

Abstract
In this work, we assess the usefulness of static 15N NMR techniques for the determination of the 15N chemical shift anisotropy (CSA) tensor parameters and 15N-1H dipolar splittings in powder protein samples. By using five single labeled samples of the villin headpiece subdomain protein in a hydrated lyophilized powder state, we determine the backbone 15N CSA tensors at two temperatures, 22 and –35°C, in order to get a snapshot of the variability across the residues and as a function of temperature. All sites probed belonged to the hydrophobic core and most of them were part of α-helical regions. The values of the anisotropy (which include the effect of the dynamics) varied between 130 and 156 ppm at 22°C, while the values of the asymmetry were in the 0.32–0.082 range. The Leu-75 and Leu-61 backbone sites exhibited high mobility based on the values of their temperature-dependent anisotropy parameters. Under the assumption that most differences stem from dynamics, we obtained the values of the motional order parameters for the 15N backbone sites. While a simple one-dimensional line shape experiment was used for the determination of the 15N CSA parameters, a more advanced approach based on the “magic sandwich” SAMMY pulse sequence (Nevzorov & Opella, J. Magn. Reson. (2003) 164, 182-186) was employed for the determination of the 15N-1H dipolar patterns, which yielded estimates of the dipolar couplings. Accordingly, the motional order parameters for the dipolar interaction were obtained. It was found that the order parameters from the CSA and dipolar measurements are highly correlated, validating that the variability between the residues is governed by the differences in dynamics. The values of the parameters obtained in this work can serve as reference values for developing more advanced magic-angle spinning recoupling techniques for multiple labeled samples.
Keywords: static NMR, 15N CSA, SAMMY, protein dynamics
Graphical abstract
Introduction
15N chemical shift anisotropy (CSA) and 1H-15N dipolar interactions play an important role in studies of protein structure and dynamics.(1, 2) Protein dynamics at 15N sites have been extensively probed by a variety of NMR approaches on uniformly labeled protein samples in solution, solid and liquid crystalline states. (2-8) Factors contributing to 15N chemical shifts are less understood than those for 1H and 13C nuclei and can vary from the type of the amino acid to torsion angles, hydrogen bonding patterns and others.(9-13) In many solution NMR backbone dynamics studies, a single value of CSA is used for all residues and several works have looked into the validity of this approximation. For example, Loth et al.(14) and Yao et al.(3) found a considerable extent of residue-specific variations. Major variations apart from dynamical contributions appear to be caused by secondary structures.(3, 4, 6, 15)
The rapid development of solid-state NMR techniques with the incorporation of fast magic-angle spinning, sample deuteration and proton detection has led to the possibility of detailed site-resolved investigations of dynamics based on dipolar and CSA interactions in the solid state.(5, 16-18) The methodology for backbone dynamics studies in the solid state is still under development and several recoupling techniques have been used to re-introduce the anisotropic interaction of interests that are affected by dynamics, such as backbone CSA and dipolar tensors.(4-6, 16) The accuracy of the obtained tensors and motional parameters relies on the technical aspects of the recoupling sequences, which may be sensitive to RF field inhomogeneity and/or homonuclear decoupling efficiency. Important works include the investigations of re-assembled Thioredoxin by Yang et al.,(6) in which the dynamics were probed via 15N CSA line shapes, dipolar tensors and longitudinal relaxation times. Wylie et al.(4, 15) looked at 15N CSA anisotropy in the immunoglobin binding domain B1 of protein G, and the works by Schanda et al.(5) and Chevelkov et al.(16) emphasized the importance of deuteration to circumvent the need for high power homonuclear decoupling. Schanda et al.(5) obtained a very accurate set of motional order parameters using a deuterated sample of microcrystalline ubiquitin via rotational-echo double resonance.
While for most protein samples in the solid state the future lies in multiple isotopic labeling and advanced MAS techniques, not all samples may be amenable to deuteration and multiple labels. Studies of single labeled protein samples can be performed under static conditions, and, as such, can provide a reference point for the more advanced MAS methodology. It is also interesting to assess the inherent limits of studies of the interactions and dynamics of single labeled powdered protein samples. Static measurements are extensively used for studies of oriented membrane proteins, but few works have looked into unoriented powder samples.(19-22)
Our goal is to assess dipolar 15N-1H splittings and 15N CSA tensors for several sites in a protein hydrophobic core under static conditions using lyophilized hydrated powder samples. In this work, we will utilize a small model protein, which can be synthesized using solid-state peptide synthesis with the incorporation of single labels. The structure of the villin headpiece subdomain protein (HP36)(23-25) along with the hydrophobic sites probed in this work is shown in Figure 1.
Figure 1.
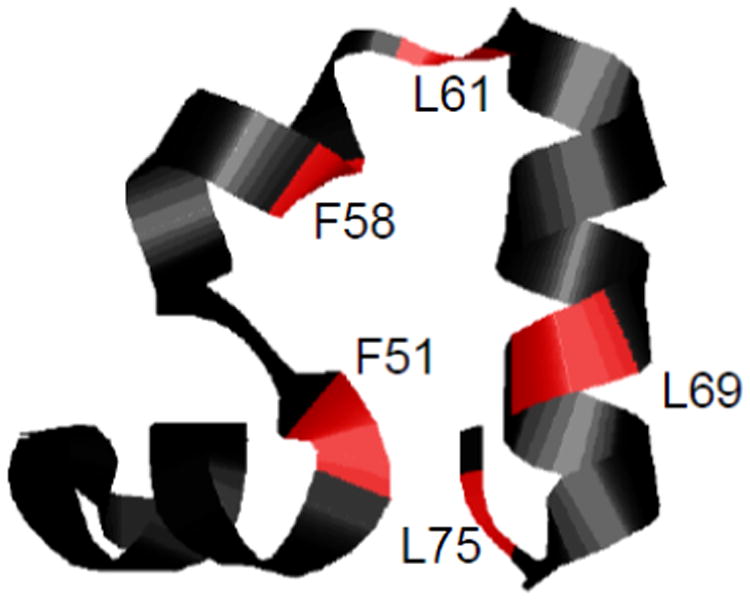
Ribbon diagram of the HP36 structure with the 15N backbone sites probed in this work shown in red.
Polarization inversion spin exchange at the magic-angle (PISEMA) pulse sequence(26) has evolved as a method of choice for static studies of membrane proteins, as it allows for the accurate determination of dipolar couplings and relative orientations of CSA and the dipolar tensors. This two-dimensional pulse sequence relies on the off-resonance Lee-Goldburg scheme(27) for homonuclear decoupling, with dipolar coupling evolving in the indirect dimension and 15N chemical shift tensor in the direct dimension. The dipolar splittings obtained via this technique are not accurate if the chemical shift of the protons is off-resonance.(28) Thus, for the powder samples SAMMY pulse sequences were suggested,(29) which circumvent this problem via the “magic sandwich” element.(30)
We use the SAMMY sequence to obtain the effective dipolar coupling constants and static 15N CSA line shapes to obtain the components of the 15N CSA tensor. Our results identify the limits of SAMMY techniques for powder protein samples and provide reference values of the 15N CSA and 15N-1H dipolar interactions. The comparison of the tensorial parameters across all residues as well as their temperature dependence provides further insight into the contributions of dynamics and enables an estimation of the order parameters for both CSA and dipolar interactions. Our data can be useful for both the refinement of more advanced magic-angle spinning-based techniques and the development of quantum-mechanical-based approaches for the predictions of the CSA and dipolar interaction parameters.
Experimental
Sample preparation
The protein samples were prepared using solid-state peptide synthesis (performed by Thermofisher Scientific Co, Rockford, IL) with the incorporation of isotopically labeled amino acids at selected sites. Powdered N-15 labeled fluorenylmethyloxycarbonyl (FMOC)-Phenylalanine and (FMOC)-Leucine amino acids were purchased from Cambridge Isotopes Laboratories (Andover, MA). The peptides were purified by reversed-phase HPLC and the identity and purity were confirmed by mass spectroscopy and reversed-phase HPLC. Lyophilized powders were dissolved in water and the pH was adjusted to about 6 using NaOH/HCl. The hydrated state was achieved by exposing lyophilized powder to water vapor in a sealed chamber until the water content reached about 35–40% by weight, which is a typical amount of water for protein powder samples.(31, 32) Hydrated in these fashion, the samples in amounts of 15–20 mg were packed in 5 mm NMR tubes (cut to 21 mm length) using Teflon tape to center the sample volume in the coil.
NMR measurements
All experiments were performed on a 400 MHz NMR spectrometer with a Bruker DRX console where the Larmor frequencies of 1H and 15N were 400.1 and 40.5 MHz, respectively, using a wideline low-E probe(33) with a 5-mm inner diameter coil built at the National High Magnetic Field Laboratory. Temperature calibration was performed by recording static lead nitrate line shapes (34) and using the freezing point of D2O, 3.8°C, as the fixed point for the calibration. The 15N chemical shifts were referenced using the 15N-glycine amino acid as the secondary reference with the value of the chemical shift taken as 33.4 ppm, This value originates from the unified scale with the ratio 15N/1H of 0.10132912, which is based on the TMS reference as well as referencing with respect to liquid NH3 of 0 ppm, as reported by Bertani et al.(35)
I. 15N CSA
Spectra were recorded using cross-polarization (4 us 90° pulse, 0.8 ms contact time) followed by a Hahn echo with a 20 us echo delay. The typical acquisition time was 10 ms and the recycle delay was in the 0.8 to 1 s range. The number of scans varied between 10∙k and 60∙k. A SPINAL64 decoupling scheme with 62.5 kHz power was used. The SPINAL setup was implemented with the super-cycle of QQ̄Q̄QQ̄QQQ̄ with Q=18010180350180151803451802018034018015180345, where the subscripts refer to the phase angles of the 180° pulses. (36) The proton decoupling field of 62.5 kHz used in the SPINAL sequence was calibrated experimentally by the measurement of 1H 180° pulse length via indirect observation of the 15N signals through cross-polarization. This decoupling field is expected to be sufficient for covering the 1H chemical shift range on a static peptide sample at 9.4 T. (37) The cooling of the probe for measurements at –35°C was achieved using liquid nitrogen. Spectra were processed with 200 Hz exponential line broadening.
II. SAMMY
SAMMY experiments were performed according to the method of Nevzorov et al. (29) In order to minimize the rf-offset effects, magnetization was kept in either the x-y plane or along the z axis by the use of “magic sandwiches”,(30) which suppress homonuclear interactions. The rf fields for both 1H and 15N channels during the cross-polarization and SAMMY block were 62.5 kHz. The 1H 90° pulse length was 4 μs and the cross-polarization contact time was 1 ms. Proton decoupling during the evolution in the direct dimension was 62.5 kHz. A recycle delay of 0.7 s was used. The number of scans varied between 10∙k and 12∙k. The number of points in the direct (CSA) dimension was 256 and the number of increments in the dipolar dimension was in the 26–38 range. This led to experimental times between 3.5 and 4.5 days per sample. The scaling factor for the dipolar dimension was experimentally determined by performing the dipolar encoded HETCOR on the NAV crystal sample.(38) An exponential line broadening of 100 Hz in the direct dimension was used for processing.
Fitting of 15N CSA line shapes
The fitting of one-dimensional 15N CSA line shapes in order to obtain the tensorial parameters was performed using mean-squared error minimization, employing internal Matlab routines based on the Levenberg-Marquardt algorithm. In the simplest model, the fitting parameters consisted of three components of the CSA tensors. In this scenario, line broadening was fixed at 200 Hz, which was the value used for processing the experimental spectra. An additional fitting parameter describing the apparent T2 contribution was employed in the second scenario, which resulted in variable line broadening in the fitted spectra. Note that in general transverse relaxation can be anisotropic, but in this case an isotropic approximation was employed phenomenologically. The errors in the fitting parameters were determined using Monte-Carlo simulations. Random Gaussian noise with variance obtained from the signal-free parts of the experimental spectra was added to the B-spline smoothed experimental signal and refitted for 200 repetitions. The errors reported represent twice the standard deviation.
Results and Discussion
We emphasize that each of the protein samples had only one backbone site labeled with 15N, thus all of the results are site-specific. All of the sites belong to the hydrophobic core (Figure 1), minimizing the effects of inter-molecular interactions.
15N CSA tensors
Static 15N CSA tensors were measured at two temperatures, 22°C and –35°C. The line shapes (Figure 2) were fitted to obtain the tensorial parameters defined via the Haeberlen convention:(39) the isotropical chemical shift δiso =(δxx + δyy + δzz)/3 with the principle components ordering defined by |δzz−δiso|≥|δxx−δiso|≥|δyy−δiso|, anisotropy Δδ = δzz−(δxx+δyy)/2 and asymmetry η =(δxx − δyy)/(δzz − δiso). The resulting values are listed in Table 1, where the reduced anisotropy parameter, δ =δzz−δiso, is also included since these have been reported in many protein studies.
Figure 2.
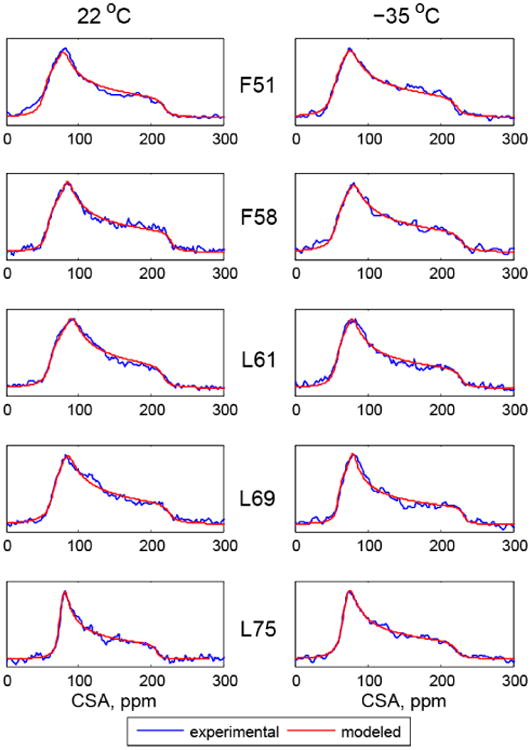
Normalized experimental (blue) and simulated (red) 15N CSA line shapes. As explained in the text, compared with experimental processing an extra line broadening of 300 Hz was used for all residues other than L75 for which 100 Hz was used.
Table I.
15N CSA tensor parameters at 22°C. As explained in the text, compared with experimental processing an extra line broadening of 300 Hz was used for all residues other than L75 for which 100 Hz was used.
| Residues | δiso, ppm Solid | Δδ, ppm | η | δ, ppm | δiso, ppm solution* |
|---|---|---|---|---|---|
| F51 | 115.3 ± 0.6 | 151.8 ± 1.4 | 0.264 ± 0.012 | 101.2 ± 0.9 | 114.3 |
| F58 | 122.5 ± 0.4 | 156.2 ± 1.0 | 0.254 ± 0.010 | 104.1 ± 0.7 | 122.4 |
| L61 | 121.6 ± 0.5 | 141.1 ± 1.2 | 0.322 ± 0.010 | 94.1 ± 0.8 | 122.5 |
| L69 | 124.0 ± 0.4 | 150.7 ± 1.1 | 0.225 ± 0.010 | 100.5 ± 0.7 | 123.8 |
| L75 | 119.5 ± 0.6 | 130.5 ± 1.9 | 0.082 ± 0.014 | 87.1 ± 0.9 | 121.2 |
A correction of –0.7 ppm was applied to the chemical shifts reported by McKnight et al.(23) so that both solid and solution shifts are effectively referenced relative to TMS.
Wylie et al.(4, 40) used a model in which ROCSA (recoupling of chemical shift anisotropy(41)) CSA line shapes were fitted using an additional fitting parameter describing an effective T2 contribution. We examined the effect of this alternative model in which variable line broadening (lb) was used as an additional fitting parameter. For all residues except L75, this procedure yields on average an additional 300 Hz broadening to the 200 Hz broadening used in the processing of experimental line shapes. The data for L75 are consistent with the extra 100 Hz broadening. The choice of line broadening parameters has a significant influence only on the resulting values of the asymmetries, which are systematically lower for a larger line broadening choice. (Supporting Information S1)
Several insights emerge from the analysis of the static 15N line shapes. First, the isotropic chemical shifts at 22°C are within about 0.5 ppm of those measured for the same protein in solution at pH 5.0 and 30°C, with the exception of L75 for which the difference is 2 ppm(23) (Table 1). The data confirmed our previous suggestions(42, 43) that protein structures are similar in solution and in hydrated powder states.
The L75 residue has the smallest anisotropy (Δδ=130.5±1.9 ppm) at 22°C, followed by L61 (Δδ=141.1±1.2), while the rest of the residues are in the 150-–156 ppm range. These values fall in the range reported for ROSCA and slow magic-angle measurements performed on the B1 domain of protein G.(4, 15) Since the anisotropy parameter is expected to be reduced for mobile residues, the value of 130.5±1.9 ppm for L75 implies that this site is the most mobile out of the five residues probed here. The backbone 15N site of L75 is located at the C-terminus end and this has been shown to be mobile in solution NMR dynamics studies using laboratory frame relaxation measurements.(44)
As the temperature is lowered to –35°C, the isotropic chemical shifts remain comparable to those at room temperature, suggesting that no major changes in structure take place (Figure 3). Thus, the changes in anisotropy provide information about the dynamics. The L75 and L61 residues show the largest variations of the order of 18–20 ppm, while the F51 and F58 sites experience almost no change in anisotropy. We note that in order to obtain the parameters of the rigid tensors, the temperatures would have to be lowered beyond –35°C, probably down to at least –70°C.(45, 46) Therefore, our measurements of the two temperatures aim to get a snapshot of the temperature dependence of the tensors rather than compare the truly rigid and mobile states. The average value of anisotropy for the rigid tensors was found to be 173 ppm for the α-helical residues in the B3 domain of protein G.(3) We use this value to estimate the values of the order parameters , which are shown for the two temperatures in Figure 4. All residues with the exception of L61 belong to α-helices. L61 is at the very beginning of the turn and thus its rigid value of Δδ could be somewhat smaller than 173 ppm, which would lead to the underestimation of the order parameter value. The changes in Δδ upon lowering the temperature translate into an increase in of the order of 0.1 for L75 and L61 and in the 0.02–0.06 range for the rest of the residues. F51 and F58 are the most solvent-protected residues and their values of the order parameters are least dependent on temperature, which may suggest the role of the hydration layer in these fluctuations.(18) It is worth noting that the asymmetry η changes slightly for all residues but L61. The significant variation in η (changing from 0.32 at 22°C to 0.20 at – 35°C, Figure 3) suggests more complex motions at this site with a possibility of multiple motional axes.
Figure 3.
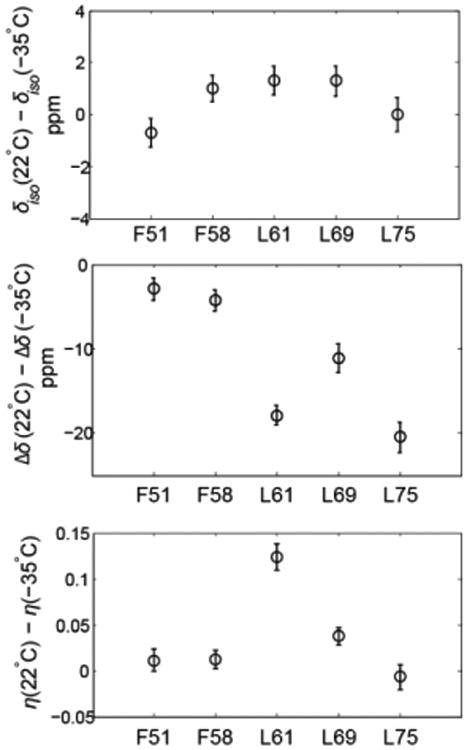
Changes in the values of δiso, Δδ and η between 22 and –35°C.
Figure 4.
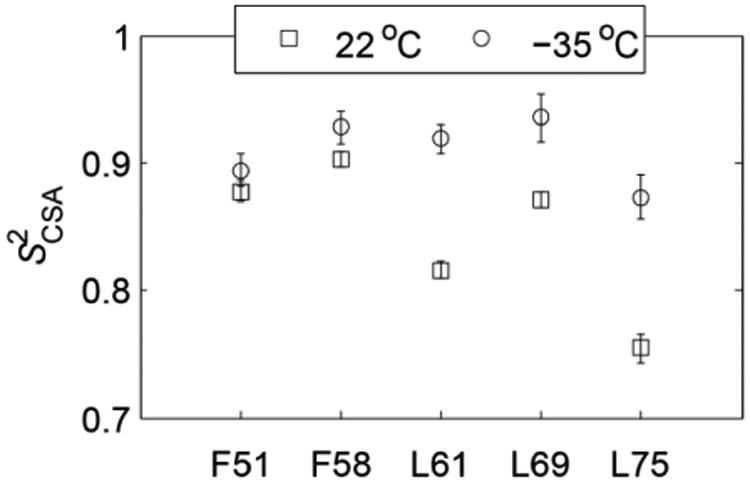
Order parameters at 22°C (squares) and –35°C (circles), calculated with Δδrigid =173 ppm.
It is important to note that the ROCSA technique requires careful optimization(4, 40) in order to obtain accurate values of η. Our simple measurements free of scaling factors are important for providing the reference values of the tensorial parameters in protein solid samples. It has been noted(4) that single labeled samples can be more prone to variations in RF inhomogeneity and protein preparations. However, for our lyophilized samples packed into 5 mm NMR tubes using the same mass of the samples and centered in the coil, these variations are minimal.
We have previously investigated the dynamics of the HP36 hydrophobic core using 2H static NMR line shapes and relaxation measurements of methyl-bearing side chains.(42, 47-50) The core has exhibited flexibility on multiple time scales and showed the presence of glassy behavior characterized by non-Arrhenius relaxation with non-exponential magnetization build-up curves at low temperatures. The 2H line shapes measurements indicated extensive μs-ms time scale motions at room temperatures which were quenched by either lowering the temperature or by dehydrating the protein. The 15N CSA coupling constant is on the order of 6.5 kHz at 9.4 T, while the effective quadrupolar coupling constant for a methyl group averaged over fast rotations is about 53 kHz.(50) Unlike the 2H NMR line shapes, the 15N line shapes do not appear to be strongly modulated by motions on the order of the coupling constant to a first approximation, which suggests that the main source of motions is due to fast dynamics, i.e. faster than 150 μs. Thus, in the presence of concerted motions of the core, the glassiness observed in the previous 2H NMR relaxation measurements would be expected to manifest via non-exponential 15N relaxation. Based on the signal-to-noise ratio in our 15N line shapes, static 15N relaxation measurements would be feasible for these powder samples, especially at a higher field strength. We also note that 2H relaxation indicated the presence of the conformational exchange at temperatures above 250 K. Thus, it is possible that in order to observe the glassiness at the backbone sites it would be necessary to perform the measurements at temperatures much lower than 250 K. Lewandowski et al.(18) have recently determined that 15N backbone sites undergo dynamical transitions at around 220 K. Thus, static 15N relaxation measurements can also be useful in reporting on these dynamical transitions at backbone sites. Most importantly, we emphasize that 15N measurements on backbone sites provide complimentary view of the core dynamics compared to the 2H NMR measurements performed on side chains. Combination of these two approaches can provide a global view of the core dynamics in static powder samples.
Dipolar 15N-1H couplings
Two-dimensional SAMMY line shapes are shown in Figure 6. In principle, the SAMMY experiment is capable of yielding both the magnitudes of the heteronuclear couplings and the angles defining the relative orientations of the 15N CSA and 15N-1H dipolar tensors. However, the extraction of the angles requires high-sensitivity data that can delineate the fine features distinguishing one orientation from another. The quality of the data in our low-sensitivity samples is not sufficient for fitting the values of the angles. This issue is explored in more detail in Supporting Information S2 using F58 as an example.
Figure 6.
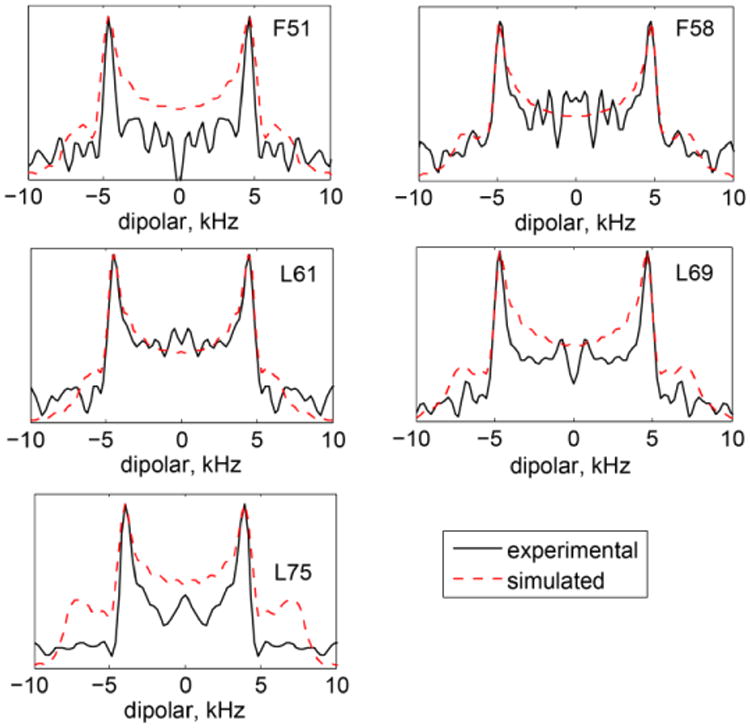
Experimental (black solid lines) and simulated (red dashed lines) one-dimensional slices taken at the maximum intensities of the two-dimensional SAMMY data. Simulated data correspond to the best-fit values of as described in the text.
We, therefore, confine our analysis to the magnitude of the effective dipolar splittings that define the widths of the SAMMY spectra. Assuming that the fluctuations of the N-H bond vectors are axially symmetric and in the fast regime, the magnitude of the dipolar splittings can be represented by the product of the rigid splitting in the absence of motions and the square of the order parameters reflecting the amplitude of motions. The most commonly used rigid value of dipolar coupling is reported as 11.7 kHz.(51) However, a value as low as 10.5 kHz has also been reported in the literature.(6, 52) To estimate the values of , we perform simulations of the SAMMY patterns using the CSA parameters obtained from the line shape data. We then extract one-dimensional slices across the dipolar dimension at the maximum overall spectral intensity and compare the experimental and simulated splittings for a range of values. The best-fit one-dimensional line shapes obtained with this procedure are shown in Figure 6. A typical value for the relative orientation of the backbone 15N CSA and 15N-1H dipolar tensors for the α-helical residues is 17° (53) with the range of 16-22° reported by the recent study of Wylie et al.(40) However, in the presence of motions around a common axis, the effective relative orientation can be averaged to zero. We also checked that the resulting fitted values of are not affected by the choice of the angle between 0 and 17°.
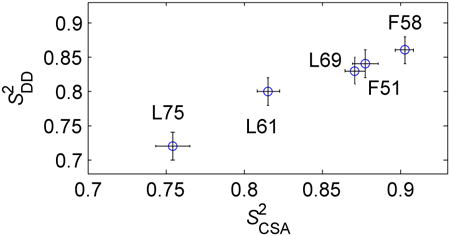
The values of and are highly correlated (Figure 7), but the values are systematically smaller by about 4%. The fluctuations in both these interactions are caused primarily by motions of the N-H bond vector. However, the 1H-15N dipolar interaction is more susceptible to vibrational motions of the light hydrogen atom compared to the CSA interaction.(54) Thus, the 4% reduction in the values of the dipolar order parameters may reflect the presence of these vibrational modes. An alternative explanation for the slight discrepancy between the values of and is the uncertainty in the rigid value of the dipolar coupling constant. In order to match and , it is sufficient to assume that the rigid value of the dipolar coupling constant is 11.2 kHz. Additionally, the discrepancies in the overall pattern between the experimental and simulated dipolar slices in Figure 6 for some of the residues might point out that the effect of the motions is more complex than a simple scaling introduced by the order parameter.
Figure 7.
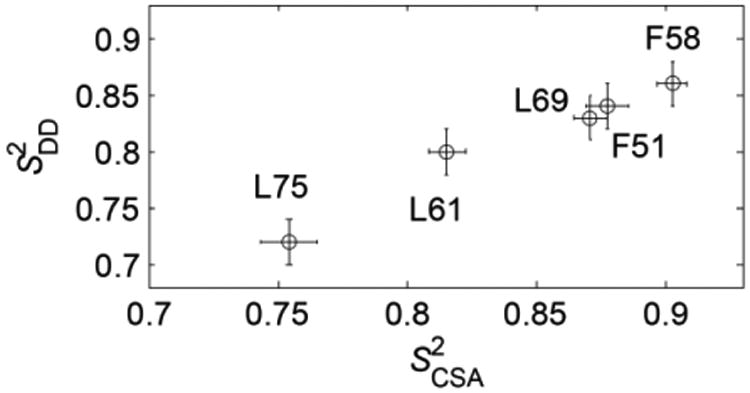
A plot of versus at 22°C. The Pearson correlation coefficient is r = 0.985.
Conclusions
We probed the HP36 backbone 15N CSA and 15N-15H dipolar tensors of several hydrophobic core leucine and phenylalanine residues by static NMR and assessed the limits of static techniques for these samples. The precision of the 15N CSA tensor parameters is sufficient to detect clear variations between the residues at room temperature and assess the dependence of the parameters on temperature. Out of the five residues probed, L75 and L61 were determined to be the most mobile based on the values of anisotropies and their dependence on temperature. As most of the residues belong to the same secondary structure region, we assumed the same rigid value for the anisotropy and obtained order parameters .
Our work represents the first investigation to test the usefulness of the SAMMY sequence for lyophilized protein samples. The sensitivity is not sufficient to determine the relative orientations of the 15N CSA and 15N-15H dipolar tensors. However, the overall narrowing of the SAMMY patterns compared with the rigid case reports on contribution due to the dynamics and allows for the estimation of the motional order parameters, assuming a single value for the dipolar coupling constant in the rigid case. We observed an excellent correlation between the values of and calculated in this manner. However, the values of were systematically 4% lower, possibly providing evidence for the vibrational averaging due to motions of the amide hydrogen atoms.
Lastly, the data provide some insights into the comparison of the hydrophobic core structure and dynamics in solution versus lyophilized powder states. The isotropic chemical shifts are overall consistent between the two states. L75, located at the C-terminus, appears to be highly dynamic in both solution and solid phases and also displays the largest variation in the isotropic chemical shift between the two phases. The results for the backbone fluctuations of the hydrophobic core residues will be combined with our previous investigations of HP36 dynamics in solution and solid states(44, 47, 49, 55, 56) as well as with future molecular dynamics simulations to assess a global picture of concerted hydrophobic core fluctuations. The CSA and dipolar tensor parameters obtained in this work are also useful as reference values for developing more advanced magic-angle spinning recoupling techniques for multiply-labeled samples, as these simple static experiments are relatively free of experimental artifacts and do not have large scaling factors associated with them.
Supplementary Material
Figure 5.
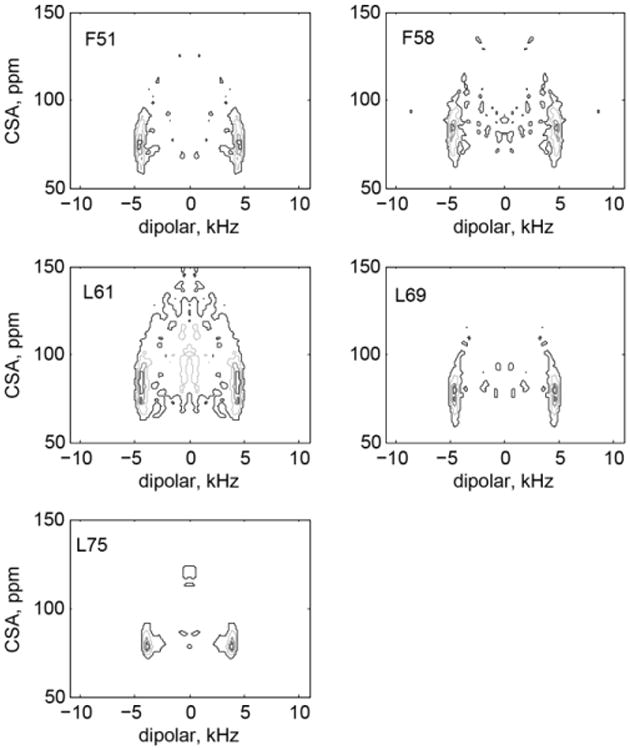
Experimental two-dimensional SAMMY line shapes at 22°C. Contour levels are selected at 0.4, 0.6, 0.8 and 0.9 of the maximum intensity.
Highlights.
We measured 15N CSA tensor parameters and 1H-15N dipolar coupling constants for backbone sites of a globular protein under static conditions.
All probed sites belonged to the hydrophobic core and were shown to have order parameters less than 1 based on the values of 15N CSA anisotropy and SAMMY-based dipolar splittings.
The values of the tensorial parameters obtained from these simple static measurements can serve as reference values useful in developing more advanced magic-angle spinning recoupling techniques.
Acknowledgments
This work was supported by National Science Foundation grant MCB-1122154 to L.V. and D.O. as well as by National Institutes of Health grant P20GM103395. Experiments were performed at the National High Magnetic Field Laboratory, which is supported by National Science Foundation Cooperative Agreement No. DMR-1157490, the State of Florida and the U.S. Department of Energy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berjanskii MV, Wishart DS. A simple method to predict protein flexibility using secondary chemical shifts. J Am Chem Soc. 2005;127:14970–14971. doi: 10.1021/ja054842f. [DOI] [PubMed] [Google Scholar]
- 2.Palmer AG. NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 3.Yao LS, Grishaev A, Cornilescu G, Bax A. Site-Specific Backbone Amide N-15 Chemical Shift Anisotropy Tensors in a Small Protein from Liquid Crystal and Cross-Correlated Relaxation Measurements. J Am Chem Soc. 2010;132:4295–4309. doi: 10.1021/ja910186u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wylie BJ, Franks WT, Rienstra CM. Determinations of N-15 chemical shift anisotropy magnitudes in a uniformly N-15, C-13-labeled microcrystalline protein by three-dimensional magic-angle spinning nuclear magnetic resonance spectroscopy. J Chem Phys B. 2006;110:10926–10936. doi: 10.1021/jp060507h. [DOI] [PubMed] [Google Scholar]
- 5.Schanda P, Meier BH, Ernst M. Quantitative Analysis of Protein Backbone Dynamics in Microcrystalline Ubiquitin by Solid-State NMR Spectroscopy. J Am Chem Soc. 2010;132:15957–15967. doi: 10.1021/ja100726a. [DOI] [PubMed] [Google Scholar]
- 6.Yang J, Tasayco ML, Polenova T. Dynamics of Reassembled Thioredoxin Studied by Magic Angle Spinning NMR: Snapshots from Different Time Scales. J Am Chem Soc. 2009;131:13690–13702. doi: 10.1021/ja9037802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krushelnitsky A, Reichert D. Solid-state NMR and protein dynamics. Prog Nucl Magn Reson Spectrosc. 2005;47:1–25. [Google Scholar]
- 8.Lakomek NA, Carlomagno T, Becker S, Griesinger C, Meiler J. A thorough dynamic interpretation of residual dipolar couplings in ubiquitin. J Biomol NMR. 2006;34:101–115. doi: 10.1007/s10858-005-5686-0. [DOI] [PubMed] [Google Scholar]
- 9.Le HB, Oldfield E. Ab initio studies of amide-N-15 chemical shifts in dipeptides: Applications to protein NMR spectroscopy. J Phys Chem. 1996;100:16423–16428. [Google Scholar]
- 10.Cornilescu G, Bax A. Measurement of proton, nitrogen, and carbonyl chemical shielding anisotropies in a protein dissolved in a dilute liquid crystalline phase. J Am Chem Soc. 2000;122:10143–10154. [Google Scholar]
- 11.Brender JR, Taylor DM, Ramamoorthy A. Orientation of amide-nitrogen-15 chemical shift tensors in peptides: A quantum chemical study. J Am Chem Soc. 2001;123:914–922. doi: 10.1021/ja001980q. [DOI] [PubMed] [Google Scholar]
- 12.Xu XP, Case DA. Probing multiple effects on N-15, C-13 alpha, C-13 beta, and C-13′ chemical shifts in peptides using density functional theory. Biopolymers. 2002;65:408–423. doi: 10.1002/bip.10276. [DOI] [PubMed] [Google Scholar]
- 13.Cai L, Fushman D, Kosov DS. Density functional calculations of N-15 chemical shifts in solvated dipeptides. J Biomol NMR. 2008;41:77–88. doi: 10.1007/s10858-008-9241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loth K, Pelupessy P, Bodenhausen G. Chemical shift anisotropy tensors of carbonyl, nitrogen, and amide proton nuclei in proteins through cross-correlated relaxation in NMR spectroscopy. J Am Chem Soc. 2005;127:6062–6068. doi: 10.1021/ja042863o. [DOI] [PubMed] [Google Scholar]
- 15.Wylie BJ, Sperling LJ, Frericks HL, Shah GJ, Franks WT, Rienstra CM. Chemical-shift anisotropy measurements of amide and carbonyl resonances in a microcrystalline protein with slow magic-angle spinning NMR spectroscopy. J Am Chem Soc. 2007;129:5318–5320. doi: 10.1021/ja0701199. [DOI] [PubMed] [Google Scholar]
- 16.Chevelkov V, Fink U, Reif B. Accurate Determination of Order Parameters from H-1,N-15 Dipolar Couplings in MAS Solid-State NMR Experiments. J Am Chem Soc. 2009;131:14018–14022. doi: 10.1021/ja902649u. [DOI] [PubMed] [Google Scholar]
- 17.Asami S, Reif B. Proton-Detected Solid-State NMR Spectroscopy at Aliphatic Sites: Application to Crystalline Systems. Acc Chem Res. 2013;46:2089–2097. doi: 10.1021/ar400063y. [DOI] [PubMed] [Google Scholar]
- 18.Lewandowski JR, Halse ME, Blackledge M, Emsley L. Direct observation of hierarchical protein dynamics. Science. 2015;348:578–581. doi: 10.1126/science.aaa6111. [DOI] [PubMed] [Google Scholar]
- 19.Nevzorov AA, Opella SJ. Structural fitting of PISEMA spectra of aligned proteins. J Magn Reson. 2003;160:33–39. doi: 10.1016/s1090-7807(02)00138-6. [DOI] [PubMed] [Google Scholar]
- 20.Ramamoorthy A, Wei YF, Lee DK. PISEMA solid-state NMR spectroscopy. 2004;52:1–52. [Google Scholar]
- 21.Davis JH, Auger M. Static and magic angle spinning NMR of membrane peptides and proteins. Prog Nucl Magn Reson. 1999;35:1–84. [Google Scholar]
- 22.Lazo ND, Hu W, Cross TA. Low-temperature solid-state N-15 NMR characterization of polyleptide backbone librations. J Magn Reson B. 1995;107:43–50. doi: 10.1006/jmrb.1995.1056. [DOI] [PubMed] [Google Scholar]
- 23.McKnight CJ, Doering DS, Matsudaira PT, Kim PS. A thermostable 35-residue subdomain within villin headpiece. J Mol Biol. 1996;260:126–134. doi: 10.1006/jmbi.1996.0387. [DOI] [PubMed] [Google Scholar]
- 24.McKnight CJ, Matsudaira PT, Kim PS. NMR structure of the 35-residue villin headpiece subdomain. Nat Struct Biol. 1997;4:180–184. doi: 10.1038/nsb0397-180. [DOI] [PubMed] [Google Scholar]
- 25.Chiu TK, Kubelka J, Herbst-Irmer R, Eaton WA, Hofrichter J, Davies DR. High-resolution x-ray crystal structures of the villin headpiece subdomain, an ultrafast folding protein. Proc Natl Acad Sci U S A. 2005;102:7517–7522. doi: 10.1073/pnas.0502495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu CH, Ramamoorthy A, Opella SJ. High-Resolution Heteronuclear Dipolar Solid State Nmr-Spectroscopy. J Magn Reson. 1994;109:270–272. [Google Scholar]
- 27.Lee M, Goldburg WI. Nuclear-Magnetic-Resonance Line Narrowing By A Rotating Rf Field. Phys Rev. 1965;140:1261–1265. [Google Scholar]
- 28.Yamamoto K, Lee DK, Ramamoorthy A. Broadband-PISEMA solid-state NMR spectroscopy. Chem Phys Lett. 2005;407:289–293. doi: 10.1016/j.cplett.2005.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nevzorov AA, Opella SJ. A “Magic Sandwich” pulse sequence with reduced offset dependence for high-resolution separated local field spectroscopy. J Magn Reson. 2003;164:182–186. doi: 10.1016/s1090-7807(03)00240-4. [DOI] [PubMed] [Google Scholar]
- 30.Hohwy M, Nielsen NC. Elimination of high order terms in multiple pulse nuclear magnetic resonance spectroscopy: Application to homonuclear decoupling in solids. J Chem Phys. 1997;106:7571–7586. [Google Scholar]
- 31.Rupley JA, Gratton E, Careri G. Water and Globular-Proteins. Trends Biochem Sci. 1983;8:18–22. [Google Scholar]
- 3.Khodadadi S, Pawlus S, Sokolov AP. Influence of hydration on protein dynamics: combining dielectric and neutron scattering spectroscopy data. J Phys Chem B. 2008;112:14273–14280. doi: 10.1021/jp8059807. [DOI] [PubMed] [Google Scholar]
- 33.Gor'kov PL, Chekmenev EY, Li C, Cotten M, Buffy JJ, Traaseth NJ, Veglia G, Brey WW. Using low-E resonators to reduce RF heating in biological samples for static solid-state NMR up to 900 MHz. J Magn Reson. 2007;185:77–93. doi: 10.1016/j.jmr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 34.Beckmann PA, Dybowski C. A thermometer for nonspinning solid-state NMR spectroscopy. J Magn Reson. 2000;146:379–380. doi: 10.1006/jmre.2000.2171. [DOI] [PubMed] [Google Scholar]
- 35.Bertani P, Raya J, Bechinger B. N-15 chemical shift referencing in solid state NMR. Solild State Nucl Mag. 2014;61-62:15–18. doi: 10.1016/j.ssnmr.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 36.Fung BM, Khitrin AK, Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J Magn Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 37.Fu RQ. Efficient heteronuclear dipolar decoupling in NMR of static solid samples using phase-wiggled two-pulse phase modulation. Chem Phys Lett. 2009;483:147–153. [Google Scholar]
- 38.Fu R, Gordon ED, Hibbard DJ, Cotten M. High Resolution Heteronuclear Correlation NMR Spectroscopy of an Antimicrobial Peptide in Aligned Lipid Bilayers: Peptide-Water Interactions at the Water-Bilayer Interface. J Am Chem Soc. 2009;131:10830–10835. doi: 10.1021/ja903999g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haeberlen U. High-Resolution NMR in Solids: Selective Averaging. In: Waugh JS, editor. Advances in Magnetic Resonance. Academic Press; New York: 1976. [Google Scholar]
- 40.Wylie BJ, Sperling LJ, Nieuwkoop AJ, Franks WT, Oldfield E, Rienstra CM. Ultrahigh resolution protein structures using NMR chemical shift tensors. Proc Natl Acad Sci U S A. 2011;108:16974–16979. doi: 10.1073/pnas.1103728108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan JCC, Tycko R. Recoupling of chemical shift anisotropies in solid-state NMR under high-speed magic-angle spinning and in uniformly C-13-labeled systems. J Chem Phys. 2003;118:8378–8389. [Google Scholar]
- 42.Vugmeyster L, Ostrovsky D, Ford JJ, Lipton AS. Freezing of dynamics of a methyl group in a protein hydrophobic core at cryogenic temperatures by deuteron NMR spectroscopy. J Am Chem Soc. 2010;132:4038–4039. doi: 10.1021/ja909599k. [DOI] [PubMed] [Google Scholar]
- 43.Vugmeyster L, Tien D, Ostrovsky D, Fu R. Effect of subdomain interactions on methyl group dynamics in the hydrophobic core of villin headpiece protein. Prot Sci. 2014;23:145–156. doi: 10.1002/pro.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vugmeyster L, Trott O, McKnight CJ, Raleigh DP, Palmer AG. Temperature-dependent dynamics of the villin headpiece helical subdomain, an unusually small thermostable protein. J Mol Biol. 2002;320:841–854. doi: 10.1016/S0022-2836(02)00537-5. [DOI] [PubMed] [Google Scholar]
- 45.Doster W. The dynamical transition of proteins, concepts and misconceptions. Eur Biophys J. 2008;37:591–602. doi: 10.1007/s00249-008-0274-3. [DOI] [PubMed] [Google Scholar]
- 46.Frauenfelder H, Chen G, Berendzen J, Fenimore PW, Jansson H, McMahon BH, Stroe IR, Swenson J, Young RD. A unified model of protein dynamics. Proc Natl Acad Sci U S A. 2009;106:5129–5134. doi: 10.1073/pnas.0900336106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vugmeyster L, Ostrovsky D, Penland K, Hoatson GL, Vold RL. Glassy Dynamics of Protein Methyl Groups Revealed by Deuteron NMR. J Phys Chem B. 2013;117:1051–1061. doi: 10.1021/jp311112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vugmeyster L, Ostrovsky D, Lipton AS. Origin of Abrupt Rise in Deuteron NMR Longitudinal Relaxation Times of Protein Methyl Groups below 90 K. J Phys Chem B. 2013;117:6129–6137. doi: 10.1021/jp4021596. [DOI] [PubMed] [Google Scholar]
- 49.Vugmeyster L, Ostrovsky D, Khadjinova A, Ellden J, Hoatson GL, Vold RL. Slow motions in the hydrophobic core of chicken villin headpiece subdomain and their contributions to configurational entropy and heat capacity from solid-state deuteron NMR measurements. Biochemistry. 2011;50:10637–10646. doi: 10.1021/bi201515b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vugmeyster L, Ostrovsky D, Ford JJ, D BS, Lipton AS, Hoatson GL, Vold RL. Probing the dynamics of a protein hydrophobic core by deuteron solid-state nuclear magnetic resonance spectroscopy. J Am Chem Soc. 2009;131:13651–13658. doi: 10.1021/ja902977u. [DOI] [PubMed] [Google Scholar]
- 51.Cady SD, Hong M. Simultaneous extraction of multiple orientational constraints of membrane proteins by C-13-detected N-H dipolar couplings under magic angle spinning. J Magn Reson. 2008;191:219–225. doi: 10.1016/j.jmr.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 52.Roberts JE, Harbison GS, Munowitz MG, Herzfeld J, Griffin RG. Measurement Of Heteronuclear Bond Distances In Polycrystalline Solids By Solid-State Nmr Techniques. J Am Chem Soc. 1987;109:4163–4169. [Google Scholar]
- 53.Wang J, Denny J, Tian C, Kim S, Mo Y, Kovacs F, Song Z, Nishimura K, Gan Z, Fu R, Quine JR, Cross TA. Imaging membrane protein helical wheels. J Magn Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- 54.Tang S, Case DA. Vibrational averaging of chemical shift anisotropies in model peptides. J Biomol NMR. 2007;38:255–266. doi: 10.1007/s10858-007-9164-8. [DOI] [PubMed] [Google Scholar]
- 55.Vugmeyster L, McKnight CJ. Slow motions in chicken Villin Headpiece subdomain probed by cross-correlated NMR relaxation of amide NH bonds in successive residues. Biophys J. 2008;95:5941–5950. doi: 10.1529/biophysj.108.134320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vugmeyster L, Ostrovsky D. Temperature dependence of fast carbonyl backbone dynamics in chicken villin headpiece subdomain. J Biomol NMR. 2011;50:119–127. doi: 10.1007/s10858-011-9500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.