

Abstract
Molecular biomimetic models suggest that proteins in the soft matrix of nanocomposites have a multimodular architecture. Engineered proteins were used together with nanofibrillated cellulose (NFC) to show how this type of architecture leads to function. The proteins consist of two cellulose-binding modules (CBM) separated by 12-, 24-, or 48-mer linkers. Engineering the linkers has a considerable effects on the interaction between protein and NFC in both wet colloidal state and a dry film. The protein optionally incorporates a multimerizing hydrophobin (HFB) domain connected by another linker. The modular structure explains effects in the hydrated gel state, as well as the deformation of composite materials through stress distribution and crosslinking. Based on this work, strategies can be suggested for tuning the mechanical properties of materials through the coupling of protein modules and their interlinking architectures.
Keywords: biomimetics, cellulose, materials, nanocomposites, supramolecular chemistry
Biological materials provide an inspiration for materials science since they show several interesting functions such as efficient combination of stiffness and toughness, gradients in properties, or solutions for joining dissimilar structures.[1, 2] A key insight has been that biological materials are hierarchically organized over several length scales, from overall architecture down to colloidal and macromolecular interactions.[3, 4] Bioinspired design and modeling has provided insight into higher-level and microscopic structures through the use of, for example, layered reinforcements, composites produced by freeze-casting, spin coatings, and layer-by-layer depositions.[5] Composite structures are a recurring theme that lead to stiff and tough materials and they are typically comprised of two main parts: stiff elements with elongated structure and a soft adhesive matrix.[6] The soft matrix provides binding for promoting overall strength and stiffness while still allowing energy dissipation upon mechanical loading. However, the molecular- and colloidal-level interactions and structures that lead to these functions are only now being explored,[7, 8] with several essential questions remaining unanswered, such as what are their molecular interactions, how do they assemble, what are the kinetics and mechanisms of their interactions?
Herein, we test the hypothesis that, in nature, proteins mediate matrix interactions and the structural details lead to functionally tuned properties. We aimed specifically to use modular domain architecture as a biomimetic model. Matrix proteins in the pearl oyster and structural proteins in squid beak are examples of natural modular structures that contain adhesive and interaction modules bound to each other by linkers.[9, 10]
As the stiff element, cellulose has been used for constructing nanoscale composites with interlocking soft matrices of polymeric, protein, or supramolecular components.[11–14] Nanofibrillated cellulose (NFC) is a form of cellulose for which this approach is particularly interesting since it is readily made and it has a very high aspect ratio, lateral dimensions of a few nm, lengths up to micrometers, and excellent mechanical properties.[15] Sheets of NFC have been prepared for “nanopaper” films, in which the high aspect ratio of pristine NFC results in a dense “spaghetti-like” colloidal structures in which stiffness and strength is promoted by entanglement of the nanofibrils and hydrogen bonds.[16]
For molecular adhesiveness to cellulose, we used cellulose-binding modules (CBMs). Their fold is termed the “cysteine knot” because they are highly cross-linked by disulphide bonds and have a small size (36–38 amino acids). They bind to cellulose through a set of aromatic residues and hydrogen bonding.[17] Linking two CBMs together allowed us to systematically adjust the interactions by changing the length of the linkers between the two modules. We constructed a set of three “double” CBMs (dCBMs) with linkers of 12, 24, and 48 amino acids between the CBM modules. The CBM sequences were from the enzymes Cel6A and Cel7A from the fungus Trichoderma reesei (Figure 1). For more information on the constructs, see the Supporting Information.
Figure 1.
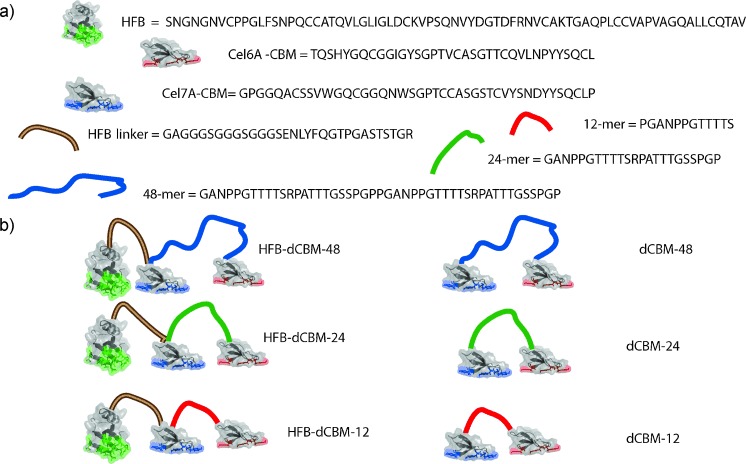
The modules and combinations used in the matrix design. a) The sequences of the modules (HFB, Cel6A CBM, and Cel7A CBM), the HFB-linker, and the three different CBM linkers (12-, 24-, and 48-mer). b) The six combinations. The dCBMs were obtained from the HFB-dCBMs through trypsin hydrolysis at the last R residue in the HFB linker.
Hydrophobins (HFB) are amphiphilic proteins that were chosen for their known propensity to form multimers and because they have very stable folded structures.[18] They form multimers through hydrophobic interactions and hydrogen bonds.[19] The HFBI hydrophobin that was used has a size of 74 amino acids and is cross-linked by four disulfide bonds. The proteins were made so that each of the three different dCBM constructs could be obtained with or without HFB, thereby giving a set of six different proteins (Figure 1). These are the three dCBM constructs with linkers of 12, 24, and 48 amino acids (named dCBM-12, dCBM-24, and dCBM-48), and the three otherwise identical constructs (HFB-dCBM-12, HFB-dCBM-24, and HFB-dCBM-48) with a HFB domain attached through a linker (Figure S1 and experimental details in the Supporting Information). Mass spectroscopy showed that the linkers in the 24- and 48-mers were O-glycosylated while the 12-mer was not. O-glycosylation typically leads to more extended conformations of the linkers.[20]
The NFC was obtained through mechanical disintegration of bleached kraft pulp as described in the Supporting Information. Analysis of the binding interaction of the dCBMs and NFC was done by using tritium labeling of the proteins. Binding isotherms are shown in Figure 2. A one-site Langmuir model was fitted to the data to give the binding parameters (Table S2 in the Supporting Information). These show a clear effect of the linkers on cooperative binding, where 12- and 24-mer linkers gave very similar properties, while the long 48-mer linker gave a clearly lower capacity.
Figure 2.
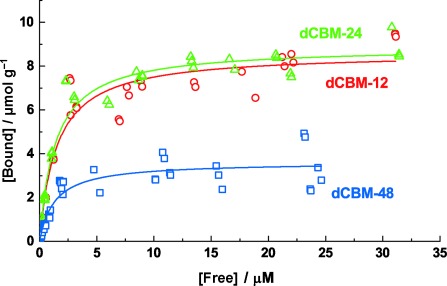
Binding isotherms of dCBM proteins on NFC.
For preparing samples, NFC and protein were mixed, with subsequent ultrasonication for efficient dispersion. Rheological measurements showed that the proteins increased the gel stiffness of NFC significantly (Figure 3 and Figure S2). The 12-mer gave higher G′ values than the 24-mer, although both bound similarly to NFC, thus showing that the modulus increases with decreasing linker length between CBMs. The 48-mer bound less, which together with the long linker could explain its lower G′ value. From these data, we conclude that the dCBD architecture results in crosslinking of NFC, with shorter linkers giving less flexibility. The presence of the HFB module lowered the G′ value compared to the corresponding non-fused dCBM. This effect could be based on steric exclusion owing to the relatively large size of HFB multimers, thereby reducing interactions between fibrils. As discussed below, the HFB-fused variants show identical binding compared to separated dCBMs, which rules out decreased binding to NFC as an explanation for this behavior. It should be emphasized that hydrophobins show highly dynamic interactions in aqueous solutions, forming large complexes as fusion proteins.[19] Cryo-TEM imaging indicated that NFC samples with proteins show fewer individually dispersed nanofibrils than samples without (Figure S3), thus indicating an association with the proteins.
Figure 3.
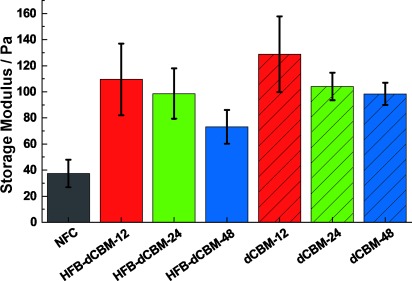
Gel formation by NFC and proteins (2 g l−1 NFC and a 42 % (w/w) protein to NFC). The average storage modulus (G′) increases the most with short linkers.
Free-standing films were made by collecting the dispersed NFC material on porous membranes by filtering followed by drying. Different protein loadings, specifically 25 %, 42 %, and 75 % of protein to NFC (w/w) with 2 g l−1 NFC, were investigated. The effects of varying overall protein concentration on the stress–strain curves are shown in Figures S4–S6. Based on this series, we chose 42 % for further experiments. Calculations based on the binding parameters (Table S2) showed that the level of loading was in all cases close to saturation of the cellulose. The protein amount in each of the films was determined by amino acid analysis. The results show that in all cases, variants with and without HFB bound identically, that is, HFB did not affect CBM binding. The amounts of dCBM-12 and dCBM-24 bound in the films were very similar, 12.1±0.9 μmol g−1 and 11.9±0.9 μmol g−1, respectively. The amount of dCBM-48 was 8.0±0.8 μmol g−1.
Tensile stress–strain results of the films are shown in Figures 4 and 5. Films without protein were used as a control and all measurements were made at controlled humidity (50 % RH) and ambient temperature (21 °C).
Figure 4.
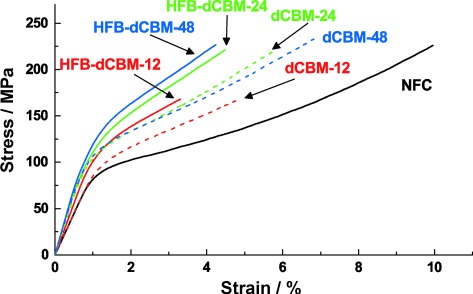
Representative stress–strain curves. The curve for the plain NFC control is shown as a solid black line; those for the HFB-dCBM proteins with 12-, 24-, and 48-mer linker lengths as solid red, green, and blue lines, respectively. Curves for the corresponding dCBMs are shown as dashed lines.
Figure 5.
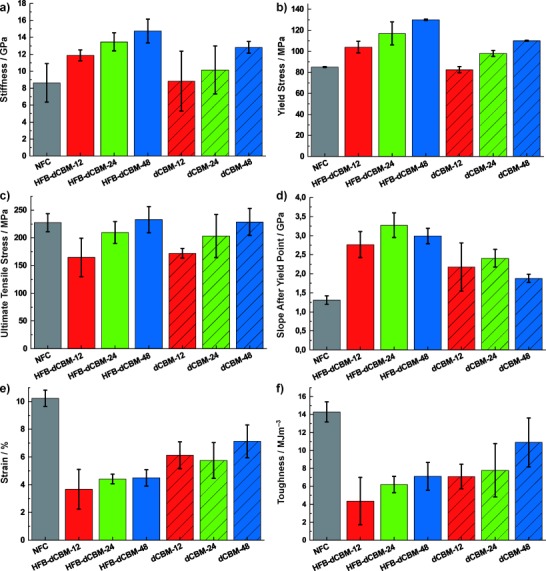
Mechanical properties of the HFB-dCBMs and dCBMs with an NFC film as a reference. a) Stiffness; b) yield strength; c) ultimate tensile strength; d) slope after yield point; e) strain-to-failure values and f) toughness. The standard deviations are shown for all data.
The curves show the same general shape that is typical for many biological composites.[6] Initially, at low deformation, the material is stiff until a yield point. After the yield point, the slope of the curve decreases and plastic deformation occurs. A large part of the toughness of the material (area under the curve) is from the region of plastic deformation. During this plastic deformation, there is a viscous flow of cellulose fibrils past each other until a stress maximum of 225 MPa, at which point catastrophic failure occurs.
We next consider how the molecular features of the matrix protein translate to mechanical functionality. Matrix-mediated interactions are seen by increases in stiffness, yield stress, and slope after the yield point. Two main observations are that the linker length between CBM modules and the presence of HFB produce significant effects. Comparing linker lengths in the 12-mer and 24-mer dCBM films is straightforward since these films contain equimolar amounts of protein. Notably the 12-mer linker led to significantly reduced ultimate tensile stress values in both the HFB-linked and dCBM versions. The short linker likely causes more local concentrations of stress and lower potential to distribute stress loads, thereby leading to catastrophic rupture of the structures. Our interpretation is that a longer linker allows more optimal crosslinking by being better able to span interfibril distances. The dCBM-48 bound to the cellulose to a lesser extent than the two constructs with shorter linkers. Although the 48-mer gave a stiffer material with a higher yield point than the other proteins, the slope after the yield point is lower than for the 24-mer, which may be due to the lower protein content or that fact that the longer linker was less capable of counteracting the viscous flow during plastic deformation. As a consequence, fewer local strain concentrations occur and the ultimate strength remained high.
Notably, the interactions between the HFB motifs were operational in the films, as seen by cleaving the peptide bond between HFB and dCBM. The decrease in the Young’s modulus when removing the HFB part was 3.04, 3.31, and 1.93 GPa for the 12, 24, and 48-mer constructs, respectively. The decrease in yield strength was 21.5, 19, and 20 MPa, while the decrease in slope after the yield point was 0.59, 0.86, and 1.11 GPa. It is well documented that HFB forms multimeric complexes of quite large size.[21] The drive towards multimers increases with concentration, that is, during the drying process, we can expect stronger complex formation compared to the wet gel state.[19] In the dry state, we can expect that hydrophobic interactions between the proteins would allow some measure of fluidity in the interactions. However, hydrogen bonding also contributes significantly to the interactions, as can be seen from structural analysis.[18]
We have shown herein that engineering of the modular architecture of a protein leads to functional effects in both the aqueous dispersed state and dried films of NFC composites. We interpret the results through the systematic variation of molecular structures. In the hydrated gel state, a shorter linker between dCBMs was advantageous for increasing stiffness in the gels, while the HFB modules interfered with gel-forming interactions. In the dry state, the effects were the opposite. The efficient crosslinking of short linkers led to more inefficient packing interactions between fibrils, thereby resulting in lower stiffness and strength. Longer linkers generated crosslinking while still allowing better packing of the fibrils during drying and under tension. The dry-state effect of the protein can be attributed to the fibril crosslinking formed in the wet gel state. Crosslinking by hydrophobin resulted in stiffness and resistance to plastic deformation in the dry state, while in the gel state, the presence of these domains led to a loss of interactions. These arguments are based on the relatively small differences between protein variants that lead to significant differences in function. A major challenge for interpretation is posed by the complex transition from wet to dry, since water significantly affects biomolecular interactions, and this transition is still far from being understood. In addition, differences in the processing history can lead to variations in film properties. Nonetheless, based on this work, we can expect to find that the mechanical properties in natural materials can be tuned through the coupling of protein modules and an interlinking architecture. This work further shows a functional basis for the modularity found in natural materials, and suggests a biological approach for molecular-level engineering of the soft matrix in nanocomposites.
Acknowledgments
We thank Riitta Suihkonen for assistance and Arja Paananen for the help with rheology. The work was supported by the Academy of Finland (Centres of Excellence Program (2014–2019), projects 264493 and 259034, Emil Aaltonen foundation, FinCEAL, and Bioregs.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Ashby MF, Gibson LJ, Wegst U, Olive R. Proc. R. Soc. London Ser. A. 1995;450:123–140. [Google Scholar]
- 2.Miserez A, Schneberk T, Sun C, Zok FW, Waite JH. Science. 2008;319:1816–1819. doi: 10.1126/science.1154117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aizenberg J, Weaver JC, Thanawala M, Sundar V, Morse DE, Fratzl P. Science. 2005;309:275–278. doi: 10.1126/science.1112255. [DOI] [PubMed] [Google Scholar]
- 4.Wegst UGK, Bai H, Saiz E, Tomsia AP, Ritchie RO. Nat. Mater. 2015;14:23–36. doi: 10.1038/nmat4089. [DOI] [PubMed] [Google Scholar]
- 5.Espinosa HD, Juster AL, Latourte FJ, Loh OY, Gregoire D, Zavattieri PD. Nat. Commun. 2011;2:173–179. doi: 10.1038/ncomms1172. [DOI] [PubMed] [Google Scholar]
- 6.Fratzl P, Burgert I, Gupta HS. Phys. Chem. Chem. Phys. 2004;6:5575–5579. [Google Scholar]
- 7.Fantner GE, Oroudjev E, Schitter G, Golde LS, Thurner P, Finch MM, Turner P, Gutsmann T, Morse D, Hansma H, et al. Biophys. J. 2006;90:1411–1418. doi: 10.1529/biophysj.105.069344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki M, Iwashima A, Kimura M, Kogure T, Nagasawa H. Mar. Biotechnol. 2012;15:145–158. doi: 10.1007/s10126-012-9471-2. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki M, Saruwatari K, Kogure T, Yamamoto Y, Nishimura T, Kato T, Nagasawa H. Science. 2009;325:1388–1390. doi: 10.1126/science.1173793. [DOI] [PubMed] [Google Scholar]
- 10.Tan Y, Hoon S, Guerette PA, Wei W, Ghadban A, Hao C, Miserez A, Waite JH. Nat. Chem. Biol. 2015;11:488–495. doi: 10.1038/nchembio.1833. [DOI] [PubMed] [Google Scholar]
- 11.Prakobna K, Terenzi C, Zhou Q, Furó I, Berglund LA. Carbohydr. Polym. 2015;125:92–102. doi: 10.1016/j.carbpol.2015.02.059. [DOI] [PubMed] [Google Scholar]
- 12.Benítez AJ, Torres-Rendon J, Poutanen M, Walther A. Biomacromolecules. 2013;14:4497–4506. doi: 10.1021/bm401451m. [DOI] [PubMed] [Google Scholar]
- 13.McKee JR, Huokuna J, Martikainen L, Karesoja L, Nykänen A, Kontturi E, Tenhu H, Ruokolainen J, Ikkala O. Angew. Chem. Int. Ed. 2014;53:5049–5053. doi: 10.1002/anie.201401072. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2014;126:5149–5153. [Google Scholar]
- 14.Malho JM, Ouellet-Plamondon C, Rüggeberg M, Laaksonen P, Ikkala O, Burgert I, Linder MB. Biomacromolecules. 2015;16:311–318. doi: 10.1021/bm501514w. [DOI] [PubMed] [Google Scholar]
- 15.Eichhorn SJ, Dufresne A, Aranguren M, Marcovich NE, Capadona JR, Rowan SJ, Weder C, Thielemans W, Roman M, Renneckar S, et al. J. Mater. Sci. 2010;45:1–33. [Google Scholar]
- 16.Sehaqui H, Zhou Q, Ikkala O, Berglund L. Biomacromolecules. 2011;12:3638–3644. doi: 10.1021/bm2008907. [DOI] [PubMed] [Google Scholar]
- 17.Vrnai A, Mäkelä MR, Djajadi DT, Rahikainen J, Hatakka A, Viikari L. Adv. Appl. Microbiol. 2014;88:103–165. doi: 10.1016/B978-0-12-800260-5.00004-8. [DOI] [PubMed] [Google Scholar]
- 18.Hakanpää J, Szilvay GR, Kaljunen H, Maksimainen M, Linder M, Rouvinen J. Protein Sci. 2006;15:2129–2140. doi: 10.1110/ps.062326706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szilvay GR, Nakari-Setälä T, Linder MB. Biochemistry. 2006;45:8590–8598. doi: 10.1021/bi060620y. [DOI] [PubMed] [Google Scholar]
- 20.Johnson QR, Lindsay RJ, Raval SR, Dobbs JS, Nellas RB, Shen T. J. Phys. Chem. B. 2014:2050–2055. doi: 10.1021/jp410788r. [DOI] [PubMed] [Google Scholar]
- 21.Lienemann M, Gandier JA, Joensuu JJ, Iwanaga A, Takatsuji Y, Haruyama T, Master E, Tenkanen M, Linder MB. Appl. Environ. Microbiol. 2013;79:5533–5538. doi: 10.1128/AEM.01493-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information