

Abstract
Copy number variants (CNVs) have been implicated in multiple neuropsychiatric conditions, including autism spectrum disorder (ASD), schizophrenia, and intellectual disability (ID). Chromosome 15q13 is a hotspot for such CNVs due to the presence of low copy repeat (LCR) elements, which facilitate non-allelic homologous recombination (NAHR). Several of these CNVs have been overrepresented in individuals with neuropsychiatric disorders; yet variable expressivity and incomplete penetrance are commonly seen. Dosage sensitivity of the CHRNA7 gene, which encodes for the α7 nicotinic acetylcholine receptor in the human brain, has been proposed to have a major contribution to the observed cognitive and behavioral phenotypes, as it represents the smallest region of overlap to all the 15q13.3 deletions and duplications. Individuals with zero to four copies of CHRNA7 have been reported in the literature, and represent a range of clinical severity, with deletions causing generally more severe and more highly penetrant phenotypes. Potential mechanisms to account for the variable expressivity within each group of 15q13.3 CNVs will be discussed.
Keywords: Nicotinic acetylcholine receptors, CHRNA7, copy number variation, neuropsychiatric disease
Graphical abstract
1. Introduction
Copy number variation of chromosome 15q13 has been implicated in several neuropsychiatric diseases. The proximal part of chromosome 15 is one of the least stable regions in the genome, largely due to the presence of six low copy repeat (LCR) elements that cluster into six breakpoints (BP1-BP6), mediating non-allelic homologous recombination (NAHR), leading to chromosomal microdeletions and -duplications. Between BP1 and BP3, deletions are known to result in Prader-Willi syndrome when on the paternal chromosome and Angelman syndrome when on the maternal chromosome. Duplications of the same segment have been associated with autism, learning disabilities, and seizures. Telomeric to the Prader-Willi/Angelman syndrome region, at 15q13.3, CNVs have been observed in wide range of neuropsychiatric phenotypes, including cognitive deficits, schizophrenia, epilepsy, mood disorders, attention deficit hyperactivity disorder (ADHD), and autism spectrum disorder (ASD) [1].
The CNVs at 15q13.3 manifest incomplete penetrance, with deletions leading to neuropsychiatric phenotypes in approximately 80% of cases [2] and duplications having much lower penetrance [1]. The most common deletions span from BP4 to BP5, a region containing six genes: FAN1, MTMR10, TRPM1, KLF13, OTUD7A and CHRNA7 and one microRNA: hsa-miR-211. Of these genes, CHRNA7 has been suggested as playing a major role in the neuropsychiatric phenotypes observed in patients.
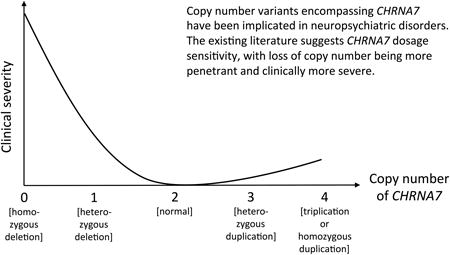
In this review, we will discuss the entire spectrum of 15q13 CNVs, including homozygous deletions, heterozygous deletions, heterozygous duplications, and triplications, as well as their associated phenotypes. Cases were selected from the literature based on the report of phenotypic data beyond that used for ascertainment. We summarize a total of 363 patients from 54 publications, assuming that patients were not reported repeatedly in multiple publications. We propose that CHRNA7 is a dosage-sensitive gene, with more severe phenotypes caused by loss of copy number, and relatively milder effects by gain in copy number.
2. CHRNA7 as a candidate gene
CHRNA7 encodes for the α7 subunit of nicotinic acetylcholine receptors (nAChRs). Nicotinic acetylcholine receptors represent a family of ligand-gated ion channels and are members of the cys-loop receptor superfamily (reviewed by Schaaf, 2014) [3]. These ion channels have high expression in the brain, with highest expression of CHRNA7 in the hippocampus. Receptors comprised of α7 subunits are typically homopentameric, although they have also been shown to interact with β2 subunits [4] and potentially with subunits encoded by CHRFAM7A (discussed in section 6.4). Homopentameric α7 nAChRs, located both pre- and post-synaptically, are important in mediating signal transduction at synapses and in regulating both inhibitory and excitatory neurotransmitter release [5]. Additionally, α7 nAChRs play a role in synaptic plasticity, learning, and memory through neuronal calcium signaling [6]. As an ion channel involved in the regulation of neurotransmitters, CHRNA7 has been considered a strong candidate gene for seizure phenotypes, which is corroborated by deletions of CHRNA7 being found in 1% of patients with idiopathic generalized epilepsies (IGE). Furthermore, CHRNA7 has been suggested as a candidate for some of the other neurodevelopmental and neuropsychiatric phenotypes observed in patients with 15q13.3 CNVs.
3. 15q13.3 as a hotspot for genomic rearrangements
Chromosome 15q13 contains LCR elements clustering into BP1 to BP6. As a result, 15q13 is highly susceptible to NAHR, which likely results in many of the CNVs observed in the region. Distal to the Prader-Willi/Angelman syndrome region at BP1 to BP3, various recurrent microdeletions and microduplications occur between BP3 and BP5 (Figure 1). The largest of these rearrangements are mediated by BP3 and BP5, spanning ∼3.4 Mb to ∼3.9 Mb. While BP3-BP4 deletions are known to occur, they will not be discussed here, as they do not include CHRNA7 [7]. CNVs between BP3 and BP5 have a lower prevalence than BP4-BP5 CNVs, with only 14 heterozygous deletion individuals and four duplication individuals reported in the literature [2,7–17]. The fewer number of individuals with BP3-BP5 CNVs is likely due to the lower amount of shared homology between BP3 and BP5 when compared to BP4 and BP5. The LCRs making up BP4 and BP5 share a large amount of homology, with three regions (oriented in opposite directions relative to each other) of 95 kb, 140 kb, and 218 kb having 99.6% identity. These provide the substrate for NAHR [15]. Additionally, a frequently observed inversion within BP4 results in segments of BP4 and BP5 being in the same orientation, which increases the intergenerational likelihood of an NAHR event. In contrast, there are fewer shared homologous regions between BP3 and BP5. CNVs between BP3 and BP5 affect more than 20 genes, but the phenotypes of patients with these CNVs (discussed in Sections 4.1.1. and 4.2.1.1.) are remarkably similar to those with BP4-BP5 CNVs (discussed in Sections 4.1.2 and 4.2.1.2.), suggesting that the causative dosage sensitive gene or genes lies between BP4 and BP5.
Figure 1. Recurrent Copy Number Variants Found at Chromosome 15q13.3.
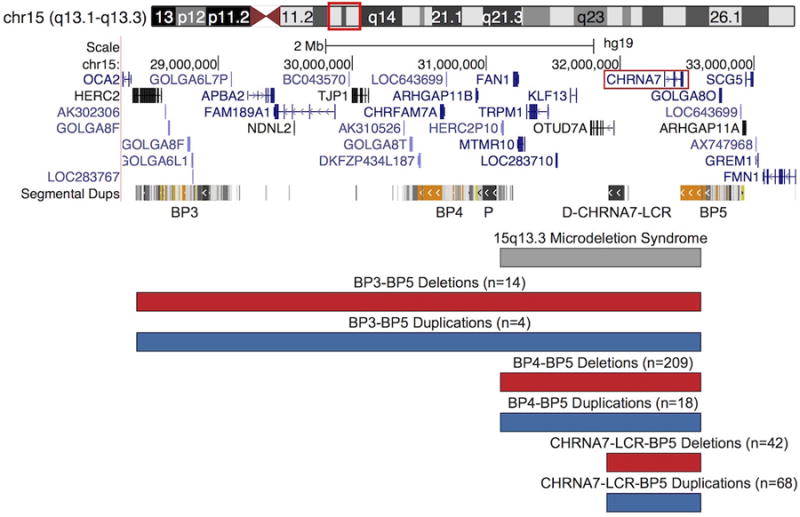
Multiple recurrent deletions (red) and duplications (blue), likely the result of NAHR, occur at 15q13.3 due to the presence of LCRs that cluster into six breakpoints (BP1-BP6) along 15q13, with BP3-BP5 shown. The proximal CHRNA7-LCR (P) and distal CHRNA7-LCR (D-CHRNA7-LCR) are also shown. DECIPHER coordinates for 15q13.3 microdeletion syndrome are shown in gray. Large deletions and duplications, up to 3.9 Mb, occur between BP3 and BP5. The CNVs occur between BP4 and BP5 typically range from 1.5 Mb to 2 Mb in size. Smaller CNVs between the D-CHRNA7-LCR and BP5 have also been found to occur, encompassing CHRNA7 +/- the first exon of OTUD7A. CHRNA7 is highlighted by a red box. The number of published cases is given in parentheses above each set of breakpoints, with homozygous CNVs being omitted. Adapted from UCSC Genome Browser.
The most extensively documented CNVs at 15q13 span ∼1.5 Mb to ∼2 Mb between by BP4 and BP5. Sharp et al. (2008) first described deletions between BP4 and BP5 in five individuals with intellectual disability (ID) and epilepsy, and estimated the prevalence of these microdeletions to be approximately 0.3% among individuals with ID. Since then, reports on many individuals with BP4-BP5 deletions have been published, totaling to 208 heterozygous individuals identified in the literature with a wide range of clinical phenotypes [2,8–13,15,16,18–45]. Duplications of this region are less common, with only 18 cases reported in the literature [16,27,42,46–49]. The limited number reported could, however, also be due to the reduced penetrance of clinical phenotypes among individuals with such duplications. Based on differing breakpoints and size, two different classes of BP4-BP5 duplications have been described [1]. As previously mentioned, these CNVs encompass six genes, one of which is CHRNA7.
Smaller CNVs only affecting CHRNA7 and the first exon of the longer isoform of OTUD7A, a gene encoding for a deubiquitinase, have been reported in patients with phenotypes similar to those described for BP4-BP5 CNVs (discussed in Sections 4.1.3. and 4.2.1.3.) [50]. Additional LCR elements at the distal CHRNA7-LCR (D-CHRNA7-LCR) are found proximal to CHRNA7, and the breakpoints of these smaller CNVs map between exons 1 and 2 of OTUD7A. Within the D-CHRNA7-LCR, there are at least six NAHR “hotspots”, consisting of repetitive DNA elements, providing additional substrates for NAHR [1]. Based on varying breakpoints and size, five classes of small CHRNA7 duplications have been described. These smaller CNVs, as well as some BP4-BP5 CNVs, are also often accompanied by additional rearrangements within the breakpoint regions themselves, likely due to inversions in the region and the presence of CHRFAM7A. The latter is a chimeric fusion gene of CHRNA7 and an unknown genetic element, FAM7A, which shares exons 5-10 with CHRNA7, increasing the amount homology in the region [51].
The smallest CNVs, also utilizing breakpoints in the D-CHRNA7-LCR, have been found as small as ∼350 kb. These deletions only encompass CHRNA7, and as affected individuals manifest similar phenotypes to those with larger deletions, CHRNA7 has been proposed to be at least partially responsible for the cognitive and behavioral manifestations of 15q13.3 CNVs in general. For the purposes of this review, we have grouped all the copy number changes using the D-CHRNA7-LCR, which includes CNVs spanning CHRNA7 and the first exon of OTUD7A as well as those spanning only CHRNA7.
4. Phenotypes associated with changes in CHRNA7 copy number
4.1. 15q13.3 microdeletion syndrome
15q13.3 deletion syndrome (OMIM 612001) is caused by heterozygous deletions at 15q13.3, ranging in size from ∼350 kb to 3.9 Mb. The estimated frequency of 15q13.3 microdeletions in patients with ID is approximately 0.29%. This is comparable to the prevalence of Angelman syndrome (0.34%), Prader-Willi syndrome (0.22%) and Williams syndrome (0.31%) in patients with ID [15]. Additionally, deletions of CHRNA7 are found in ∼1% of individuals with IGE and have been found to be overrepresented in schizophrenia cohorts [10,32]. While it has been suggested that there may be a sex bias in 15q13.3 microdeletion syndrome [20], we are unable to observe any significant difference between the number of males (n=108) and females (n=124) carrying deletions of CHRNA7. Emphasizing the wide range of phenotypes associated with these deletions, probands have been ascertained from cohorts consisting of ID, developmental delay (DD), multiple congenital abnormalities (MCA), dysmorphic features, ASD, IGE, and clinical samples submitted for array comparative genomic hybridization (aCGH). Deletions between ∼1.5 Mb and ∼2 Mb are the most common, but both larger and smaller deletions are reported with similar clinical phenotypes. Of note, homozygous deletions involving CHRNA7 have been reported and are phenotypically more severe phenotypes (discussed in Section 4.1.4.).
4.1.1. Phenotypes associated with BP3-BP5 deletions
The largest recurrent 15q13.3 deletions occur between BP3 and BP5 and span approximately 3.4 Mb to 3.9 Mb, encompassing more than 20 genes, including CHRNA7. Fourteen individuals with heterozygous BP3-BP5 deletions have been reported in the literature [2,7–16,22]. Inheritance was determined in seven of the 14 cases, with four being de novo (28.5% of cases with known inheritance) [7,8,10,16], two inherited from an affected mother (14.3%) [14], and one inherited from a neurotypical mother (14.3%) [12,13] (Figures 2 & 3). In addition, one clinically unaffected father of a child with a homozygous 15q13.3 deletion was identified as a heterozygous BP3-BP5 deletion carrier. All of the reported individuals excluding two parents have neuropsychiatric phenotypes, suggesting almost complete penetrance of these BP3-BP5 deletions. However, this cannot be determined conclusively due to the limited number of reported cases and the lack of inheritance information for half (n=7, 50%) of the individuals.
Figure 2. CHRNA7 CNVs with Known Parental Transmission.
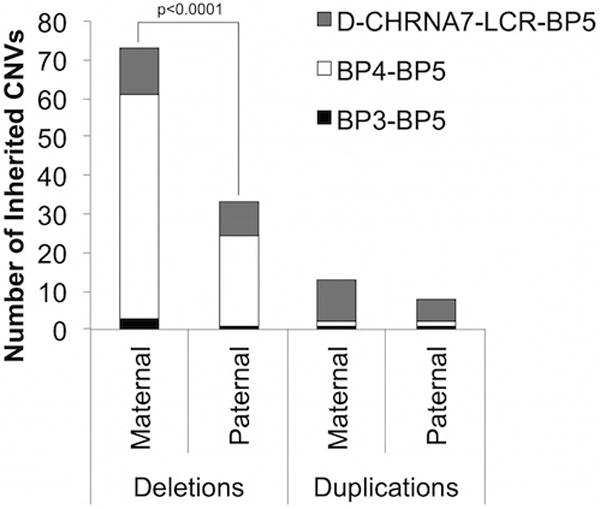
D-CHRNA7-BP5 CNVs are shown in gray (n=21 deletions, n=25 duplications), BP4-BP5 CNVs are shown in dark gray (n=81 deletions, n=3 duplications), and BP3-BP5 CNVs are shown in black (n=4 deletions, 3 duplications). Overall, CHRNA7 deletions are transmitted maternally significantly more (p<0.0001, χ2) than paternally. However, this is primarily due to BP4-BP5 deletions, which also have significantly more (p<0.0001) maternally transmitted alleles. Homozygous deletions, heterozygous deletions, duplications, and triplications are included.
CNV: copy number variant, BP: breakpoint
Figure 3. Inheritance of CHRNA7 CNVs.

The majority of 15q13.3 CNVs have unknown inheritance (black). The remaining CNVs have multiple modes of inheritance (paternal-gray, maternal-white, de novo-diagonal lines), with none being significantly different. The large number of cases with unknown inheritance may be masking any significance for modes of inheritance. For duplications, the smaller sample size may also be contributing to the lack of significance. Homozygous deletions, heterozygous deletions, duplications, and triplications are included. CNV: copy number variant, BP: breakpoint
Of affected individuals with these larger deletions (n=12), often having more than one clinical condition, nine (75%) were diagnosed with cognitive deficits, including ID, developmental delay, or learning disabilities (Table I, Figure 4). Eight individuals (66.7% of affected individuals) were also reported to have language or speech impairments. Almost half (n=5, 41.7%) had a history of seizures, epilepsy, and/or abnormal electroencephalogram (EEG) findings. Other neuropsychiatric conditions were prevalent in these patients, including ADHD or attention difficulties (n=5, 41.7%), ASD (n=3, 25%), and other abnormal behaviors, including Prader-Willi-like phenotypes and hyperphagia (n=5, 41.7%). Five cases were reported as overweight or obese. Mildly dysmorphic features were reported in seven cases (58.3%), hypotonia in four (33.3%). Other medical (n=6 50%) and neurological (n=4, 33.3%) conditions were reported which may be impacting the overall phenotypes observed (Table II).
Table I. Clinical features of heterozygous CHRNA7 deletion and duplication cases by breakpoints from the literature.
BP3-BP5 CNVs span up to ∼3.9 Mb, BP4-BP5 CNVs span ∼1.5Mb to ∼2 Mb, D-CHRNA7-LCR-BP5 CNVs span from D-CHRNA7-LCR to BP5, up to ∼680 kb.
| Heterozygous Detections | Heterozygous Duplications | |||||||
|---|---|---|---|---|---|---|---|---|
|
|
||||||||
| BP3-BP5 | BP4-BP5 | D-CHRNA7-LCR-BP5 | All Detections | BP3-BP5 | BP4-BP5 | D-CHRNA7-LCR-BP5 | All Duplications | |
|
|
||||||||
| Cognitive Deficitsa | 9/12 | 105/192 | 19/37 | 133/241 (55.2%) | 3/3 | 7/16 | 17/61 | 27/80 (33.8%) |
| Seizures/Epilopsy/EEG Abnormalities | 5/12 | 71/192 | 13/37 | 88/241 (36.9%) | 0/3 | 3/16 | 4/61 | 7/80 (8.8%) |
| Language/Speech impairment | 8/12 | 54/192 | 5/37 | 67/241 (27.8%) | 0/3 | 5/16 | 5/61 | 10/80 (12.5%) |
| Dysmorphic Features | 7/12 | 47/192 | 9/37 | 63/241 (26.1%) | 0/3 | 4/16 | 3/61 | 7/80 (8.8%) |
| Abnormal Behavior | 5/12 | 45/192 | 7/37 | 58/241 (24 1%) | 0/3 | 4/16 | 7/61 | 11/80 (13.6%) |
| ASD | 3/12 | 22/192 | 6/37 | 31/241 (12.9%) | 2/3 | 7/16 | 9/61 | 18/80 (22 5%) |
| ADHDb | 5/11 | 18/192 | 5/37 | 28/241 (11.6%) | 2/3 | 5/16 | 11/61 | 18/80 (22 5%) |
| Mood Disordersc | 0/12 | 25/192 | 4/37 | 29/241 (12%) | 0/3 | 3/16 | 11/61 | 17/80 (21.3%) |
| Schizophrenia | 0/12 | 26/192 | 1/37 | 27/241 (11.2%) | 0/3 | 1/16 | 17/61 | 18/80 (22.5%) |
| Hypotonia | 4/12 | 14/192 | 4/37 | 22/241 (9.1%) | 1/3 | 2/16 | 7/61 | 10/80 (12.5%) |
| Eye Pathology | 4/12 | 9/192 | 3/37 | 16/241 (6.6%) | 0/3 | 0/16 | 1/61 | 1/80 (1.3%) |
BP: breakpoint, EEG: electroencephalogram, ASD: autism spectrum disorder, ADHD: attention deficit hyperactivity disorder
Homozygous deletions and triplications not included.
Cognitive deficits include intellectual disability, developmental delay, and learning difficulties;
Includes poor attention and hyperactivity;
Mood disorders includes anxiety, bipolar disorder, depression, and unspecified mood disorders.
Figure 4. Clinical features of individuals with heterozygous CHRNA7 CNVs.

Cognitive deficits were the most prevalent phenotype in both duplication and deletion patients. Seizures (including epilepsy and EEG abnormalities) were considerably more common in patients with CHRNA7 deletions. ASD, ADHD or attention difficulties, mood disorders, and schizophrenia were more common in individuals with CHRNA7 duplications. Cognitive deficits (including ID, DD, and learning difficulties) were the only features to occur in near half of all individuals with heterozygous CNVs, emphasizing the variable expressivity associated with CHRNA7 changes in copy number. Mood disorders includes anxiety, bipolar disorder, depression, and unspecified mood disorders. Phenotypically normal individuals, homozygous deletions and triplications not included.
Table II. Complete phenotypic information for CHRNA7 CNV patients.
Three hundred and sixty three individuals with CHRNA7 CNVs were identified. Patients are listed by deletion or duplication and breakpoints. Number of individuals, including number affected and number with no reported phenotype is indicated in the appropriate box. Phenotypes only include those reported in more than two individuals, except for BP3-BP5 duplications due to the small number in the literature. Phenotypic data includes only individuals with reported phenotypes. Phenotypes are based on what was reported in the literature, but it is possible that unreported conditions may also be present in patients.
| Breakpoints | Number of Cases | Inheritance | Phenotypes | References |
|---|---|---|---|---|
| Deletions | ||||
|
| ||||
| 14 Total | 50% (n=7) Unknown | 75% (n=9) Cognitive Deficitsa | [2], [7], [8], [9], [10], [11], [13], [14], [16], [22] | |
| 12 Affected | 28.6% (n=4) De Novo | 66.7% (n=8) Language Impairment | ||
| 2 No Reported Phenotype | 14.3% (n=2) Maternal (Affected) | 58.3% (n=7) Dysmorphic Features | ||
| 7.1% (n=1) Maternal (Unaffected) | 50% (n=6) Medical Condition | |||
| BP3-BP5 | 41.7% (n=5) Seizures b | |||
| 41.7% (n=5) Overweight c | ||||
| 41.7% (n=5) ADHD d | ||||
| 41.7% (n=5) Abnormal Behavior | ||||
| 33.3% (n=4) Neurological Condition | ||||
| 33.3% (n=4) Hypotonia | ||||
| 33.3% (n=4) Eye Pathologies | ||||
| 25% (n=4) Short Stature | ||||
| 25% (n=4) ASD | ||||
|
| ||||
| 208 Total | 58.2% (n=121) Unknown | 55% (n=105) Cognitive Deficits | [2], [8], [9], [10], [11], [12], [13], [15], [16], [18], [19], [20], [21], [22], [23], [24], [26], [27], [28], [29], [30], [31],[32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44], [45], [46], [49] | |
| 191 Affected | 14.4% (n=39) Maternal (Unaffected) | 37.2% (n=71) Seizures | ||
| 17 No Reported Phenotype | 8.7% (n=18) De Novo | 28.3% (n=54) Language Impairment | ||
| 5.3% (n=11) Paternal (Unaffected) | 24.6% (n=47) Dysmorphic Features | |||
| 5.8% (n=12) Maternal (Affected) | 24.1% (n=46) Abnormal Behavior | |||
| 3.4% (n=7) Paternal (Affected) | 13.6% (n=26) Schizophrenia | |||
| 13.1% (n=25) Mood Disorder e | ||||
| 11.5% (n=22) ASD | ||||
| 9.4% (n=18) ADHD | ||||
| BP4-BP5 | 7.32% (n=14) Hypotonia | |||
| 7.32% (n=14) Medical Condition | ||||
| 6.8% (n=13) Neurological Condition | ||||
| 5.2% (n=10) Short Stature | ||||
| 4.7% (n=9) Eye Pathology | ||||
| 4.2% (n=8) Microcephaly | ||||
| 3.1% (n=6) Sleeping Problems | ||||
| 2.6% (n=5) Macrocephaly | ||||
| 2.6% (n=5) Overweight | ||||
|
| ||||
| 52.8% (n=19) Cognitive Deficits | [9], [13], [20], [32], [50], [52], [53], [54], [78] | |||
| 42 Total | 54.8% (n=23) Unknown | 36.1% (n=13) Seizures | ||
| 36 Affected | 16.7% (n= 7) Maternal (Affected) | 25% (n=9) Dysmorphic Features | ||
| 6 No Reported Phenotype | 11.9% (n=5) Paternal (Affected) | 19.4% (n=7) Abnormal Behavior | ||
| 7.1% (n=3) Maternal (Unaffected | 16.7% (n=6) ASD | |||
| 7.1% (n=3) Paternal (Unaffected) | 13.9% (n=5) Langauge Impairment | |||
| 2.3% (n=1) De Novo | 13.9% (n=5) ADHD | |||
| D-CHRNA7-BP5 | 13.9% (n=5) Failure to Thrive | |||
| 11.1% (n=4) Mood Disorder | ||||
| 11.1% (n=4) Hypotonia | ||||
| 11.1% (n=4) Medical Condition | ||||
| 8.3% (n=3) Neurological Condition | ||||
| 8.3% (n=3) Overweight | ||||
| 8.3% (n=3) Eye Pathology | ||||
|
| ||||
| Duplications | ||||
|
| ||||
| 4 Total | 50% (n=2) Unknown | 75% (n=3) Cognitive Deficits | [16], [17] | |
| 3 Affected | 25% (n=1) Maternal (Affected) | 50% (n=2) ASD | ||
| 1 No Reported Phenotype | 25% (n=1) Paternal (Unaffected) | 50% (n=2) Mood Disorder | ||
| BP3-BP5 | 50% (n=2) ADHD | |||
| 25% (n=1) Hypotonia | ||||
| 25% (n=1) Overweight | ||||
|
| ||||
| 18 Total | 61.1% (n=11) Unknown | 43.8% (n=7) Cognitive Deficits | [16], [27], [42], [46], [47], [48], [49] | |
| 16 Affected | 27.8% (n=5) De Novo | 43.8% (n=7) ASD | ||
| 2 No Reported Phenotype | 5.6% Paternal (Unaffected) | 31.3% (n=5) Language Impairment | ||
| 5.6% Maternal (Unaffected) | 31.3% (n=5) ADHD | |||
| BP4-BP5 | 25% (n=4) Abnormal Behavior | |||
| 25% (n=4) Dysmorphic Features | ||||
| 18.8% (n=3) Seizures | ||||
| 18.8% (n=3) Mood Disorder | ||||
|
| ||||
| 67 Total | 79.1% (n=53) Unknown | 28.3% (n=17) Cognitive Deficits | [1], [31], [32], [43], [46], [49], [53], [57], [58], [59] | |
| 60 Affected | 7.5% (n=5) Maternal (Unaffected) | 28.3% (n=17) Schizophrenia | ||
| 7 No Reported Phenotype | 6% (n=4) Maternal (Affected) | 18.3% (n=11) ADHD | ||
| 4.5% (n=3) Paternal (Unaffected) | 18.3% (n=11) Mood Disorder | |||
| 3% (n=2) Paternal (Affected) | 15% (n=9) ASD | |||
| D-CHRNA7-BP5 | 11.7% (n=7) Hypotonia | |||
| 8.3% (n=5) Language Impairment | ||||
| 6.7% (n=4) Seizures | ||||
| 5% (n=3) Neurological Condition | ||||
| 5% (n=3) Dysmorphic Features | ||||
BP: breakpoint, EEG: electroencephalogram, ASD: autism spectrum disorder, ADHD: attention deficit hyperactivity disorder,
Cognitive deficits include intellectual disability, developmental delay, and learning difficulties;
Seizures includes epilepsy and abnormal EEG findings;
Overweight includes obesity;
ADHD includes hyperactivity and attention difficulties;
Mood disorders include anxiety, bipolar disorder, depression, and unspecified mood disorders.
4.1.2. Phenotypes associated with BP4-BP5 deletions
The largest number of 15q13.3 deletion cases have breakpoints in BP4 and BP5, and are ∼1.5 Mb to ∼2 Mb in size. From our literature review, we identified 208 individuals with heterozygous BP4-BP5 deletions with sufficient clinical information beyond ascertainment data [2–8,11,12,14–41]. Of these 208 cases, 121 deletions were of unknown inheritance (Figures 2 & 3). Of the cases with known inheritance (n=87, 20.7% (n=18) were de novo. A total of 57.5% of BP4-BP5 deletion cases were inherited from unaffected parents by report: 12.6% (n=11) from an unaffected father and 44.8% (n=39) from an unaffected mother. Neuroaffected parents transmitted 8% (n=7) paternally and 13.8% (n=12) maternally. For total parental inheritance, significantly (p<0.0001) more BP4-BP5 deletions were transmitted maternally (n=51, 73.9%) than paternally (n=18, 26.1%). Five cases had an additional genetic aberration that may be contributing to their phenotypes, including 16q duplications [37, 8], and mosaic Turner syndrome [39] (Table II).
Of the neuroaffected individuals (n=192, 92.3%), with individuals often having more than one clinical feature, the phenotypes show considerable variability (Table I, Figure 4). Just over half (n=105, 54.7% of affected cases) have diagnosed cognitive deficits. About one third (n=71, 37%) have a history of seizures, epilepsy, and/or EEG abnormalities. Fifty-four cases (28.1%) have diagnosed language or speech impairments. Other neuropsychiatric phenotypes include schizophrenia (n=26, 13%), ASD or autistic features (n=22, 11.5%), and ADHD or attention difficulties (n=18, 9.4%). Abnormal behaviors, including aggression, inhibition, and impulsiveness were observed in almost a quarter of the cases (n=46, 24%). Mood disorders (n=25, 13%) were prevalent in these individuals, including anxiety, bipolar disorder, and depression. Dysmorphic features were present in 47 cases (24.5%), however these were typically mild, and no consistent pattern of dysmorphia was seen. Fourteen (7.3%) individuals were reported to have hypotonia. Additional medical conditions (n=14, 7.3%) and neurological conditions (n=13, 6.8%) were reported.
4.1.3. Phenotypes associated with D-CHRNA7-LCR-BP5 deletions
Smaller deletions encompassing CHRNA7 were first reported by Shinawi et al. (2009). They documented ten individuals with heterozygous ∼680 kb deletions, spanning CHRNA7 and the first exon of OTUD7A. These deletions share the distal breakpoints of the larger deletions (BP5), but have proximal breakpoints at the distal CHRNA7-LCR (D-CHRNA7-LCR) [50]. This case series substantiated the idea that haploinsufficiency of CHRNA7 is a major contributor to the phenotypes associated with 15q13.3 microdeletion syndrome. The deletion of the first exon of OTUD7A was felt to be less likely pathogenic, as OTUD7A encodes a deubiquitinase, and most enzymes can tolerate a 50% decrease in protein abundance without a major decrease in flux due to enzyme kinetics. Subsequently, even smaller deletions of <500 kb that only include CHRNA7 were identified [52], and will be included in the analysis here. Individuals with these smallest CHRNA7 deletions reported to date have similar phenotypes to those with the ∼680 kb deletions, as well as the larger BP4-BP5 and BP3-BP5 deletions.
A total of 42 individuals with small deletions encompassing only CHRNA7 +/- the first exon of OTUD7A, were found through our literature review, ranging from 380 kb to 693 kb [9,12,20,32,49,50,52–54, 78]. Inheritance was determined for almost half (n=19, 45.2%) of the cases, with one being de novo (5.3% of known cases), eight paternally inherited (42.1%), and 10 (52.6%) maternally inherited (Figures 2 & 3). Three mothers and three fathers carrying the deletion were neurotypical (n=6, 14%). Seven of the probands had additional genetic aberrations, five of which have been implicated in neuropsychiatric disorders, including one 5p15.1 duplication, one 1q21.1 deletion [20], one with Charcot Marie Tooth Type 1A, one 2q24.3-q31.1 deletion, and one with a duplication at Xq26.2 [50] (Table II).
The most prevalent phenotype observed in neuroaffected individuals with DCHRNA7-LCR-BP5 deletions (n=37), with individuals often having more than one clinical condition, were cognitive deficits (n=19, 51.4% of affected cases) (Table I, Figure 4). A history of seizures, epilepsy, and/or EEG abnormalities was reported for 13 individuals (35.1%). Other neuropsychiatric conditions included ASD or autistic features (n=6, 16.2%), ADHD or attention difficulties (n=5, 13.5%), mood disorders, including anxiety and bipolar disorder (n=4, 10.8%), and schizophrenia (n=1, 2.7%). Language or speech impairments were reported in five (13.5%) cases. Seven cases (19%) exhibited abnormal behaviors, including aggression, hyperphagia, and inhibition. Dysmorphic features, usually mild, were observed in nine (24.3%) cases. Additional cardiovascular (n=5, 13.5%), neurological (n=3, 8.1%), and medical conditions (n=4, 10.8%) were also reported (Table II).
4.1.4. Phenotypes associated with homozygous deletions of CHRNA7
Homozygous 15q13.3 deletion involving CHRNA7 causes a severe to profound neurological disorder. It has been reported in a total of 11 cases in the literature [9,18,20,28,52,54–56]. Three (27.3%) of the 11 published cases are compound heterozygotes [9,56], one with BP4-BP5 and BP3-BP5 deletions, and two BP4-BP5 and D-CHRNA7-LCR-BP5 deletions. Of the other eight (72.7%%) patients, two are homozygous for D-CHRNA7-LCR-BP5 deletions [52,54], and six are homozygous for BP4-BP5 deletions [18,20,28,55] (Figures 2 & 3). The general clinical presentation of homozygous 15q13.3 microdeletion is that of a neonatal encephalopathy. All 11 reported individuals had cognitive deficits and hypotonia. Ten individuals (91%) had additional neurological conditions, including abnormal brain MRI, areflexia, subarachnoid cysts, and dysmorphic corpus callosum. Nine cases (81.8%) had a history of seizures, epilepsy, or EEG abnormalities. Language or speech impairments (n=9, 81.8%) and ASD (n=1, 9.1%) were also reported. Eye pathologies, including visual impairment, optic nerve atrophy, and abnormal visual evoked potentials (VEP) were observed in eight (72.7%) patients, all of which had homozygous deletions of at least 1.5 Mb. This is a much higher prevalence of eye phenotypes than what is seen in heterozygous deletion individuals (n=16, 6%). The size of the homozygous deletions associated with eye phenotypes is critical, as it includes TRPM1, mutations of which are known as the cause of autosomal recessive congenital stationary night blindness [55]. The more severe phenotype in these patients may be explained by compounding effects due to the loss of multiple genes. Genotype-phenotype correlation suggests that homozygous loss of TRPM1 is responsible for the visual impairment of these patients, as individuals with homozygous small D-CHRNA7-LCR deletions not including TRPM1 do not typically manifest eye pathologies.
4.2. Elucidation of phenotypes and pathogenicity associated with 15q13.3 gains
4.2.1. Phenotypes associated with published duplications of 15q13.3
While loss of CHRNA7 has been found to have high penetrance, gains of CHRNA7 have been more enigmatic. Like 15q13 deletions, the genomic size of these duplications varies due to different breakpoints in the multiple LCRs in the region. The smallest published duplications are mediated by D-CHRNA7-LCR and BP5 while the largest are mediated by BP3 and BP5. Duplications involving CHRNA7 were first reported by van Bon et al. (2009), and since then it has been reported that CHRNA7 duplications occur in approximately 0.57% of samples submitted for clinical chromosomal microarray analysis [3], 1.25% cases with ADHD [43], and 1% of cases with IGE [10]. These duplications also occur in up to 1 in 180 controls [10]. While CHRNA7 duplications are currently considered of unknown clinical significance, there is evidence in the literature suggesting that they may be pathogenic, however with decreased penetrance [1]. One would assume that increased gene dosage of CHRNA7 leads to greater protein abundance of α7 subunits, however, that has yet to be shown.
While small CHRNA7 duplications have been found in control individuals, these control subjects were not included in our analysis due to lack of clinical data. Transmitting parents, with and without phenotypes, were included. In total, 90 individuals with duplications involving CHRNA7 were available for review [1,16,17,27,31,32,42,43,46–49,53,57–59].
4.2.1.1. Phenotypes associated with BP3-BP5 duplications
Only four individuals with BP3-BP5 duplications have been reported to date, making this the rarest 15q13.3 CNV to be reported in the literature (Table II). One of the three had an additional genetic aberration, a 200 kb deletion at 14q31.1, implicated in ASD [16]. Two were of unknown inheritance, while one was inherited from a phenotypically normal father [16] and one from a phenotypically affected mother [13] (Figures 2 & 3).
Three out of the four cases were reported to have neuropsychiatric phenotypes (Table I, Figure 4). All three affected individuals had cognitive deficits. ASD, ADHD, and mood disorders were each diagnosed in two cases. Hypotonia was reported in only one case, as was being overweight. It appears that the range of phenotypes observed in BP3-BP5 duplication patients overlaps with what is seen in both individuals with deletions of 15q13.3 and smaller duplications of 15q13.3.
4.2.1.2. Phenotypes associated with BP4-BP5 duplications
Eighteen BP4-BP5 duplications have been identified in the current literature (Table II). Over half (n=11, 61.1%) of these are of unknown inheritance, making it difficult to conclude any patterns in transmission (Figures 2 & 3). Of the cases with known inheritance (n=7), five cases (83.3%) were de novo [16,27,46] while two were inherited from a phenotypically normal parent [46,48].
The phenotypes of affected patients (n=16) seem to mirror those associated with CHRNA7 deletions and both larger and smaller duplications (Table I, Figure 4). Almost half (n=7, 43.8% of affected patients) have cognitive deficits, while the same number have a diagnosis of ASD. Five cases (31.3%) have a diagnosis of ADHD or attention difficulties. Language or speech impairment was observed in five cases (31.3%). Other neuropsychiatric phenotypes included seizures, epilepsy, and/or EEG abnormalities (n=3, 18.8%), mood disorders (n=3, 18.8%), and schizophrenia (n=1, 6.25%). Finally, dysmorphic features were reported in four cases (25%).
4.2.1.3. Phenotypes associated with D-CHRNA7-LCR–BP5 duplications
The most common duplications of CHRNA7, with an estimated prevalence of one in 174 to one in 186 individuals, encompass only the gene itself or include the first exon of OTUD7A, with 67 cases identified in the literature for which clinical data are available [1,3,31,32,46,47,53,57–59]. Of these, 53 (79.1%) have unknown inheritance (Figures 2 & 3). For the cases with known inheritance, none were de novo. Eight duplications were inherited from an unaffected parent (57.1% of cases with known inheritance; maternal n=5, 35.7%; paternal n=3, 21.4%) [1,46,57,58], showing incomplete penetrance for this CNV. Two cases were transmitted from an affected father (14.3%) [1], while four were transmitted from an affected mother (28.6%) [1,59]. Fourteen cases had additional genetic aberrations associated with neuropsychiatric disease, including five ARHGAP11B deletions associated with ASD, which may be due to their location within BP4 on 15q13 [57,58] (Table II, Figure 1).
Only seven of 67 cases (10.4%) had reportedly normal phenotypes, all of which were transmitting parents. Of the affected individuals (n=60, 89.6%), often having multiple clinical features, no single phenotype occurred in more than a third of the cases. The most prevalent phenotypes were cognitive deficits and schizophrenia, affecting 17 individuals each (28.3% of affected cases) (Table I, Figure 4). Other neuropsychiatric phenotypes included ADHD (n=11, 18.3%), mood disorders (n=11, 18.3%), and ASD (n=9, 15%). Language or speech impairment was reported in five cases (8.3%). Seven cases (11.6%) exhibited abnormal behaviors, including disruptive behavior disorder, pica, and alcohol abuse. Seven cases (11.6%) were reported to have hypotonia and dysmorphic features were reported in only three cases (5%). In a study of individuals with Alzheimer disease, the small duplication of CHRNA7 was found in six of 276 cases (2%) and one of 332 controls (0.3%) [31].
4.2.1.4. Phenotypes associated with triplications of 15q13.3
Complex genomic rearrangements resulting in a triplication of CHRNA7 have been observed in one published family, totaling to four individuals, three of which had confirmed neuropsychiatric phenotypes [60]. The CHRNA7 triplication lies within a duplication-inverted triplication-duplication (DUP-TRP/INV-DUP) type rearrangement [61]. The proband and his twin both inherited the triplication maternally, while their mother inherited the CNV paternally (Figures 2 & 3). Neuropsychiatric phenotypes were present in three of four individuals with the triplication, including cognitive deficits, ASD, ADHD or attention difficulties, and mood disorders. Language or speech impairment was present in only one child, as were seizures that had onset at 20 years old in the mother. Alcoholism, drug addiction, and abusive behaviors were present in the maternal grandfather with the triplication, suggesting neuropsychiatric disease may have been present, although full clinical information was not available. Additionally, three of the four triplication cases were reported to have mildly dysmorphic features.
4.2.2. Caveats of estimated pathogenicity of CHRNA7 gains
Most of the current studies have identified duplications at 15q13.3 in large cohorts of clinical chromosomal microarray (CMA) samples. Cognitive delays, DD/ID, ASD, and MCA represent some of the most common indications for having CMA performed as a clinical test. Thus, the phenotypic spectrum presented here may simply be a result of ascertainment bias. In order to answer the question of pathogenic relevance of CHRNA7 gains, one would have to screen large unbiased populations, and then perform a detailed neurobehavioral assessment in those individuals identified to carry a CHRNA7 duplication. Similarly, this could be done for children who were identified as CHRNA7 duplication carriers prenatally [1,62].
5. Shared clinical phenotypes of patients with CHRNA7 CNVs
Patients with both CHRNA7 gains and losses have been found to manifest a similar range of neuropsychiatric phenotypes, suggesting that the human brain is sensitive to CHRNA7 dosage. Dosage sensitivity is often observed in synaptopathies, such as the neuropsychiatric phenotypes associated with both deletions and duplications of SHANK3 and MECP2 [63]. We have assessed the common phenotypes between the heterozygous deletions and duplications of CHRNA7 (Table I). Overall, almost half (n=160, 44.1%) have cognitive deficits, making this the most often observed phenotype. A history of seizures, epilepsy, or EEG abnormalities was reported for 26.4% (n=96) of cases. Language and speech impairment is also common, affecting 21.2% (n=77) of published cases. Other neuropsychiatric conditions include abnormal behaviors (n=69, 19%), ASD (n=49, 13.5%), ADHD or attention difficulties (n=46, 12.7%), mood disorders (n=46, 12.4%), and schizophrenia (n=45, 12.4%), highlighting the variable expressivity associated with changes in CHRNA7 copy number.
Due to the fewer cases of CHRNA7 duplications reported in the literature, it is challenging to determine whether certain phenotypes are more frequently seen with deletions or duplications, respectively. However, it appears that gains of CHRNA7 may have a less severe phenotype than losses (Table I). In particular, cognitive deficits (55.2% of all affected deletions cases, 34.2% of all affected duplications cases), language or speech impairment (27.8% of deletion cases, 12.7% of duplication cases) and seizures (36.9% of deletion cases, 8.9% of duplications cases) occur at higher frequencies for CHRNA7 deletions. In contrast, ASD occurs more frequently with CHRNA7 gains (22.8%) than with losses (12.9%), as do ADHD (22.8% in duplication patients, 11.6% in deletion patients) and schizophrenia (22.8% in duplications, 11.2% in deletions).
6. Potential explanations for incomplete penetrance and variable expressivity of 15q13.3 CNVs
Individuals with CNVs involving CHRNA7 manifest a range of phenotypes that is primarily neurobehavioral or neuropsychiatric, but the penetrance of these phenotypes is incomplete, and among those who are clinically affected, there is great variability in severity and expressivity. One would assume that altered dosage of CHRNA7 leads to changes in α7 protein abundance, subsequently changing calcium signaling in respective domains of the central nervous system. But the genetic and/or environmental factors that serve as modifiers of the CHRNA7-related phenotypes have yet to be identified. Several hypotheses have been proposed.
6.1. Additional CNVs may contribute to phenotypic variability in patients with CHRNA7 CNVs
For ID and DD, a “two-hit hypothesis” has been proposed, such that the accumulation of multiple large copy number variants is associated with more severe clinical phenotypes [64]. Additional CNVs in patients with CHRNA7 copy number changes may contribute to the phenotypes that are observed. Three published CHRNA7 duplication patients have additional genetic aberrations of SHANK2, for which deletions and point mutations have been implicated in ASD [57,58]. Several other patients with CHRNA7 deletions and duplications carry additional CNVs implicated in ASD. However, additional genetic abnormalities are only reported in 8.5% (n=27) of affected individuals and were seen in three unaffected individuals, suggesting that additional genetic aberrations are not the sole explanation for the variability observed.
6.2. Single nucleotide polymorphisms as a possible source of phenotypic variability in patients with CHRNA7 CNVs
Researchers have hypothesized that in CHRNA7 deletion cases single nucleotide polymorphisms (SNPs) on the remaining allele may be contributing to the phenotypic variation seen in patients. Common variation has been found to have an additive effect in ASD [65], so it is possible that these common variants could be transmitted from unaffected parents and contribute to a phenotype when in the presence of a CHRNA7 CNV. However, only very few SNPs have been identified in CHRNA7. In the coding region of the gene, one published rare (<1% MAF) SNP in the last exon has been found in one ASD proband and his mother, however it occurred at the same frequency in controls and was not predicted as deleterious [48]. Twenty-one SNPs have been found in the promoter region of CHRNA7, several of which have a functional effect on gene transcription in patients with schizophrenia [66]. Most of these polymorphisms result in decreased transcription [67].
The major challenge in identifying SNPs of CHRNA7 is represented by the gene structure, with exons 5-10 of the gene and the 3′ UTR region being duplicated in the fusion gene CHRFAM7A. Therefore sequencing the region shared between the two genes is usually uninformative which makes the identification of deleterious CHRNA7 mutations or variants that may play a role as modifiers of CNV-related phenotypes quite difficult.
6.3. Epigenetic changes may contribute to phenotypic variability in patients with CHRNA7 CNVs
Epigenetic changes may also contribute to phenotypic variability in CHRNA7 CNV cases, as it has been shown that chromatin interactions with the Prader-Willi Imprinting Center may modulate CHRNA7 expression [68]. The same study showed decreased CHRNA7 expression in the brains of individuals with Rett syndrome and autism. Similarly, in 15q11-13 duplication syndrome (most frequently BP2-BP3), which accounts for 1-3% of ASD cases, CHRNA7 transcription has shown to be reduced in both induced pluripotent stem cell (iPSC) models [69] and human neuronal cell line models [70], suggesting that dysregulation of the gene could contribute to neuropsychiatric phenotypes. The CHRNA7 promoter is subject to methylation, which can be altered due to point mutations in the promoter region, and the gene's mRNA levels are correlated with the amount of promoter methylation.
6.4. Modifier genes may contribute to phenotypic variability in patients with CHRNA7 CNVs
Modifier genes may account for variation in phenotypes observed among individuals with CHRNA7 CNVs. A hypothesized modifier gene for CHRNA7 CNVs is the fusion gene CHRFAM7A, as recently been reviewed by Sinkus et al. (2015). CHRFAM7A is an ancestral, human specific fusion gene resulting from the partial duplications of CHRNA7 and FAM7A, the latter of which is a novel genetic element of unknown function [67]. CHRFAM7A has been suggested as a possible autism candidate gene itself and has expression about one magnitude lower than CHRNA7 in the brain [71,72]. The duplicated CHRNA7 genomic sequence, exons 5 through 10, which is shared with the fusion gene, is 99.9% similar, resulting in the genes being overall 76% similar and sharing four transmembrane domains, but having different Ntermini, with CHNRA7-FAM7A lacking part of the ligand binding site [73]. The gene is also in opposite orientation to CHRNA7, which may impact its expression. In both neuroaffected and control populations, the CHRFAM7A locus copy number is variable, with 1% individuals having zero copies, 10%-20% having one copy, and most individuals having two or three copies. Furthermore, CHRFAM7A is incredibly polymorphic, with up to 30 known polymorphisms. One polymorphism, a two base pair deletion in exon 6 (CHRFAM7AΔ2bp), has been associated with schizophrenia and inversely associated with IGE [74,75].
Functional studies suggest that CHRFAM7A may impact α7 receptors. It has been suggested that CHRFAM7A may confer a dominant negative effect on CHRNA7, as it has the ability to be incorporated into α7 receptors, which are typically homopentameric. The initial hypothesis had been explored in Xenopus oocytes and non-neuronal cell lines [72], and it was more recently shown that the CHRFAM7A protein product, CHRNA7-FAM7A, and the polymorphic protein product, CHRNA7-FAM7AΔ2bp, form functional receptors with α7 subunits also in mouse and rat cells, when overexpressed [76]. While the CHRNA7-FAM7A nAChR subunits lack part of the ligand-binding site, the CHRNA7-FAM7AΔ2bp nAChR subunit completely lacks the ligand-binding site. The hypothesized dominant negative effect occurs through CHRNA7-FAM7A or CHRNA7-FAM7AΔ2bp subunits interacting with α7 subunits and decreasing the total number of functional ligand-binding sites. Furthermore, CHRNA7-FAM7A has been shown to interact with other receptors, including α3 and α4 receptors, and it is possible that it may interact with the larger family of cys-loop receptors, such as 5HT3 and GABA receptors. If the functionality of these receptors was also impacted by CHRFAM7A copy number and interactions, it could also explain part of the phenotypic variability seen in patients. However, this has yet to be shown in human cells. It is possible that the varying copy number of CHRFAM7A or CHRFAM7AΔ2bp may have a role in modulating the phenotypes observed in individuals with CHRNA7 CNVs, as the protein levels of α7 subunits and their functionality may be altered in these patients.
7. Clinical implications
Deletions of chromosome 15q13.3 represent a rare, but well-defined cause of neurocognitive deficits and psychiatric disease. The incomplete penetrance and variable expressivity of the associated phenotypes, even within the same family, represent a significant challenge in counseling affected families, both in a pre- and postnatal setting. Even more challenging appears to be the situation for individuals affected with copy number gains involving CHRNA7, because of their uncertain pathogenicity. Due to the high prevalence of small CHRNA7 duplications in the population, these may represent one of the most common genomic events with an effect on cognition and behavior. However, it has yet to be shown that the duplications lead to (a) an actual increase in α7 protein abundance, (b) physiological differences, such as altered calcium signaling, and (c) neurophysiological changes and clinical/behavioral changes in cohorts that are ideally free of ascertainment bias.
With the increasing number of conditions associated with 15q13.3 CNVs and the increasing number of patients being identified, the demand for therapeutics is becoming more apparent. As membrane-bound receptor, α7 is amenable to pharmacotherapeutic modulation by agonists, antagonists, or allosteric modulators [77]. Nicotinic agonist drugs have already been approved and marketed for smoking cessation and have been utilized in neuropsychiatric diseases including depression, ADHD, schizophrenia, Alzheimer disease, and Parkinson disease [3]. These disorders all have alterations of α7 protein levels and/or CHRNA7 copy number implicated in their pathogenicity. Due to the lack of efficacy or side affects of some of the developed drugs, nicotinic positive allosteric modulators have also been considered, as they lack intrinsic activity and only become active when acetylcholine is present. Additionally, the sensitivity of nicotinic positive allosteric modulators is higher, as the binding sites for these modulators differ between different nAChRs. Some of the respective drugs (or combinations thereof) could be considered to be repurposed for an indication of specific CHRNA7 CNVs. Carefully designed, placebo controlled studies would be warranted.
8. Conclusions
Variations in copy number of CHRNA7 have been found to be associated with a wide range of phenotypes, including cognitive deficits, language and speech impairments, ASD, and mood disorders. The CHRNA7 gene appears to be critically dosage-sensitive with most profound phenotypes in individuals with zero copies of the gene, moderately severe neuropsychiatric disease in those with heterozygous deletions, and incompletely penetrant disease in individuals with more than two copies of the gene. The variable expressivity and incomplete penetrance of 15q13.3 CNVs suggest that there may be other factors contributing to the phenotypes observed in patients, such as additional genetic abnormalities, epigenetic factors, and/or modifier genes and CNVs, with the fusion gene CHRFAM7A being one of the prime candidates. Future studies will need to focus on the underlying molecular mechanisms of both CHRNA7 deletions and duplications, in concert with varying copy number of potential modifier genes or CNVs.
Acknowledgments
This work was generously supported by the Doris Duke Charitable Foundation Grant #2011034, by the Joan and Stanford Alexander Family and by the NIGMS training grant T32 GM08307 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. We thank Jiani Yin, Janson White, and Evan Jones for their proofreading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Szafranski P, Schaaf CP, Person RE, Gibson IB, Xia Z, Mahadevan S, et al. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: benign or pathological? Hum Mutat. 2010;31:840–50. doi: 10.1002/humu.21284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lowther C, Costain G, Stavropoulos D, Melvin R, Silversides C, Andrade D, et al. Genetics in Medicine : Official Journal of the American College of Medical Genetics. 2014. Delineating the 15q13.3 microdeletion phenotype: a case series and comprehensive review of the literature; pp. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaaf C. Nicotinic acetylcholine receptors in human genetic disease. Genetics in Medicine : Official Journal of the American College of Medical Genetics. 2014;16:649–56. doi: 10.1038/gim.2014.9. [DOI] [PubMed] [Google Scholar]
- 4.Murray T, Liu Q, Whiteaker P, Wu J, Lukas R. Nicotinic acetylcholine receptor alpha7 subunits with a C2 cytoplasmic loop yellow fluorescent protein insertion form functional receptors. Acta Pharmacologica Sinica. 2009;30:828–41. doi: 10.1038/aps.2009.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Albuquerque E, Pereira E, Alkondon M, Rogers S. Mammalian Nicotinic Acetylcholine Receptors: From Structure to Function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benfante R, Antonini R, De Pizzol Expression of the a7 nAChR subunit duplicate form (CHRFAM7A) is down-regulated in the monocytic cell line THP-1 on treatment with LPS. Journal of Neuroimmunology. 2011;230:74–84. doi: 10.1016/j.jneuroim.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Rosenfeld J, Stephens L, Coppinger J, Ballif B, Hoo J, French B, et al. Deletions flanked by breakpoints 3 and 4 on 15q13 may contribute to abnormal phenotypes. European Journal of Human Genetics : EJHG. 2011;19:547–554. doi: 10.1038/ejhg.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ben-Shachar S, Lanpher B, German JR, Qasaymeh M, Potocki L, Nagamani SC, et al. Microdeletion 15q13.3: a locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J Med Genet. 2009;46:382–8. doi: 10.1136/jmg.2008.064378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Endris V, Hackmann K, Neuhann T, Grasshoff U, Bonin M, Haug U, et al. Homozygous loss of CHRNA7 on chromosome 15q13.3 causes severe encephalopathy with seizures and hypotonia. American Journal of Medical Genetics, Part A. 2010;152:2908–2911. doi: 10.1002/ajmg.a.33692. [DOI] [PubMed] [Google Scholar]
- 10.Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160–2. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Männik K, Parkel S, Palta P, Zilina O, Puusepp H, Esko T, et al. A parallel SNP array study of genomic aberrations associated with mental retardation in patients and general population in Estonia. European Journal of Medical Genetics. 2011;54:136–43. doi: 10.1016/j.ejmg.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Pagnamenta AT, Khan H, Walker S, Gerrelli D, Wing K, Bonaglia MC, et al. Rare familial 16q21 microdeletions under a linkage peak implicate cadherin 8 (CDH8) in susceptibility to autism and learning disability. J Med Genet. 2011;48:48–54. doi: 10.1136/jmg.2010.079426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–72. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–42. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 15.Sharp A, Mefford H, Li K, Baker C, Skinner C, Stevenson R, et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nature Genetics. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Bon BW, Mefford HC, Menten B, Koolen DA, Sharp AJ, Nillesen WM, et al. Further delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcome. J Med Genet. 2009;46:511–23. doi: 10.1136/jmg.2008.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lionel AC, Crosbie J, Barbosa N, Goodale T, Thiruvahindrapuram B, Rickaby J, et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci Transl Med. 2011;3:95ra75. doi: 10.1126/scitranslmed.3002464. [DOI] [PubMed] [Google Scholar]
- 18.LePichon J, Bittel D, Graf W, Yu S. A 15q13.3 homozygous microdeletion associated with a severe neurodevelopmental disorder suggests putative functions of the TRPM1, CHRNA7, and other homozygously deleted genes. American Journal of Medical Genetics, Part A. 2010;152:1300–1304. doi: 10.1002/ajmg.a.33374. [DOI] [PubMed] [Google Scholar]
- 19.Levinson DF, Duan J, Oh S, Wang K, Sanders AR, Shi J, et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry. 2011;168:302–16. doi: 10.1176/appi.ajp.2010.10060876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masurel-Paulet a, Andrieux J, Callier P, Cuisset J, Le Caignec Delineation of 15q13.3 microdeletions. Clinical Genetics. 2010;78:149–161. doi: 10.1111/j.1399-0004.2010.01374.x. [DOI] [PubMed] [Google Scholar]
- 21.McMahon JM, Scheffer IE, Nicholl JK, Waters W, Eyre H, Hinton L, et al. Detection of microchromosomal aberrations in refractory epilepsy: a pilot study. Epileptic Disord. 2010;12:192–8. doi: 10.1684/epd.2010.0326. [DOI] [PubMed] [Google Scholar]
- 22.Muhle H, Mefford HC, Obermeier T, von Spiczak S, Eichler EE, Stephani U, et al. Absence seizures with intellectual disability as a phenotype of the 15q13.3 microdeletion syndrome. Epilepsia. 2011;52:e194–8. doi: 10.1111/j.1528-1167.2011.03301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6:e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicholl J, Waters W, Suwalski S, Brown S, Hull Y, Harbord MG, et al. Epilepsy with cognitive deficit and autism spectrum disorders: prospective diagnosis by array CGH. Am J Med Genet B Neuropsychiatr Genet. 2013;162B:24–35. doi: 10.1002/ajmg.b.32114. [DOI] [PubMed] [Google Scholar]
- 25.Pagnamenta AT, Wing K, Sadighi Akha E, Knight SJ, Bölte S, Schmötzer G, et al. A 15q13.3 microdeletion segregating with autism. Eur J Hum Genet. 2009;17:687–92. doi: 10.1038/ejhg.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Popovici C, Busa T, Missirian C, Milh M, Moncla A, Philip N. Mosaic 15q13.3 deletion including CHRNA7 gene in monozygotic twins. European Journal of Medical Genetics. 2013;56:274–7. doi: 10.1016/j.ejmg.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–35. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spielmann M, Reichelt G, Hertzberg C, Trimborn M, Mundlos S, Horn D, et al. Homozygous deletion of chromosome 15q13.3 including CHRNA7 causes severe mental retardation, seizures, muscular hypotonia, and the loss of KLF13 and TRPM1 potentially cause macrocytosis and congenital retinal dysfunction in siblings. European Journal of Medical Genetics. 2011;54:e441–e445. doi: 10.1016/j.ejmg.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Dibbens LM, Mullen S, Helbig I, Mefford HC, Bayly MA, Bellows S, et al. Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: precedent for disorders with complex inheritance. Hum Mol Genet. 2009;18:3626–31. doi: 10.1093/hmg/ddp311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guilmatre A, Dubourg C, Mosca ALL, Legallic S, Goldenberg A, Drouin-Garraud V, et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry. 2009;66:947–56. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinzen EL, Need AC, Hayden KM, Chiba-Falek O, Roses AD, Strittmatter WJ, et al. Genome-wide scan of copy number variation in late-onset Alzheimer's disease. J Alzheimers Dis. 2010;19:69–77. doi: 10.3233/JAD-2010-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–41. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kevelam SH, Jansen FE, Binsbergen Ev, Braun KP, Verbeek NE, Lindhout D, et al. Copy number variations in patients with electrical status epilepticus in sleep. J Child Neurol. 2012;27:178–82. doi: 10.1177/0883073811416006. [DOI] [PubMed] [Google Scholar]
- 34.Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17:142–53. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirov A, Dimova P, Todorova A, Mefford H, Todorov T, Saraylieva G, et al. 15q13.3 microdeletions in a prospectively recruited cohort of patients with idiopathic generalized epilepsy in Bulgaria. Epilepsy Res. 2013;104:241–5. doi: 10.1016/j.eplepsyres.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 36.Lacaze E, Gruchy N, Penniello-Valette M, Plessis G, Richard N, Decamp M, et al. De novo 15q13.3 microdeletion with cryptogenic west syndrome. American Journal of Medical Genetics, Part A. 2013;161:2582–2587. doi: 10.1002/ajmg.a.36085. [DOI] [PubMed] [Google Scholar]
- 37.Banka S, Fitzgibbon GJ, Gaunt L, Rankin WJ, Clayton-Smith J. A novel 800 kb microduplication of chromosome 16q22.1 resulting in learning disability and epilepsy may explain phenotypic variability in a family with 15q13 microdeletion. Am J Med Genet A. 2011;155A:1453–7. doi: 10.1002/ajmg.a.34034. [DOI] [PubMed] [Google Scholar]
- 38.Coppola A, Bagnasco I, Traverso M, Brusco A, Di Gregorio E, Del Gaudio L, et al. Different electroclinical picture of generalized epilepsy in two families with 15q13.3 microdeletion. Epilepsia. 2013;54:e69–73. doi: 10.1111/epi.12130. [DOI] [PubMed] [Google Scholar]
- 39.Costain G, Bassett AS. Clinical applications of schizophrenia genetics: genetic diagnosis, risk, and counseling in the molecular era. Appl Clin Genet. 2012;5:1–18. doi: 10.2147/TACG.S21953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cubells J, Deoreo E, Harvey P, Garlow S, Garber K, Adam M, et al. Pharmaco-genetically guided treatment of recurrent rage outbursts in an adult male with 15q13.3 deletion syndrome. American Journal of Medical Genetics, Part A. 2011;155:805–810. doi: 10.1002/ajmg.a.33917. [DOI] [PubMed] [Google Scholar]
- 41.de Kovel CG, Trucks H, Helbig I, Mefford HC, Baker C, Leu C, et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133:23–32. doi: 10.1093/brain/awp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart LR, Hall AL, Kang SHLH, Shaw CA, Beaudet AL. High frequency of known copy number abnormalities and maternal duplication 15q11-q13 in patients with combined schizophrenia and epilepsy. BMC Med Genet. 2011;12:154. doi: 10.1186/1471-2350-12-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams NM, Franke B, Mick E, Anney RJ, Freitag CM, Gill M, et al. Genome-wide analysis of copy number variants in attention deficit hyperactivity disorder: the role of rare variants and duplications at 15q13.3. Am J Psychiatry. 2012;169:195–204. doi: 10.1176/appi.ajp.2011.11060822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valbonesi S, Magri C, Traversa M, Faraone S, Cattaneo A, Milanesi E, et al. Copy number variants in attention-deficit hyperactive disorder : identification of the 15q13 deletion and its functional role. 2014:1–12. doi: 10.1097/YPG.0000000000000056. [DOI] [PubMed] [Google Scholar]
- 45.Stefansson H, Rujescu D, Cichon S, Pietiläinen OP, Ingason A, Steinberg S, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–6. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller D, Shen Y, Weiss L, Korn J, Anselm I, Bridgemohan C, et al. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J Med Genet. 2009;46:242–248. doi: 10.1136/jmg.2008.059907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams NM, Zaharieva I, Martin A, Langley K, Mantripragada K, Fossdal R, et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet. 2010;376:1401–8. doi: 10.1016/S0140-6736(10)61109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bacchelli E, Battaglia A, Cameli C, Lomartire S, Tancredi R, Thomson S, et al. Analysis of CHRNA7 rare variants in autism spectrum disorder susceptibility. Am J Med Genet A. 2015 doi: 10.1002/ajmg.a.36847. [DOI] [PubMed] [Google Scholar]
- 49.Van Den Bossche MJ, Johnstone M, Strazisar M, Pickard BS, Goossens D, Lenaerts ASS, et al. Rare copy number variants in neuropsychiatric disorders: Specific phenotype or not? Am J Med Genet B Neuropsychiatr Genet. 2012;159B:812–22. doi: 10.1002/ajmg.b.32088. [DOI] [PubMed] [Google Scholar]
- 50.Shinawi M, Schaaf CP, Bhatt SS, Xia Z, Patel A, Cheung SW, et al. A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat Genet. 2009;41:1269–71. doi: 10.1038/ng.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gault J, Robinson M, Berger R, Drebing C, Logel J, Hopkins J, et al. Genomic Organization and Partial Duplication of the Human α7 Neuronal Nicotinic Acetylcholine Receptor Gene (CHRNA7) Genomics. 1998;52:173185. doi: 10.1006/geno.1998.5363. [DOI] [PubMed] [Google Scholar]
- 52.Hoppman-Chaney N, Wain K, Seger P, Superneau D, Hodge J. Identification of single gene deletions at 15q13.3: Further evidence that CHRNA7 causes the 15q13.3 microdeletion syndrome phenotype. Clinical Genetics. 2012;83:345–351. doi: 10.1111/j.1399-0004.2012.01925.x. [DOI] [PubMed] [Google Scholar]
- 53.Galizia EC, Srikantha M, Palmer R, Waters JJ, Lench N, Ogilvie CM, et al. Array comparative genomic hybridization: results from an adult population with drug-resistant epilepsy and co-morbidities. Eur J Med Genet. 2012;55:342–8. doi: 10.1016/j.ejmg.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao J, DeWard S, Madan-Khetarpal S, Surti U, Hu J. A small homozygous microdeletion of 15q13.3 including the CHRNA7 gene in a girl with a spectrum of severe neurodevelopmental features. American Journal of Medical Genetics Part A. 2011;155A:2795–800. doi: 10.1002/ajmg.a.34237. [DOI] [PubMed] [Google Scholar]
- 55.Masurel-Paulet A, Drumare I, Holder M, Cuisset J, Vall'ee L, Defoort S, et al. Further delineation of eye manifestations in homozygous 15q13.3 microdeletions including TRPM1: A differential diagnosis of ceroid lipofuscinosis. American Journal of Medical Genetics, Part A. 2014;164:1537–1544. doi: 10.1002/ajmg.a.36471. [DOI] [PubMed] [Google Scholar]
- 56.Prasun P, Hankerd M, Kristofice M, Scussel L, Sivaswamy L, Ebrahim S. Compound heterozygous microdeletion of chromosome 15q13.3 region in a child with hypotonia, impaired vision, and global developmental delay. American Journal of Medical Genetics, Part A. 2014;164:1815–1820. doi: 10.1002/ajmg.a.36535. [DOI] [PubMed] [Google Scholar]
- 57.Chilian B, Abdollahpour H, Bierhals T, Haltrich I, Fekete G, Nagel I, et al. Dysfunction of SHANK2 and CHRNA7 in a patient with intellectual disability and language impairment supports genetic epistasis of the two loci. Clinical Genetics. 2013 doi: 10.1111/cge.12105. [DOI] [PubMed] [Google Scholar]
- 58.Leblond C, Heinrich J, Delorme R, Proepper C, Betancur C, Huguet G, et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genetics. 2012;8:e1002521. doi: 10.1371/journal.pgen.1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Melchior L, Bertelsen B, Debes N, Groth C, Skov L, Mikkelsen J, et al. Microduplication of 15q13.3 and Xq21.31 in a family with tourette syndrome and comorbidities. American Journal of Medical Genetics, Part B: Neuropsychiatric Genetics. 2013;162:825–831. doi: 10.1002/ajmg.b.32186. [DOI] [PubMed] [Google Scholar]
- 60.Soler-Alfonso C, Carvalho C, Ge J, Roney E, Bader P, Kolodziejska K, et al. European Journal of Human Genetics : EJHG. 2014. CHRNA7 triplication associated with cognitive impairment and neuropsychiatric phenotypes in a three-generation pedigree; pp. 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carvalho CM, Ramocki MB, Pehlivan D, Franco LM, Gonzaga-Jauregui C, Fang P, et al. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat Genet. 2011;43:1074–81. doi: 10.1038/ng.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moreno-De-Luca D, Sanders SJ, Willsey AJ, Mulle JG, Lowe JK, Geschwind DH, et al. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol Psychiatry. 2013;18:1090–5. doi: 10.1038/mp.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zoghbi H, Bear M. Synaptic Dysfunction in Neurodevelopmental Disorders Associated with Autism and Intellectual Disabilities. Cold Spring Harb Perspect Biol. 2012;4:a009886. doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. 2012;367:1321–31. doi: 10.1056/NEJMoa1200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, Willsey AJ, et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism. 2012;3:9. doi: 10.1186/2040-2392-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leonard S, Gault J, Hopkins J, Logel J, Vianzon R, Short M, et al. Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch Gen Psychiatry. 2002;59:1085–96. doi: 10.1001/archpsyc.59.12.1085. [DOI] [PubMed] [Google Scholar]
- 67.Sinkus ML, Graw S, Freedman R, Ross RG, Lester HA, Leonard S. The Human CHRNA7 and CHRFAM7A Genes: A Review of the Genetics, Regulation, and Function. Neuropharmacology. 2015 doi: 10.1016/j.neuropharm.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yasui DH, Scoles HA, Horike SI, Meguro-Horike M, Dunaway KW, Schroeder DI, et al. 15q11.2-13.3 chromatin analysis reveals epigenetic regulation of CHRNA7 with deficiencies in Rett and autism brain. Hum Mol Genet. 2011;20:4311–23. doi: 10.1093/hmg/ddr357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Germain ND, Chen PFF, Plocik AM, Glatt-Deeley H, Brown J, Fink JJ, et al. Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1. Mol Autism. 2014;5:44. doi: 10.1186/2040-2392-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meguro-Horike M, Yasui DH, Powell W, Schroeder DI, Oshimura M, Lasalle JM, et al. Neuron-specific impairment of inter-chromosomal pairing and transcription in a novel model of human 15q-duplication syndrome. Hum Mol Genet. 2011;20:3798–810. doi: 10.1093/hmg/ddr298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Casey J, Magalhaes T, Conroy J, Regan R, Shah N, Anney R, et al. A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Human Genetics. 2012;131:565–579. doi: 10.1007/s00439-011-1094-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Araud T, Graw S, Berger R, Lee M, Neveu E, Bertrand D, et al. The chimeric gene CHRFAM7A, a partial duplication of the CHRNA7 gene, is a dominant negative regulator of α7*nAChR function. Biochem Pharmacol. 2011;82:904–14. doi: 10.1016/j.bcp.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sinkus M, Lee M, Gault J, Logel J, Short M, Freedman R, et al. A 2-base pair deletion polymorphism in the partial duplication of the alpha7 nicotinic acetylcholine gene (CHRFAM7A) on chromosome 15q14 is associated with schizophrenia. Brain Res. 2009;1291:1–11. doi: 10.1016/j.brainres.2009.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rozycka A, Dorszewska J, Steinborn B, Lianeri M, Winczewska-Wiktor A, Sniezawska A, et al. Association study of the 2-bp deletion polymorphism in exon 6 of the CHRFAM7A gene with idiopathic generalized epilepsy. DNA Cell Biol. 2013;32:640–7. doi: 10.1089/dna.2012.1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Flomen R, Collier D, Osborne S, Munro J, Breen G, St Clair Association study of CHRFAM7A copy number and 2 bp deletion polymorphisms with schizophrenia and bipolar affective disorder. American Journal of Medical Genetics Part B, Neuropsychiatric Genetics : The Official Publication of the International Society of Psychiatric Genetics. 2006;141B:571–5. doi: 10.1002/ajmg.b.30306. [DOI] [PubMed] [Google Scholar]
- 76.Wang Y, Xiao C, Indersmitten T, Freedman R, Leonard S, Lester H. The duplicated α7 subunits assemble and form functional nicotinic receptors with the full-length α7. The Journal of Biological Chemistry. 2014 doi: 10.1074/jbc.M114.582858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deutsch SI, Urbano MR, Burket JA, Herndon AL, Winebarger EE. Pharmacotherapeutic implications of the association between genomic instability at chromosome 15q13.3 and autism spectrum disorders. Clin Neuropharmacol. 2011;34:203–5. doi: 10.1097/WNF.0b013e31823a1247. [DOI] [PubMed] [Google Scholar]
- 78.Mikhail F, Lose E, Robin N, Descartes M, Rutledge K, Rutledge S, et al. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. American Journal of Medical Genetics, Part A. 2011;155:2386–2396. doi: 10.1002/ajmg.a.34177. [DOI] [PubMed] [Google Scholar]