152(1):1–10. doi: 10.1016/j.livsci.2012.11.017. [DOI] [Google Scholar]
- 7.Warmuth V, Manica A, Eriksson A, Barker G, Bower M. Autosomal genetic diversity in non-breed horses from eastern Eurasia provides insights into historical population movements. Anim Genet. 2013;44(1):53–61. doi: 10.1111/j.1365-2052.2012.02371.x. [DOI] [PubMed] [Google Scholar]
- 8.Ku CS, Naidoo N, Teo SM, Pawitan Y. Regions of homozygosity and their impact on complex diseases and traits. Hum Genet. 2011;129(1):1–15. doi: 10.1007/s00439-010-0920-6. [DOI] [PubMed] [Google Scholar]
- 9.Bosse M, Megens HJ, Madsen O, Paudel Y, Frantz LA, Schook LB, Crooijmans RP, Groenen MA. Regions of homozygosity in the porcine genome: consequence of demography and the recombination landscape. PLoS Genet. 2012;8(11):e1003100. doi: 10.1371/journal.pgen.1003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purfield DC, Berry DP, McParland S, Bradley DG. Runs of homozygosity and population history in cattle. BMC Genet. 2012;13:70. doi: 10.1186/1471-2156-13-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gibson J, Morton NE, Collins A. Extended tracts of homozygosity in outbred human populations. Hum Mol Genet. 2006;15(5):789–795. doi: 10.1093/hmg/ddi493. [DOI] [PubMed] [Google Scholar]
- 12.Gu J, Orr N, Park SD, Katz LM, Sulimova G, MacHugh DE, Hill EW. A genome scan for positive selection in thoroughbred horses. PLoS ONE. 2009;4(6):e5767. doi: 10.1371/journal.pone.0005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freedman AH, Gronau I, Schweizer RM, Ortega-Del Vecchyo D, Han E, Silva PM, Galaverni M, Fan Z, Marx P, Lorente-Galdos B. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 2014;10(1):e1004016. doi: 10.1371/journal.pgen.1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirin M, McQuillan R, Franklin CS, Campbell H, McKeigue PM, Wilson JF. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE. 2010;5(11):e13996. doi: 10.1371/journal.pone.0013996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L, Smolej-Narancic N, Janicijevic B, Polasek O, Tenesa A, Macleod AK, Farrington SM, Rudan P, Hayward C, Vitart V, Rudan I, Wild SH, Dunlop MG, Wright AF, Campbell H, Wilson JF. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83(3):359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nothnagel M, Lu TT, Kayser M, Krawczak M. Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans. Hum Mol Genet. 2010;19(15):2927–2935. doi: 10.1093/hmg/ddq198. [DOI] [PubMed] [Google Scholar]
- 17.Nalls M, Guerreiro R, Simon-Sanchez J, Bras J, Traynor B, Gibbs J, Launer L, Hardy J, Singleton A. Extended tracts of homozygosity identify novel candidate genes associated with late-onset Alzheimer’s disease. Neurogenetics. 2009;10(3):183–190. doi: 10.1007/s10048-009-0182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alkuraya FS. The application of next-generation sequencing in the autozygosity mapping of human recessive diseases. Hum Genet. 2013;132(11):1197–1211. doi: 10.1007/s00439-013-1344-x. [DOI] [PubMed] [Google Scholar]
- 19.Lencz T, Lambert C, DeRosse P, Burdick KE, Morgan TV, Kane JM, Kucherlapati R, Malhotra AK. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc Natl Acad Sci U S A. 2007;104(50):19942–19947. doi: 10.1073/pnas.0710021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qanbari S, Pausch H, Jansen S, Somel M, Strom TM, Fries R, Nielsen R, Simianer H. Classic selective sweeps revealed by massive sequencing in cattle. PLoS Genet. 2014;10(2):e1004148. doi: 10.1371/journal.pgen.1004148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ai H, Huang L, Ren J. Genetic diversity, linkage disequilibrium and selection signatures in Chinese and Western pigs revealed by genome-wide SNP markers. PLoS ONE. 2013;8(2):e56001. doi: 10.1371/journal.pone.0056001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boyko AR, Quignon P, Li L, Schoenebeck JJ, Degenhardt JD, Lohmueller KE, Zhao K, Brisbin A, Parker HG, Cargill M. A simple genetic architecture underlies morphological variation in dogs. PLoS Biol. 2010;8(8):e1000451. doi: 10.1371/journal.pbio.1000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Axelsson E, Ratnakumar A, Arendt M-L, Maqbool K, Webster MT, Perloski M, Liberg O, Arnemo JM, Hedhammar Å, Lindblad-Toh K. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495(7441):360–364. doi: 10.1038/nature11837. [DOI] [PubMed] [Google Scholar]
- 24.Pfahler S, Distl O. Effective Population Size, Extended Linkage Disequilibrium and Signatures of Selection in the Rare Dog Breed Lundehund. PLoS ONE. 2015;10(4):e0122680. doi: 10.1371/journal.pone.0122680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferenčaković M, Hamzić E, Gredler B, Solberg TR, Klemetsdal G, Curik I, Sölkner J. Estimates of autozygosity derived from runs of homozygosity: empirical evidence from selected cattle populations. J Anim Breed Genet. 2013;130(4):286–293. doi: 10.1111/jbg.12012. [DOI] [PubMed] [Google Scholar]
- 26.Ferencakovic M, Hamzic E, Gredler B, Curik I, Sölkner J. Runs of homozygosity reveal genome-wide autozygosity in the Austrian Fleckvieh cattle. Agric. Conspec. Sci. 2011;76(4):325–329. [Google Scholar]
- 27.Qanbari S, Gianola D, Hayes B, Schenkel F, Miller S, Moore S, Thaller G, Simianer H. Application of site and haplotype-frequency based approaches for detecting selection signatures in cattle. BMC Genomics. 2011;12(1):318. doi: 10.1186/1471-2164-12-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qanbari S, Pimentel E, Tetens J, Thaller G, Lichtner P, Sharifi A, Simianer H. A genome-wide scan for signatures of recent selection in Holstein cattle. Anim Genet. 2010;41(4):377–389. doi: 10.1111/j.1365-2052.2009.02016.x. [DOI] [PubMed] [Google Scholar]
- 29.Kim E-S, Cole JB, Huson H, Wiggans GR, Van Tassell CP, Crooker BA, Liu G, Da Y, Sonstegard TS. Effect of artificial selection on runs of homozygosity in US Holstein cattle. PLoS ONE. 2013;8(11):e80813. doi: 10.1371/journal.pone.0080813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacEachern S, Hayes B, McEwan J, Goddard M. An examination of positive selection and changing effective population size in Angus and Holstein cattle populations (Bos taurus) using a high density SNP genotyping platform and the contribution of ancient polymorphism to genomic diversity in Domestic cattle. BMC Genomics. 2009;10(1):181. doi: 10.1186/1471-2164-10-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacLeod IM, Larkin DM, Lewin HA, Hayes BJ, Goddard ME. Inferring demography from runs of homozygosity in whole-genome sequence, with correction for sequence errors. Mol Biol Evol. 2013;30(9):2209–2223. doi: 10.1093/molbev/mst125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aberle KS, Hamann H, Drögemüller C, Distl O. Genetic diversity in German draught horse breeds compared with a group of primitive, riding and wild horses by means of microsatellite DNA markers. Anim Genet. 2004;35(4):270–277. doi: 10.1111/j.1365-2052.2004.01166.x. [DOI] [PubMed] [Google Scholar]
- 33.Cunningham E, Dooley J, Splan R, Bradley D. Microsatellite diversity, pedigree relatedness and the contributions of founder lineages to thoroughbred horses. Anim Genet. 2001;32(6):360–364. doi: 10.1046/j.1365-2052.2001.00785.x. [DOI] [PubMed] [Google Scholar]
- 34.Khanshour A, Conant E, Juras R, Cothran EG. Microsatellite analysis of genetic diversity and population structure of Arabian horse populations. J Hered. 2013;104(3):386–98. doi: 10.1093/jhered/est003. [DOI] [PubMed] [Google Scholar]
- 35.Mavrogiannis LA, Antonopoulou I, Baxová A, Kutílek S, Kim CA, Sugayama SM, Salamanca A, Wall SA, Morriss-Kay GM, Wilkie AO. Haploinsufficiency of the human homeobox gene ALX4 causes skull ossification defects. Nat Genet. 2001;27(1):17–18. doi: 10.1038/83703. [DOI] [PubMed] [Google Scholar]
- 36.Kayserili H, Uz E, Niessen C, Vargel I, Alanay Y, Tuncbilek G, Yigit G, Uyguner O, Candan S, Okur H. ALX4 dysfunction disrupts craniofacial and epidermal development. Hum Mol Genet. 2009;18(22):4357–4366. doi: 10.1093/hmg/ddp391. [DOI] [PubMed] [Google Scholar]
- 37.Foos TM, Wu J-Y. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochem Res. 2002;27(1–2):21–26. doi: 10.1023/A:1014890219513. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Paramasivam M, Thomas A, Bai J, Kaminen-Ahola N, Kere J, Voskuil J, Rosen G, Galaburda A, Loturco J. DYX1C1 functions in neuronal migration in developing neocortex. Neuroscience. 2006;143(2):515–522. doi: 10.1016/j.neuroscience.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 39.Taipale M, Kaminen N, Nopola-Hemmi J, Haltia T, Myllyluoma B, Lyytinen H, Muller K, Kaaranen M, Lindsberg PJ, Hannula-Jouppi K. A candidate gene for developmental dyslexia encodes a nuclear tetratricopeptide repeat domain protein dynamically regulated in brain. Proc Natl Acad Sci. 2003;100(20):11553–11558. doi: 10.1073/pnas.1833911100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ishibashi K, Kanno E, Itoh T, Fukuda M. Identification and characterization of a novel Tre2/Bub2/Cdc16 (TBC) protein that possesses Rab3A GAP activity. Genes Cells. 2009;14(1):41–52. doi: 10.1111/j.1365-2443.2008.01251.x. [DOI] [PubMed] [Google Scholar]
- 41.O’Brien AMP, Utsunomiya YT, Mészáros G, Bickhart DM, Liu GE, Van Tassell CP, Sonstegard TS, Da Silva MV, Garcia JF, Sölkner J. Assessing signatures of selection through variation in linkage disequilibrium between taurine and indicine cattle. Genet Sel Evol. 2014;46(1):19. doi: 10.1186/1297-9686-46-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drgonova J, Zimonjic DB, Hall FS, Uhl GR. Effect of KEPI (Ppp1r14c) deletion on morphine analgesia and tolerance in mice of different genetic backgrounds: when a knockout is near a relevant quantitative trait locus. Neuroscience. 2010;165(3):882–895. doi: 10.1016/j.neuroscience.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gong J-P, Liu Q-R, Zhang P-W, Wang Y, Uhl G. Mouse brain localization of the protein kinase C-enhanced phosphatase 1 inhibitor KEPI (kinase C-enhanced PP1 inhibitor) Neuroscience. 2005;132(3):713–727. doi: 10.1016/j.neuroscience.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 44.Newgard CB, Brady MJ, O’Doherty RM, Saltiel AR. Organizing glucose disposal: emerging roles of the glycogen targeting subunits of protein phosphatase-1. Diabetes. 2000;49(12):1967–1977. doi: 10.2337/diabetes.49.12.1967. [DOI] [PubMed] [Google Scholar]
- 45.Dymek EE, Smith EF. A conserved CaM-and radial spoke–associated complex mediates regulation of flagellar dynein activity. J Cell Biol. 2007;179(3):515–526. doi: 10.1083/jcb.200703107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Urbanska P, Song K, Joachimiak E, Krzemien-Ojak L, Koprowski P, Hennessey T, Jerka-Dziadosz M, Fabczak H, Gaertig J, Nicastro D. The CSC proteins FAP61 and FAP251 build the basal substructures of radial spoke 3 in cilia. Mol Biol Cell. 2015;26(8):1463–1475. doi: 10.1091/mbc.E14-11-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang Y, Dong Y, Zhao J, Li W. YES1 activation elicited by heat stress is anti-apoptotic in mouse pachytene spermatocytes. Biol Reprod. 2013;89(6):131. doi: 10.1095/biolreprod.113.112235. [DOI] [PubMed] [Google Scholar]
- 48.Zhou Y, Qin D, Tang A, Zhou D, Qin J, Yan B, Diao R, Jiang Z, Cai Z, Gui Y. Developmental expression pattern of a novel gene, TSG23/Tsg23, suggests a role in spermatogenesis. Mol Hum Reprod. 2009;15(4):223–230. doi: 10.1093/molehr/gap015. [DOI] [PubMed] [Google Scholar]
- 49.Petrou P, Chiotaki R, Dalezios Y, Chalepakis G. Overlapping and divergent localization of Frem1 and Fras1 and its functional implications during mouse embryonic development. Exp Cell Res. 2007;313(5):910–920. doi: 10.1016/j.yexcr.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 50.Hunt MC, Rautanen A, Westin MA, Svensson LT, Alexson SE. Analysis of the mouse and human acyl-CoA thioesterase (ACOT) gene clusters shows that convergent, functional evolution results in a reduced number of human peroxisomal ACOTs. FASEB J. 2006;20(11):1855–1864. doi: 10.1096/fj.06-6042com. [DOI] [PubMed] [Google Scholar]
- 51.Watkins PA, Maiguel D, Jia Z, Pevsner J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J Lipid Res. 2007;48(12):2736–2750. doi: 10.1194/jlr.M700378-JLR200. [DOI] [PubMed] [Google Scholar]
- 52.Nastasi T, Bongiovanni A, Campos Y, Mann L, Toy JN, Bostrom J, Rottier R, Hahn C, Conaway JW, Harris AJ. Ozz-E3, a muscle-specific ubiquitin ligase, regulates β-catenin degradation during myogenesis. Dev Cell. 2004;6(2):269–282. doi: 10.1016/S1534-5807(04)00020-6. [DOI] [PubMed] [Google Scholar]
- 53.Gahlmann R, Kedes L. Cloning, structural analysis, and expression of the human fast twitch skeletal muscle troponin C gene. J Biol Chem. 1990;265(21):12520–12528. [PubMed] [Google Scholar]
- 54.Mirotsou M, Zhang Z, Deb A, Zhang L, Gnecchi M, Noiseux N, Mu H, Pachori A, Dzau V. Secreted frizzled related protein 2 (Sfrp2) is the key Akt-mesenchymal stem cell-released paracrine factor mediating myocardial survival and repair. Proc Natl Acad Sc. 2007;104(5):1643–1648. doi: 10.1073/pnas.0610024104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fan G-C, Gregory KN, Zhao W, Park WJ, Kranias EG. Regulation of myocardial function by histidine-rich, calcium-binding protein. Am J Physiol Heart Circ Physiol. 2004;287(4):H1705–H1711. doi: 10.1152/ajpheart.01211.2003. [DOI] [PubMed] [Google Scholar]
- 56.Gregory KN, Ginsburg KS, Bodi I, Hahn H, Marreez YM, Song Q, Padmanabhan PA, Mitton BA, Waggoner JR, Del Monte F. Histidine-rich Ca binding protein: a regulator of sarcoplasmic reticulum calcium sequestration and cardiac function. J Mol Cell Cardiol. 2006;40(5):653–665. doi: 10.1016/j.yjmcc.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 57.Metzger J, Tonda R, Beltran S, Agueda L, Gut M, Distl O. Next generation sequencing gives an insight into the characteristics of highly selected breeds versus non-breed horses in the course of domestication. BMC Genomics. 2014;15(1):562. doi: 10.1186/1471-2164-15-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hamann H, Jude R, Sieme H, Mertens U, Töpfer‐Petersen E, Distl O, Leeb T. A polymorphism within the equine CRISP3 gene is associated with stallion fertility in Hanoverian warmblood horses. Anim Genet. 2007;38(3):259–264. doi: 10.1111/j.1365-2052.2007.01594.x. [DOI] [PubMed] [Google Scholar]
- 59.Matsui Y, Zsebo KM, Hogan BL. Embryonic expression of a haematopoietic growth factor encoded by the SI locus and the ligand for c-kit. Nature. 1990;347(6294):667–669. doi: 10.1038/347667a0. [DOI] [PubMed] [Google Scholar]
- 60.Seitz JJ, Schmutz SM, Thue TD, Buchanan FC. A missense mutation in the bovine MGF gene is associated with the roan phenotype in Belgian Blue and Shorthorn cattle. Mamm Genome. 1999;10(7):710–712. doi: 10.1007/s003359901076. [DOI] [PubMed] [Google Scholar]
- 61.Wilkinson S, Lu ZH, Megens H-J, Archibald AL, Haley C, Jackson IJ, Groenen MA, Crooijmans RP, Ogden R, Wiener P. Signatures of diversifying selection in European pig breeds. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wehrle-Haller B. The role of Kit-ligand in melanocyte development and epidermal homeostasis. Pigment Cell Res. 2003;16(3):287–296. doi: 10.1034/j.1600-0749.2003.00055.x. [DOI] [PubMed] [Google Scholar]
- 63.Beechey C, Loutit J, Searle A. Panda, a new steel allele. Mouse News Lett. 1986;74(92):52. [Google Scholar]
- 64.Huang EJ, Manova K, Packer AI, Sanchez S, Bachvarova RF, Besmer P. The murine steel panda mutation affects kit ligand expression and growth of early ovarian follicles. Dev Biol. 1993;157(1):100–109. doi: 10.1006/dbio.1993.1115. [DOI] [PubMed] [Google Scholar]
- 65.Galan J, De Felici M, Buch B, Rivero M, Segura A, Royo J, Cruz N, Real L, Ruiz A. Association of genetic markers within the KIT and KITLG genes with human male infertility. Hum Reprod. 2006;21(12):3185–3192. doi: 10.1093/humrep/del313. [DOI] [PubMed] [Google Scholar]
- 66.Mau C, Poncet PA, Bucher B, Stranzinger G, Rieder S. Genetic mapping of dominant white (W), a homozygous lethal condition in the horse (Equus caballus) J Anim Breed Genet. 2004;121:374–383. doi: 10.1111/j.1439-0388.2004.00481.x. [DOI] [Google Scholar]
- 67.Andrews S. FastQC: A quality control tool for high throughput sequence data. Reference Source. 2010.
- 68.Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Korneliussen TS, Moltke I, Albrechtsen A, Nielsen R. Calculation of Tajima’s D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinformatics. 2013;14(1):289. doi: 10.1186/1471-2105-14-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11(8):R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blankenberg D, Kuster GV, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010;Chapter 19:Unit 19.10:1–21. doi: 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15(10):1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reimand J, Kull M, Peterson H, Hansen J, Vilo J. g: Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35(suppl 2):W193–W200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reimand J, Arak T, Vilo J. g:Profiler--a web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39(Web Server issue):W307–315. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41(D1):D377–386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(suppl 2):W214–W220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 83.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]