

Abstract
N-Phosphine oxide substituted imidazolylidenes (PoxIms) have been synthesized and fully characterized. These species can undergo significant changes to the spatial environment surrounding their carbene center through rotation of the phosphine oxide moiety. Either classical Lewis adducts (CLAs) or frustrated Lewis pairs (FLPs) are thus formed with B(C6F5)3 depending on the orientation of the phosphine oxide group. A strategy to reactivate FLPs from CLAs by exploiting molecular motions that are responsive to external stimuli has therefore been developed. The reactivation conditions were successfully controlled by tuning the strain in the PoxIm–B(C6F5)3 complexes so that reactivation only occurred above ambient temperature.
Keywords: frustrated Lewis pairs, hydrogenation, N-heterocyclic carbenes, phosphine oxides
Given the growing importance of economic and sustainable chemical processes, chemists have been continuously devoting their efforts to the development of homogeneous systems for molecular transformations with earth-abundant elements.[1] As such, much attention has recently been paid to the utilization of frustrated Lewis pairs (FLPs) owing to the promising advantages of employing harmless and abundant main-group elements.[2] FLPs are recognized as weakly bound noncovalent complexes comprising an electron acceptor (Lewis acid, LA) and an electron donor (Lewis base, LB), in which the formation of classical Lewis adducts (CLAs) is encumbered by steric repulsion between LA and LB.[2, 3] Whereas the typical chemical features of both the LA and the LB are usually quenched through the formation of CLAs, FLPs exhibit a much higher reactivity and can activate the enthalpically strong H–H bond. Since the landmark report from Stephan et al.,[4] the chemistry of FLPs has been rapidly developed with a variety of LBs, such as phosphines,[2e] carbenes,[2f] and nitrogen bases.[2g] Furthermore, several intermolecular systems showing thermally induced frustration have been reported.[5–7] Thermally induced FLPs can be considered as “reactivated” frustrated pairs derived from quenched CLA precursors, but such processes were previously generally induced at ambient temperature.[6, 7a]–[e] To date, few reports have focused on the development of a strategy to control the reactivation conditions of FLPs from CLAs by using external stimuli. As CLAs are often isolable and shelf-stable, well-controlled thermally induced systems would enable the generation of active FLPs whenever necessary. Therefore, the development of a method to restore the FLP reactivity from a quenched CLA under accurately controlled reaction conditions rather than under ambient conditions should expand both the utility and versatility of Lewis pair systems.[7f]–[h]
Herein, we describe a strategy to control the reactivation of an active FLP species from shelf-stable CLAs by making use of external stimuli-responsive molecular motions. Our strategy for designing a novel FLP system focused on the utilization of N-heterocyclic carbenes (NHCs)[8] and B(C6F5)3,[9] both of which have played important roles in modern organic synthesis. Pioneering reports by Stephan et al.[10] and Tamm et al.[11] disclosed that NHCs bearing N-tBu groups reacted with B(C6F5)3 to give FLPs; however, the reported NHC-based FLPs easily lost their activity through a rearrangement to abnormal carbene–borane adducts or decomposition, thereby significantly limiting their utility.[2f] An intriguing progress has recently been reported by Tamm and co-workers, who found that the treatment of 1,3-di-tert-butylimidazolin-2-ylidene (ItBu) with tris[3,5-bis(trifluoromethyl)phenyl]borane, B(XyF6)3, resulted in complexation to give [ItBu–B(XyF6)3], which reacted with 0.5 equivalents of H2 at room temperature to give [ItBu–H][(XyF6)3B–H–B(XyF6)3] along with the loss of one ItBu.[7a] Herein, we designed a novel NHC keeping the following considerations in mind (Figure 1): 1) The substituent on one of the nitrogen atoms should include both large (RL) and small (RS) units on the connecting atom (RC); 2) drastic changes in the spatial environment surrounding the carbene center should be induced by rotation around the N–RC bond to permit for the formation of both CLA and FLP species; and 3) the interconversion between the CLA and the FLP should be brought about by external stimuli.
Figure 1.
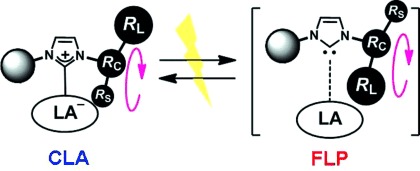
Design of a CLA that can be reactivated into an FLP.
These designs led us to prepare the carbenes PoxIm (Figure 2 a). The synthesis of the carbene precursors, PoxIm⋅HOTf (1 a–1 c), followed a pathway that was previously developed by Kostyuk et al.[12] and Hofmann et al.[13] to prepare N-phosphanyl-substituted imidazolium salts. Without isolation of these air-sensitive imidazolium species, oxidation of the N-phosphanyl group with aqueous H2O2 gave 1 a–1 c, which were isolated as bench-stable solids.[14] The structures of these products were confirmed by NMR spectroscopy and high-resolution mass spectrometry, and 1 a was also analyzed by X-ray crystallography (Supporting Information, Figure S1). The molecular structure of 1 a shows that the phosphine oxide group is almost in the same plane as the imidazolium ring, and that the C–Ha and P=O bonds are oriented in the same direction (∡C-N-P-O=5.4(5)°). Treatment of 1 a–1 c with KOtBu in THF resulted in the deprotonation of Ha and yielded PoxIm derivatives 2 a–2 c in excellent yields. All of these carbenes can be isolated as colorless crystals after recrystallization from THF/hexane solution at −30 °C. The molecular structure of 2 a is shown in Figure 2 b. The P=O bond and the lone pair of the carbene unit are oriented in an anti fashion (∡C-N2-P-O=175.9(1)°), presumably to avoid the repulsion between the lone pairs of the phosphine oxide and the carbene. The P–N2 bond length is 1.719(1) Å, which is slightly shorter than that found in 3-(di-tert-butylphosphino)-1-(2,6-diisopropylphenyl)imidazole-2-ylidene (NHCPDipp, 1.7485(16) Å).[12, 13] In the 13C NMR spectra of 2 a–2 c, the resonances of the carbene carbon were observed at approximately 220 ppm with 2JC,P values of 25.0 Hz, which are almost the same as those frequently encountered with unsaturated NHCs.[8]
Figure 2.
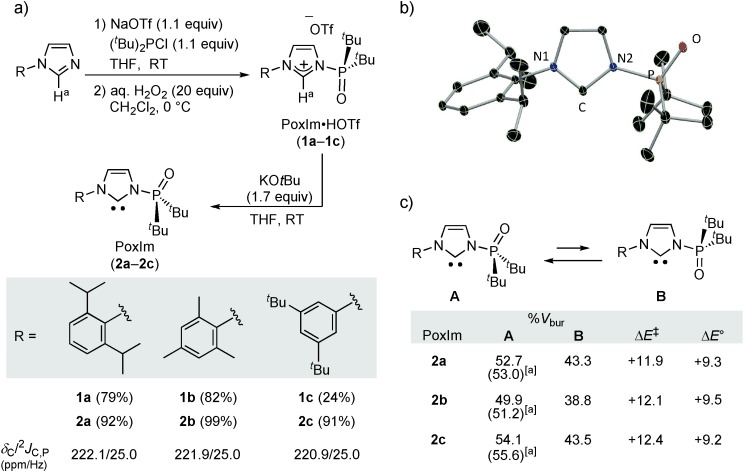
a) Synthesis of PoxIm⋅HOTf salts (1) and PoxIm carbenes (2). Yields of isolated products are given. The δC/2JC,P (ppm/Hz) values given are those of 2 measured in C6D6. b) Molecular structure of 2 a; ellipsoids set at 30 % probability. Hydrogen atoms are omitted for clarity. c) Conformation exchange of PoxIm between conformers A and B. DFT calculations were conducted at the B3LYP/6-31G++(d,p) level of theory (gas-phase, 25 °C). Calculated ∡C-N-P-O values [°] for conformer A: 2 a: 178.5, 2 b: 180.0, 2 c: 178.6; for conformer B: 2 a: 19.4, 2 b: 23.0, 2 c, 22.2. ΔE° (kcal mol−1) is the relative free energy of B with respect to A. ΔE≠ (kcal mol−1) is the calculated activation energy barrier from A to B. [a] The %Vbur values were determined from crystallographic parameters.
The impact of the rotation of the phosphine oxide around the N–P bond on the spatial environment surrounding the carbene center was evaluated based on the percent buried volume (%Vbur),[14, 15] which was calculated using the structural parameters of 2 a–2 c obtained from DFT and X-ray analyses (Figure 2 c). These compounds exhibit a relatively large degree of steric bulkiness in conformer A. Variations in the free energy were then scanned by changing the angle of ∡C-N-P-O starting from conformer A (∡C-N-P-O≈180°). The theoretical activation energy barrier of the rotation (ΔE≠) is approximately +12 kcal mol−1 at 25 °C, showing that the rotation would take place easily. Furthermore, the existence of conformer B (∡C-N-P-O≈20°), for which the %Vbur value is smaller than for conformer A, was predicted to be possible as a metastable state; however, the relative energy of conformer B is much higher than that of conformer A (ΔE°=+9.2–9.5 kcal mol−1). These results indicate that PoxIms exist predominantly as conformer A at 25 °C and can change their steric bulkiness by rotation of the phosphine oxide.
Treatment of 2 a with B(C6F5)3 in CD2Cl2 at −90 °C resulted in the quantitative formation of 3 a, in which the phosphine oxide coordinates to B(C6F5)3 (Figure 3). In the 31P NMR spectrum, the resonance of the phosphine oxide is observed at 79.2 ppm and has thus been shifted downfield through the complexation in comparison with that of 2 a (63.0 ppm).[16] Furthermore, the chemical shift of the carbene carbon is 219.5 ppm (2JC,P=30.2 Hz), indicating that the complexation does not occur at the carbene center as the chemical shift is almost the same as that of 2 a (220.5 ppm, 2JC,P=25.0 Hz). The results of the 1H, 11B, and 19F NMR analyses also support the formation of 3 a at −90 °C.[14] Elevating the temperature to 25 °C led to the conversion of 3 a into carbene borane complex (B-Pox) 4 a in 72 % yield. When the reaction of 2 a with B(C6F5)3 was conducted at room temperature in toluene, 4 a was isolated as a white solid in 95 % yield (Figure 3). Owing to the restricted rotation around the carbene–borane and N–P bonds at room temperature on the NMR timescale, each of the four iPr and two tBu groups is observed independently in the 1H and 13C NMR spectra, while fifteen resonances are observed in the 19F NMR spectrum. The resonance of the phosphine oxide is observed at 76.0 ppm in the 31P NMR spectrum. Furthermore, the structure of 4 a was confirmed by X-ray crystallography, illustrating the rotation of the phosphine oxide from an anti (∡C-N2-P-O=175.9(1)° in 2 a) to a syn arrangement (∡C-N2-P-O=7.2(2)° in 4 a). The %Vbur value of the PoxIm moiety in 4 a was calculated to be 30.3 based on experimental parameters, which is a significant decrease from the %Vbur value of free 2 a (53.0, see above).[15d],[e] The C–B bond length is 1.696(3) Å, and thus slightly longer than those measured for both [IPr–B(C6F5)3] (1.663(5) Å),[10] and [ItBu–B(XyF6)3] (1.675(4) Å).[7a] Furthermore, the boron atom in 4 a is located 0.43 Å out of the imidazolium plane, which is comparable with the arrangement in [ItBu–B(XyF6)3] (0.47 Å), but clearly differs from that of [IPr–B(C6F5)3] (0.23 Å). No significant interaction is observed between the O and B atoms in the solid structure (B⋅⋅⋅O=3.234(3) Å).
Figure 3.
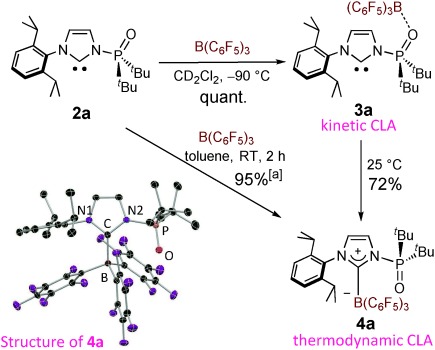
Two types of complexation between 2 a and B(C6F5)3 giving 3 a and 4 a, respectively. The yields were determined by NMR spectroscopy. [a] Yields of isolated products. The molecular structure of 4 a is also shown; ellipsoids are set at 30 % probability, and hydrogen atoms are omitted for clarity. Selected bond distances [Å] and angles [°]: C–B 1.696(3), N2–P 1.779(2), P–O 1.470(2); C-N2-P 131.7(2), C-N2-P-O 7.2(2).
Heterolytic cleavage of H2 was observed at room temperature when H2 gas was pressurized into a solution of 2 a and B(C6F5)3 in CH2Cl2 within three minutes of mixing to give 5 a in 45 % yield with the concomitant formation of 4 a in 38 % yield (Figure 4). This result indicates the in situ generation of an FLP species that yielded 5 a and 4 a through H2 activation and the formation of a carbene–borane bond, respectively. It is noteworthy that the FLP comprising 2 a and B(C6F5)3 can activate H2 at 25 °C once it has been generated in situ. Based on these results, 3 a should be formed in situ prior to the generation of the FLP (kinetic CLA→FLP), and the FLP would be converted into 4 a along with rotation of the phosphine oxide (FLP→thermodynamic CLA).[3]
Figure 4.
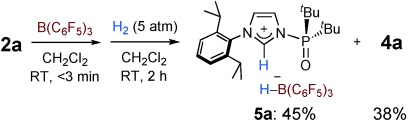
H2 activation at room temperature with a combination of 2 a and B(C6F5)3. Yields were determined by NMR spectroscopy.
In the solid state, no decomposition of 4 a was observed after 16 weeks at 20–30 °C even in the presence of air and moisture (25–35 % humidity). This stability of 4 a evokes the reactivity of a quenched Lewis acid–base adduct; however, under thermal conditions, the FLP reactivity was perfectly restored. For example, at 60 °C, 4 a activated H2 quantitatively for three hours in CH2Cl2, thus affording 5 a, which was isolated in 99 % yield (Figure 5). 5 a was also produced quantitatively at 40 °C (72 h); however, at 25 °C (72 h), no product was observed. These results demonstrate the occurrence of a thermally induced frustration process in this system at temperatures above room temperature (see also Scheme S1).[7]
Figure 5.
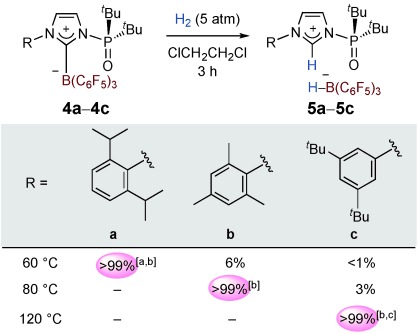
Thermally induced frustration systems with 4 a–4 c. The yields were determined by NMR spectroscopy. [a] CH2Cl2 was used as the solvent. [b] Yields of isolated products. [c] C6H5Br was used as the solvent.
The temperature required to induce the reactivation of FLPs from B-Pox was successfully controlled by tuning the strain in the B-Pox component (see below). As demonstrated in Figure 5, complex 4 b activated H2 to yield 5 b quantitatively in 1,2-dichloroethane at 80 °C, whereas 5 b was only formed in 6 % yield at 60 °C after three hours. In the case of 4 c, heterolytic H2 cleavage took place efficiently in C6H5Br at 120 °C to give 5 c quantitatively, whereas only 3 and 7 % of 5 c were formed in 1,2-dichloroethane after three hours at 80 and 100 °C, respectively. These results suggest that B-Pox complexes with a larger strain can operate at lower reaction temperatures to activate H2 than complexes with less strain; the degree of strain was evaluated based on the values of r (distance between B and the imidazolium plane), r′ (distance between P and the imidazolium plane), and the length of the C–B bond (Figure 6 a).
Figure 6.
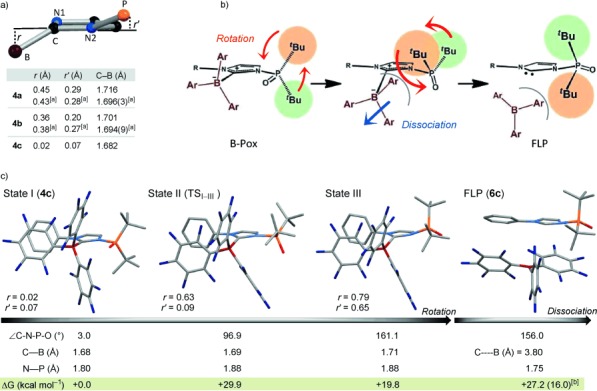
a) Comparison of the molecular structures of 4 a–4 c. The values are those of the DFT optimized structures (at the M06-2X/6-311G(d,p) level of theory for the gas-phase molecules). [a] Value determined from crystallographic parameters. b) Possible reactivation of FLPs from B-Pox. Ar=C6F5. c) Selected properties of the optimized structures of states I–III and 6 c and their relative Gibbs free energies at 120 °C. [b] Calculated activation energy barrier (kcal mol−1) from III to 6 c.
The FLP reactivation process in this work is illustrated in Figure 6 b. First, rotation of the phosphine oxide would occur followed by dissociation of B(C6F5)3 from the carbene center. In these processes, the B(C6F5)3 moiety is driven away from the carbene center by the steric demand of the tBu groups along with the rotation of the phosphine oxide.
To confirm the impact of the rotation of the phosphine oxide on the reactivation of the FLPs, variable-temperature (VT) NMR experiments were conducted with 4 c in C6D5Br from 30 to 90 °C, which revealed that the phosphine oxide, N-aryl, and B(C6F5)3 groups in 4 c can rotate more easily at higher temperatures (Figures S12–S14). The influence of the rotation of the phosphine oxide on the structure of 4 c was evaluated by DFT calculations at the M06-2X/6-311G(d,p) level of theory by scanning the ∡C-N-P-O angle starting from state I (∡C-N-P-O=3.0°), which is the optimized structure of 4 c (Figure 6 c). State II is a saddle point, and state III is metastable; both were found through the scanning process and have molecular structures drastically different from state I. The highest-energy state was found to be state II (ΔG=+29.9 kcal mol−1, ∡C-N-P-O=96.9°) where the boron atom is located 0.63 Å out of the imidazolium plane while the phosphorus atom remains almost in the plane. In state III, (ΔG=+19.8 kcal mol−1, ∡C-N-P-O=161.1°), the boron and phosphorus atoms are located at r=0.79 and r′=0.65 Å, respectively, indicating the increase in strain that is due to the steric repulsion between the tBu groups on the phosphine oxide and the C6F5 groups. This strain can be somewhat reduced through elongation of the N–P bond, and such a change of the N–P bond length is obvious for state III (4.4 % elongation from I). Significant structural changes were also observed upon further scanning from state III, which imply that maintaining the carbene–borane bond is no longer beneficial (Figure S18). According to these results, the phosphine oxide moiety in 4 c would rotate mainly within approximately ±100° at the temperature in which the FLP species is not fully reactivated, rationalizing the dynamic behavior observed by VT NMR spectroscopy, and further rotation would prompt decomplexation.
A potential-energy surface scan with respect to the distance between the carbene carbon and boron atoms was conducted from both state I and III of 4 c. A metastable energy state (6 c) was found at C⋅⋅⋅B=3.8 Å from state III, which can be regarded as an encounter complex consisting of 2 c and B(C6F5)3 (Figure 6 c and Figure S21), whereas such a state was not found from state I.[14] These results again manifest the importance of the rotation of the phosphine oxide for the reactivation of the FLP species. The theoretical activation energy barrier for the reactivation process to give 6 c (I→III→6 c) is ΔGsum≠=+35.8 kcal mol−1 at 120 °C. Under the experimental conditions, dissociation of B(C6F5)3 might partially proceed during the rotation of the phosphine oxide from state II to III.
For comparison, FLP reactivation was attempted for [NHCPDipp–B(C6F5)3] (7) in CD2Cl2 with H2 (5 atm), resulting in no reaction at 60 °C, whereas harsher reaction conditions (100 °C) led to its decomposition (Scheme S2). These results also demonstrate the importance of the phosphine oxide moiety for reactivating the FLP species from 4 a.
The hydrogenation of N-benzylidene benzenesulfonamide (8) was conducted with 4 c and H2 (5 atm) at 120 °C to demonstrate that the present system could be applied in organic synthesis (Figure 7 a).[1a, 2a],[b] After eight hours, N-phenylsulfonyl benzylamine (9) had been formed quantitatively along with 5 c in 95 % yield. At 150 °C, this reaction reached completion within three hours to furnish 9 and 5 c in >99 and 78 % yield, respectively. To elucidate the ability of 5 c to hydrogenate 8, the direct hydrogenation of 8 was also conducted with isolated 5 c, which was complete after 15 hours and gave 9 in 46 % yield with the concomitant formation of H2 in 15 % yield (Figure 7 b).[14] These results show that 5 c can mediate the hydrogenation of 8 to produce 9, but it is also plausible that FLP species that consist of 8 (and/or 9) and B(C6F5)3 and are formed by the in situ switching of the Lewis base after reactivation of the FLP comprising 2 c and B(C6F5)3 might be involved as active species. Thus far, the hydrogenation of unsaturated compounds with an NHC-based FLP system has been considered to be very challenging owing to the high proton affinity of NHCs.[2f] Our results show that NHC-based FLPs can indeed be employed for the hydrogenation of unsaturated compounds under appropriate reaction conditions.
Figure 7.
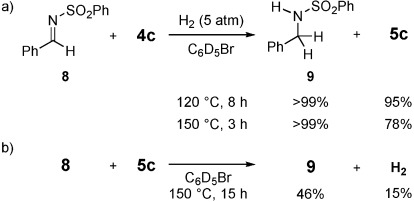
a) Reactions of 4 c with 8 in the presence of H2. b) Reaction of 5 c with 8. Yields were determined by 1H NMR spectroscopy.
In summary, molecular motions that are responsive to external stimuli can be used to reactivate frustrated Lewis pairs from shelf-stable carbene borane complexes. Key to success was the design of N-phosphine oxide substituted imidazolylidenes (PoxIms) that can undergo drastic changes to the spatial environment surrounding its carbene center. Depending on the orientation of the phosphine oxide group, either classical Lewis adducts or frustrated Lewis pairs are formed with B(C6F5)3. The reactivation conditions were successfully controlled to require higher than ambient temperature by tuning the strain in the carbene borane complexes. The impact of the rotation of the phosphine oxide on FLP reactivation was evaluated experimentally as well as theoretically, which confirmed this rotation to be critical. We believe that the present system should contribute to the realization of challenging molecular transformations by making use of the high reactivity of NHC-based FLPs.
Acknowledgments
This work was supported by Grants-in-Aid for Young Scientists (A) (25708018), Encouragement for Young Scientists (B) (15 K17824), and a Grant-in-Aid for Scientific Research on Innovative Areas (15H00943, 15H05803, and 23105546) from MEXT. Y.H. acknowledges support from the Frontier Research Base for Global Young Researchers, Osaka University on the program of MEXT.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Stephan DW. Chem. Commun. 2010;46:8526. doi: 10.1039/c0cc03313h. For examples of hydrogenation reactions, see. [DOI] [PubMed] [Google Scholar]
- 1b.Bullock RM. Science. 2013;342:1054. doi: 10.1126/science.1247240. [DOI] [PubMed] [Google Scholar]
- 1c.Kenward AL, Piers WE. Angew. Chem. Int. Ed. 2008;47:38. doi: 10.1002/anie.200702816. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2008;120:38. [Google Scholar]
- 1d.Paradies J. Angew. Chem. Int. Ed. 2014;53:3552. doi: 10.1002/anie.201309253. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2014;126:3624. [Google Scholar]
- 2a.Stephan DW. Acc. Chem. Res. 2015;48:306. doi: 10.1021/ar500375j. For selected recent reviews, see. [DOI] [PubMed] [Google Scholar]
- 2b.Stephan DW, Erker G. Angew. Chem. Int. Ed. 2015;54:6400. doi: 10.1002/anie.201409800. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2015;127:6498. [Google Scholar]
- 2c.Chase PA, Gille AL, Gilbert TM, Stephan DW. Dalton Trans. 2009:7179. doi: 10.1039/b908737k. [DOI] [PubMed] [Google Scholar]
- 2d.Erker G. C. R. Chim. 2011;14:831. [Google Scholar]
- 2e.Stephan DW, Erker G. Top. Curr. Chem. 2013;332:85. doi: 10.1007/128_2012_392. [DOI] [PubMed] [Google Scholar]
- 2f.Kolychev EL, Theuergarten E, Tamm M. Top. Curr. Chem. 2013;334:121. doi: 10.1007/128_2012_379. [DOI] [PubMed] [Google Scholar]
- 2g.Sumerin V, Chernichenko K, Schulz F, Leskelä M, Rieger B, Repo T. Top. Curr. Chem. 2013;332:111. doi: 10.1007/128_2012_391. [DOI] [PubMed] [Google Scholar]
- 3.Rokob TA, Pápai I. Top. Curr. Chem. 2013;332:157. doi: 10.1007/128_2012_399. [DOI] [PubMed] [Google Scholar]
- 4.Welch GC, San Juan RR, Masuda JD, Stephan DW. Science. 2006;314:1124. doi: 10.1126/science.1134230. [DOI] [PubMed] [Google Scholar]
- 5.Rokob TA, Hamza A, Stirling A, Pápai I. J. Am. Chem. Soc. 2009;131:2029. doi: 10.1021/ja809125r. [DOI] [PubMed] [Google Scholar]
- 6a.Wang X, Kehr G, Daniliuc CG, Erker G. J. Am. Chem. Soc. 2014;136:3293. doi: 10.1021/ja413060u. For recent examples of intramolecular weakly bound Lewis pairs based on phosphines or amines, see. [DOI] [PubMed] [Google Scholar]
- 6b.Holtrichter-Rößmann T, Rösener C, Hellmann J, Uhl W, Würthwein E-U, Fröhlich R, Wibbeling B. Organometallics. 2012;31:3272. [Google Scholar]
- 6c.Mömming CM, Kehr G, Wibbeling B, Fröhlich R, Erker G. Dalton Trans. 2010;39:7556. doi: 10.1039/c0dt00015a. [DOI] [PubMed] [Google Scholar]
- 6d.Spies P, Kehr G, Bergander K, Wibbeling B, Fröhlich R, Erker G. Dalton Trans. 2009:1534. doi: 10.1039/b815832k. [DOI] [PubMed] [Google Scholar]
- 6e.Spies P, Erker G, Kehr GG, Bergander K, Fröhlich R, Grimme S, Stephan DW. Chem. Commun. 2007:5072. doi: 10.1039/b710475h. for a review, see. [DOI] [PubMed] [Google Scholar]
- 6f.Kehr G, Schwendemann S, Erker G. Top. Curr. Chem. 2013;332:45. doi: 10.1007/128_2012_373. [DOI] [PubMed] [Google Scholar]
- 7a.Kolychev E, Bannenberg T, Freytag M, Daniliuc CG, Jones PG, Tamm M. Chem. Eur. J. 2012;18:16938. doi: 10.1002/chem.201202840. [DOI] [PubMed] [Google Scholar]
- 7b.Geier SJ, Stephan DW. J. Am. Chem. Soc. 2009;131:3476. doi: 10.1021/ja900572x. [DOI] [PubMed] [Google Scholar]
- 7c.Ullrich M, Lough AJ, Stephan DW. Organometallics. 2010;29:3647. [Google Scholar]
- 7d.Voss T, Mahdi T, Otten E, Fröhlich R, Kehr G, Stephan DW, Erker G. Organometallics. 2012;31:2367. [Google Scholar]
- 7e.Jiang C, Stephan DW. Dalton Trans. 2013;42:630. doi: 10.1039/c2dt30720k. [DOI] [PubMed] [Google Scholar]
- 7f.Jiang C, Blacque O, Fox T, Berke H. Organometallics. 2011;30:2117. [Google Scholar]
- 7g.Sumerin V, Schulz F, Nieger M, Leskelä M, Repo T, Rieger B. Angew. Chem. Int. Ed. 2008;47:6001. doi: 10.1002/anie.200800935. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2008;120:6090. for a recent report describing an intriguing concept for the protection of a reactive Lewis acidic moiety with a bulky Lewis base, see. [Google Scholar]
- 7h.Herrington TJ, Ward BJ, Doyle LR, McDermott J, White AJP, Hunt PA, Ashley AE. Chem. Commun. 2014;50:12753. doi: 10.1039/c4cc05905k. [DOI] [PubMed] [Google Scholar]
- 8a.Cazin CS. N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis. Heidelberg: Springer; 2010. For selected reviews, see. [Google Scholar]
- 8b.Glorius F. N-Heterocyclic Carbenes in Transition Metal Catalysis. Berlin: Springer; 2007. [Google Scholar]
- 8c.Nolan SP. N-Heterocyclic Carbenes in Synthesis. Weinheim: Wiley-VCH; 2006. [Google Scholar]
- 8d.Dröge T, Glorius F. Angew. Chem. Int. Ed. 2010;49:6940. doi: 10.1002/anie.201001865. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2010;122:7094. [Google Scholar]
- 8e.de Frémont P, Marion N, Nolan SP. Coord. Chem. Rev. 2009;253:862. [Google Scholar]
- 8f.Hahn FE, Jahnke MC. Angew. Chem. Int. Ed. 2008;47:3122. doi: 10.1002/anie.200703883. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2008;120:3166. see also. [Google Scholar]
- 8g.Arduengo AJ, III, Krafczyk R, Schmutzler R. Tetrahedron. 1999;55:14523. [Google Scholar]
- 9.Erker G. Dalton Trans. 2005:1883. doi: 10.1039/b503688g. [DOI] [PubMed] [Google Scholar]
- 10.Chase PA, Stephan DW. Angew. Chem. Int. Ed. 2008;47:7433. doi: 10.1002/anie.200802596. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2008;120:7543. [Google Scholar]
- 11.Holschumacher D, Bannenberg T, Hrib CG, Jones PG, Tamm M. Angew. Chem. Int. Ed. 2008;47:7428. doi: 10.1002/anie.200802705. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2008;120:7538. [Google Scholar]
- 12.Marchenko AP, Koidan HN, Pervak II, Huryeva AN, Zarudnitskii EV, Tolmachev AA, Kostyuk AN. Tetrahedron Lett. 2012;53:494. [Google Scholar]
- 13.Nägele P, Herrlich U, Rominger F, Hofmann P. Organometallics. 2013;32:181. [Google Scholar]
- 14. See the Supporting Information for details; %Vbur values were calculated using the SambVca program with the following parameters: sphere radius: 3.00 Å; distance for the metal–ligand bond: 2.00 Å; H atoms were omitted; Bondi radii scaled by 1.17.
- 15a.Poater A, Cosenza B, Correa A, Giudice S, Ragone F, Scarano V, Cavallo L. Eur. J. Inorg. Chem. 2009:1759. [Google Scholar]
- 15b.Dorta R, Stevens ED, Scott NM, Costabile C, Cavallo L, Hoff CD, Nolan SP. J. Am. Chem. Soc. 2005;127:2485. doi: 10.1021/ja0438821. [DOI] [PubMed] [Google Scholar]
- 15c.Balogh J, Slawin AMZ, Nolan SP. Organometallics. 2012;31:3259. [Google Scholar]
- 15d.Dierick S, Dewez DF, Markó IE. Organometallics. 2014;33:677. [Google Scholar]
- 15e.Collado A, Balogh J, Meiries S, Slawin AMZ, Falivene L, Cavallo L, Nolan SP. Organometallics. 2013;32:3249. [Google Scholar]
- 16.Beckett MA, Brassington DS, Light ME, Hursthouse MB. J. Chem. Soc. Dalton Trans. 2001:1768. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information