

Abstract
The interaction of a small molecule made in one cell with a large receptor made in another is the signature event of cell signaling. Understanding the structure and energy changes associated with agonist activation is important for engineering drugs, receptors and synapses. The nicotinic acetylcholine receptor (AChR) is a ~300 kD ion channel that binds the neurotransmitter acetylcholine (ACh) and other cholinergic agonists to elicit electrical responses in the central and peripheral nervous systems. This mini-review is in two sections. First, general concepts of skeletal muscle AChR operation are discussed in terms of energy landscapes for conformational change. Second, adult vs. fetal AChRs are compared with regard to interaction energies between ACh and agonist-site side chains, measured by single-channel electrophysiology and molecular dynamics simulations. The five aromatic residues that form the core of each agonist binding site can be divided into two working groups, a triad (led by αY190) that behaves similarly at all sites and a coupled pair (led by γW55) that has a large influence on affinity only in fetal AChRs.
Each endplate AChR has 5 homologous subunits, two of α(1) and one each of β, δ and either γ (fetal) or ε (adult). These nicotinic AChRs have only 2 functional agonist binding sites located in the extracellular domain, at αδ and either αγ or αε subunit interfaces. The receptor undergoes a reversible, global isomerization between structures called C and O. The C shape does not conduct ions and has a relatively low affinity for ACh, whereas O conducts cations and has a higher affinity. When both agonist sites are empty (filled only with water) the probability of taking on the O conformation (PO) is low, <10−6. When ACh molecules occupy the agonist sites the C→O opening rate constant and C↔O gating equilibrium constant increase dramatically. Following a pulse of ACh at the nerve-muscle synapse, the endplate current rises rapidly to reach a peak that corresponds to PO ~0.96.
Keywords: nicotinic, acetylcholine, neuromuscular, allosteric, gating, binding
Energy landscape
An energy landscape depicts the relationship between structure and function (Fig. 1). The x-axis represents the positions of all atoms and bonds, and the y-axis relates to the rate and equilibrium constants that together determine the cell response. As drawn, such landscapes are oversimplified because all structural rearrangements are projected onto a single dimension (‘reaction progress’), there may be multiple pathways connecting the various structures and, undoubtedly, countless small energy barriers and wells on the surface of each trajectory. Nonetheless, the simple landscape is a useful starting point for understanding receptor operation.
Figure 1. Energy landscape for adult endplate AChRs.

Without agonists, C(losed)-to-O(pen) gating is uphill (+8.3 kcal/mol; gray arrow). This energy change becomes downhill with 2 ACh molecules bound to the agonist sites, because the affinity of O is higher than that of C (−10.2 kcal/mol; the difference between the green arrows). Red arrow, C↔O oscillations that give rise to clusters of single-channel openings (see Fig. 2).
In endplate AChRs the main, stable structures (energy wells) are connected in series: C(losed)↔O(pen)↔D(esensitized). This scheme, too, is oversimplified. AChRs that have desensitized appear to recover directly to C without passing through O, so a cyclic model is likely required [1]. Also, single-channel recordings show that there are multiple C, O and D states connected in a web that may be necessary to unravel in order to fully-account for the macroscopic behaviors of some nicotinic receptors [2]. However, the fundamental energy changes that undergird agonist activation of endplate AChRs can be explored by using the basic scheme shown in Fig. 1.
The lifetime of a stable state is related exponentially to the heights of the barriers that thwart escape from the well. In the absence of agonists, the O state has a lifetime that is about a million times shorter than the others (~0.1 s vs ~5 min): It is a brief layover on the journey between C and D. The intrinsic, unliganded C↔O gating equilibrium constant (the allosteric constant) of wild-type (WT) muscle AChRs is small and constitutive openings are rare, so in whole-cell recordings without agonists it appears that the membrane current is zero. However, single-channel recordings of WT AChRs in muscle cells show that in the absence of agonists there are brief openings that occur at a low frequency [3]. These events generate a tiny cellular current that is too small to be noticed in macroscopic recordings.
The low value for the allosteric constant is a consequence of natural selection. More than a thousand mutations of muscle AChRs have been studied at the single-channel level, and most of these do nothing more than increase constitutive activity above the WT level [4, 5]. For most amino acids away from the neurotransmitter binding sites, Nature has selected side chains that are locally more stable in C vs. O. For many, the free energy changes in gating occur independently and therefore sum to enforce a small allosteric constant.
Consider a mutation of an amino acid in the transmembrane domain that makes the side chain a bit more stable in O (or less stable in C) compared to the WT, to increase the allosteric constant, but without a noticeable effect on the baseline current. The agonist binding energies (green arrows in Fig. 1) remain unchanged, so this substitution has the same quantitative effect on the stability of diliganded O. Consequently, the gating equilibrium constant in the presence of the neurotransmitter will increase to the same extent as does the allosteric constant. The midpoint of the concentration-response curve will shift to the left and the maximum will PO increase. For some natural mutations the increase in the allosteric constant changes the pharmacological profile enough to cause disease, even if the baseline current remains undetectably small in whole-cell recordings [6]. For instance, a 40-fold increase in the allosteric constant caused by the congenital myasthenic syndrome (CMS) mutation αN217K will reduce EC50 and slow the synaptic current decay even though it has no effect on agonist binding or, apparently, the macroscopic baseline current.
A receptor moves along the landscape under the influence only of thermal energy (‘spontaneously’). The small value of the allosteric constant in WT AChRs implies that the free energy of the unliganded O structure is higher (more positive; less stable) than that of C, making the adoption of the O shape uncommon under physiological conditions. However, a resting, unliganded receptor is always sampling microscopic structures near C, some of which are on the pathway to O.
An ACh molecule arrives at a neurotransmitter binding site by diffusion, having no more momentum than a few water molecules. The ligand nestles into the core of the agonist site by a process that requires some local rearrangements [7]. In mouse muscle AChRs the overall equilibrium dissociation constant for ACh binding to a resting agonist site (Kd) is ~175 μM at both αδ and αε (adult) and ~8 μM at the fetal, αγ site. The low-affinity equilibrium constant (represented by the shorter green arrow in Fig. 1) is a function of both diffusion and the local rearrangements.
The protein continuously samples conformational space randomly, but the energy profile changes when ACh is present. Instead of the exit path from C being steeply uphill, the route(s) connecting C with O becomes more favorable. Without an agonist, the uphill gating profile and thermal energy fluctuations usually result in the system rolling back down to the bottom of the C energy well. With ACh the landscape is less uphill, allowing the system to reach the CO transition state more readily. Agonists enable receptors to take on the active conformation with a higher rate and probability because microstructures along the activation pathway as well as the active, O structure are stabilized.
A bound ligand can be thought of as a reversible ‘mutation’ of the agonist site. Just as covalent amino acid substitutions can increase the relative stability of O, agonists are (transient) perturbations that tilt the energy landscape downward to favor conformational change. As a consequence, both the opening rate constant and PO are larger when there is an ACh molecule in an agonist site. Agonists increase receptor activation because they foster the crossing of a pre-existing energy landscape that is sampled spontaneously.
It is possible to estimate the amount of favorable binding energy each ACh molecule provides [8–10]. Another way to describe the above scenario of agonist action is to say that the O state has a higher affinity for ACh than the C state (in Fig. 1, the green arrow connecting the O states is the longer). The affinity difference, O vs. C, is the amount of favorable free energy provided by the ligand to tilt the energy and stabilize the O structure. It is difficult to estimate the equilibrium dissociation constant of ACh to O (Jd) in WT AChRs directly, but several approaches yield estimates of ~30 nM at αδ and αε and 0.05 nM at αγ.
The low/high affinity ratio (Kd/Jd) is called the coupling constant, and its logarithm is proportional to the extra, stabilizing free energy provided by the agonist (the difference between the green arrows). At each adult mouse muscle AChR agonist site this ratio is ~5700, so the favorable energy from each ACh molecule is (at 23°C) −0.59ln(5700)≈−5.1 kcal/mol (a 1 kcal/mol increment translates to a 5.5-fold change in equilibrium constant). In adult AChRs two ACh molecules stabilize O by ~−10.2 kcal/mol, which increases the diliganded gating equilibrium constant by a factor of 57002 and changes PO from ~10−6 to ~1. In fetal AChRs the allosteric constant is smaller but the ACh coupling constant at the αγ site is ~30 times larger (~−7.2 kcal/mol). Partial agonists have smaller coupling constants (provide less favorable energy) than does ACh, for example carbamylcholine (~3000; −4.7 kcal/mol) and choline (~300; −3.3 kcal/mol), at adult sites.
It is also possible to estimate experimentally the extent to which ACh lowers the CO transition state (TS) free energy. The slope of a log-log plot of opening rate vs. the equilibrium constant for a series of point perturbations indicates whether the local TS energy (structure) is C- vs. O-like, on a scale from 0 to 1. For a series of agonists this slope is ~0.93 [11]. Hence, with regard to the ligand, the CO barrier has a structure that is nearly the same as the stable O state. If one ACh molecule stabilizes O by −5.1 kcal/mol, it also stabilizes the TS by nearly the same extent. Two ACh molecules lower the CO barrier enough to increase the opening rate constant from ~0.001 s−1 to ~50,000 s−1, or by ~50 million-fold.
It is likely that the extra stabilizing forces from the ACh molecule begin to be felt as soon as the system departs (spontaneously) from the bottom of the C well, to start its climb towards the barrier peak. However, kinetic analyses only explore barriers and wells (including small ones; [12, 13]), not the connecting regions of the energy landscape. The favorable effect of ACh may happen suddenly when a binding site happens to adopt a particular microstructure, or it may develop gradually as the agonist and site move between several, partially-stabilized microscopic configurations. It may be that the bottom of the C-ensemble well is the only part of the landscape that does not receive extra, favorable energy from the neurotransmitter.
Some imagine that agonist binding to receptor is like a bowling ball crashing into amino acid pins at the binding site. In fact, a small, diffusing agonist molecule lacks the force to perturb mechanically. It is the chemical free energy of the agonist once it has landed that alters the internal thermal profile of the receptor, to increase the rate constant for the conformational switch and PO. At the adult nerve-muscle synapse, the favorable free energy from 2 affinity-changes for ACh (−10.2 kcal/mol) adds to the unfavorable energy of intrinstic gating (+8.3 kcal/mol) to set the diliganded gating energy (−1.9 kcal/mol). This, in turn, determines the gating equilibrium constant (25) and the peak PO of the synaptic response (0.96).
Desensitization
AChRs take on long-lived D conformations that have a high, O-like affinity for agonists but a non-conducting pore. These are usually entered from the O state and, like O, are stabilized by agonists. A receptor in the O conformation also samples randomly its local conformational space, either with or without ACh present. In muscle AChRs the CO barrier is smaller than the CD barrier, so when the system exits from O it usually returns to C rather than entering D. As a consequence, the receptor oscillates between O and C many times before D is reached (red arrow in Fig. 1) [14].
In macroscopic recordings, desensitization following a step of ACh is manifest as a sag in the whole-cell current, as the rapid C↔O oscillations slowly drain into longer-lived, non-conducting D state(s) (Fig. 2, left). In single-channel recordings the oscillations appear as ‘clusters’ of openings separated by long, silent periods in D. In some neuronal AChRs the OD barrier is the smaller, which makes the ensemble current sag faster and in single channels gives rise to only one (or a few) openings rather than prolonged clusters.
Figure 2. Clusters.

A. Activation by agonists. Left, macroscopic current (outside-out patch). A sustained step of 1 mM ACh causes a rapid increase in PO followed by a sag, as receptors enter D states [28]. Right, single-channel currents (cell-attached patch; O is down). Clusters are C↔O oscillations and the long, silent periods between clusters are sojourns in D. Aligning and averaging clusters will reproduce the macroscopic current. Bottom, close up of a cluster (30 μM ACh, adult-type mouse endplate AChRs expressed in HEK cells, 23 °C, −100 mV). B. Activation by mutations (no agonists). Top, WT endplate AChRs rarely open without agonists. Bottom, adding background mutations that increase the allosteric constant (make the gray arrow more negative, in Fig. 1) but have no effect on binding generates clusters (αY127F+αD97A+αP272A) [29]. Like agonists, these mutations ‘tilt’ the energy landscape downward, but without changing affinities (green arrows in Fig. 1).
The time constant of the decline in the macroscopic current depends on the overall shape of the energy profile, not just the O↔D desensitization rate constants. i) If a perturbation reduces PO, for example by reducing the opening rate constant, then it lowers the probability of taking on the state from which D is entered and, hence, increases the sag time constant. This happens without any effect of the mutation on the O↔D process itself. Low-efficacy agonists have longer sag time constants compared to ACh, and AChRs with a mutation that reduces efficacy similarly display slower current declines during sustained ACh application. ii) If a perturbation does nothing more than stabilize O it will slow the sag without affecting the desensitization process, which I define as the height of the OD barrier and the depth of the D well. Such perturbations generate prolonged openings and clusters, but have little to do with modifying desensitization per se. Examples are fast channel-blockers (that delay the exit from O) and some M2-9′ pore mutations. iii) If C↔O equilibrates more slowly than O↔D there will be no sag in the macroscopic current even though desensitization is occurring. The macroscopic current will rise smoothly to a stable plateau, giving the impression that there is no desensitization. The above examples should encourage authors to use the word ‘desensitization’ with care, and make clear whether they are referring empirically to the time constant of the macroscopic sag or mechanistically to the energy landscape.
Just as AChRs can adopt the O shape without any added agonist, they also can adopt D [8]. This does not happen often with unliganded, WT AChRs because they rarely reach O and usually return back to C when they do. However, a mutation that tilts the landscape downward to increase constitutive PO will also increase constitutive PD. Desensitization does not require an agonist; all it takes is getting to O, whether by a ligand or by a mutation that increases the allosteric constant (Fig. 2, right). AChRs that are active constitutively appear to desensitize and recover by similar mechanisms as do those activated by agonists.
Below, I describe agonist binding in terms of changes in free energy rather than as fold-changes in equilibrium constants [at 23°C, energy (kcal/mol)=−0.59ln(constant)]. The symbol for the extra, stabilizing free energy from the affinity change for one agonist molecule is GB1, which is proportional to the logarithm of the coupling constant (the difference in green arrow lengths in Fig. 1). Because of microscopic reversibility, this value can be calculated from the C and O equilibrium dissociation constant ratio or from the ratio of gating equilibrium constants with vs. without agonists. It turns out that in muscle AChRs, GB1 often happens to be approximately proportional to the log of the resting affinity [15]. That is, for many agonists and agonist-site mutations the length of the shorter green arrow in Fig. 1 (low-affinity binding energy) is about half that of the longer one (high-affinity binding energy). This fortuitous relationship allows resting affinity (Kd), coupling constant and GB1 to be used interchangeably.
Agonist sites
In fetal AChRs the agonist sites are αγ+αδ and in adult they are αε+αδ. Some mutations near the binding site on the non-α (complementary) side of the binding pocket knock-out function of the local site but leave the companion as WT. Using these 1-site AChRs, GB1 has been measured for several agonists at αγ, αε and αδ separately, as has the GB2 energy arising from site pairs.
In both fetal and adult AChRs and for all tested agonists, the sum of the single-site GB1 values is approximately the same as for the site pairs [16]. This indicates that the 2 sites operate almost independently with regard to the coupling constant [10]. There may be other kinds of long-distance interactions between the widely-separated binding sites, but apparently these do not have a significant influence on the energy arising from the agonist affinity change.
Consistent with agonist-site independence, GB1 at the αδ, site is the same in fetal and adult AChRs. Although the γ and ε subunits differ by ~250 side chains, none of these substitutions affect significantly the coupling constant at αδ. This observation is consistent with the view that most side chains away from the agonist site mainly influence the allosteric constant rather than agonist binding or the coupling constant. The αδ agonist site pays little attention to the subunit that is sandwiched between the two α subunits.
The αγ site provides more energy from the affinity change compared to αδ, for all tested agonists. For ACh this energy difference is ~−2 kcal/mol, and for choline it is ~−1 kcal/mol. The αε site is similar, but not identical, to αδ: αε provides about the same energy as αδ for ACh, but about 20% less for choline [17].
The response of the different sites to choline is interesting. Choline is both a precursor and breakdown product of ACh and is present at high concentrations in embryonic serum, as well as following a synaptic impulse. Because of the high GB1choline value at αγ, fetal AChRs receive a total of ~−8 kcal/mol from 2 choline molecules and ~−10 kcal/mol from 1 ACh (at αδ) and 1 choline (at αγ), which is the same for 2 ACh molecules at adult AChRs. Adult AChRs receive only ~−6 kcal/mol from choline. Hence, choline is a potent agonist of fetal AChRs but not of adult. The ability of fetal endplate AChRs to respond to choline more avidly and at lower concentrations (recall that GB1 is proportional to lnKd) may be a reason why the γ subunit is required for proper synapse maturation. Conversely, the fact that adult AChR sites have a sub-maximal coupling constant for the neurotransmitter may relate to the necessity of ignoring choline at the synapse. Perhaps choline is a valid chemical signal at juvenile synapses but just noise at adult ones.
Agonist-site functional groups
The physiology, pharmacology, chemistry and structure of neuromuscular AChR agonist sites have been investigated intensively for many years, using a wide variety of experimental approaches. In this section I focus on a narrow slice of this subject, namely recent results from my laboratory regarding the dissection of the ACh interaction energy into its components, estimated from studies of single-channel currents from mouse muscle AChRs. In most experiments the energy generated by the agonist affinity change was estimated from the gating equilibrium constants, with vs. without ligands.
The effects of mutations on the coupling constant have been measured for dozens of residues near the agonist sites and hundreds elsewhere throughout the protein. So far, only 6 amino acids in the entire protein have been found where an alanine substitution decreases this constant by ≥13-fold (makes GB1ACh more positive by ≥1.5 kcal/mol). These amino acids comprise the agonist site Core (Fig. 3). Five are the well-known aromatic ‘box’ (αY93 in loop A, αW149 in loop B, αY190 and αY198 in loop C and γW55 on complementary strand β2) and the sixth is αG147 (N-terminus of loop B) [18]. In an acetylcholine binding protein (AChBP) structure, all of the Core aromatic side chains are in close contact with the quaternary ammonium group (QA) of ACh [19]. The favorable ACh binding energy is generated mainly by Van der Waals and cation-π interactions between the aromatic rings and the QA [20].
Figure 3. The Core.

The principal side of the binding pocket (α, in AChRs) is white and the complementary side (δ, ε or γ) is tan. Aromatic triad residues are green and special pair residues are yellow. Structure is AChBP (pdb accession number 1uv6 [30]). Agonist is carbamylcholine (CCh); numbering is for mouse endplate AChRs.
There are 8 functional groups within the Core: two from each of the 3 tyrosines (benzene ring and −OH) and one from each of the 2 tryptophans (indole ring). The energy contribution from each of these was estimated from the change in the coupling constant associated with a Y→F mutation (removes the −OH) or a F/W→A mutation (removes the ring). These measurements were made using either 2-site adult and fetal AChRs or single αγ, αδ or αε sites, with similar results.
At all three sites, removal of either of the loop C tyrosine rings (αY190 or αY198) or the −OH of αY190 reduced the ACh coupling constant ~25-fold (increased GB1ACh by ~+1.9 kcal/mol). The removal of the loop B tryptophan ring (αW149) had a somewhat larger consequence (+2.3 kcal/mol at αδ/αε and +3.0 kcal/mol at αγ). These three residues, αY190, αY198 and αW149, behave similarly at all of the agonist sites. I call these amino acids the ‘aromatic triad’, which forms the core of the Core. Of the three, αY190 makes by far the largest contribution to affinity because the energetic effects of −OH and ring removal are nearly additive. The joint elimination of these two functional groups decreases the coupling constant substantially and eliminates agonist activation almost completely. In contrast, removal of the −OH from αY93 or αY198 hardly affects the coupling constant. The mechanism behind the strong, stabilizing effect of the αY190 hydroxyl remains unexplained. To a first approximation, the ACh response of the adult endplate AChR agonist sites is determined by the triad, and in particular αY190 in loop C.
Substituting glycines at all loop C positions (α188-α199) or removing this loop entirely eliminates the ability of endplate AChRs to respond to ACh [21]. Re-inserting tyrosines for the glycines at positions 190 and 198 revives the ACh response. Apparently, having the triad (αY190, αY198 and αW149) is all that is needed to bind and get energy from the agonist affinity change.
Two results suggest that ‘capping’ of loop C over the binding site is not of fundamental importance for initiating the gating isomerization in skeletal muscle AChRs. First, the deletion of loop C has almost no effect on gating in the absence of agonists. The capping event, if it occurs in AChRs, it not necessary for the global gating isomerization to take place. The second result is with regard to agonist binding. Experiments show that the ACh ‘on’ rate constant is faster to O than to C, but that the ‘off’ rate constant is much slower [10, 21, 22]. That is, the higher affinity of O is cause mainly by slower dissociation of the neurotransmitter. Closing a lid (capping) would be expected slow both ‘on’ and ‘off’ rate constants and therefore does not appear to be the structural correlate of the high O-affinity.
The other two Core aromatics are αY93 and ε/δ/γW55 (it is position 57 in δ). I call these the ‘special pair’ because, unlike the triad, they behave quite differently at the three kinds of agonist site (Fig. 4). F/A substitutions of αY93, or an A substitution of W55, have small effects (<5 fold decrease in the coupling constant) at αδ and αε. However, these mutations have much larger effects at αγ. The γW55A mutation makes GB1ACh more positive by a whopping +4.3 kcal/mol at αγ, whereas removing this indole actually makes the extra ACh binding energy more negative (favorable) by −0.5 kcal at αδ. The effect at αε is intermediate. In fetal AChRs, approximately 30% of the total energy from 2 ACh affinity-changes can be attributed to the action of γW55. The αY93A substitution has about twice the effect αγ vs αδ/αε.
Figure 4. Breaking down binding energy at the Core.
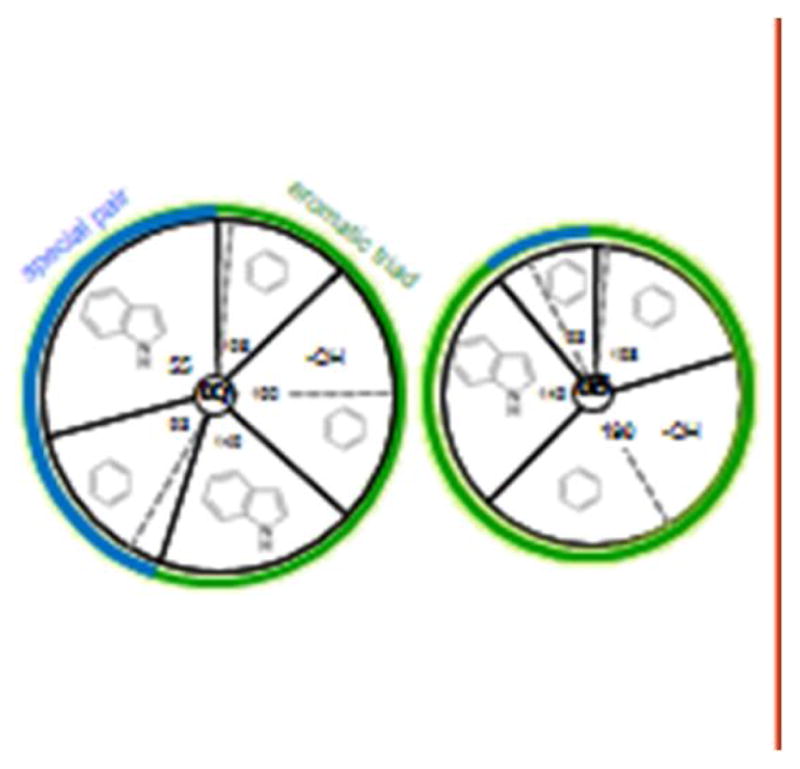
The area of each slice is approximately equal to the loss in favorable ACh binding free energy with the removal of just that group. Dashed gray lines separate −OH and ring contributions of tyrosines. The special pair makes a larger contribution at the fetal αγ site, whereas the action of the aromatic triad is similar at all sites (αε is like αδ).
Another reason why this residue pair is special is because their side chains are tightly coupled. Whereas at the αγ site αY93A and γW55A each make GB1 more positive by +2.8 and +4.5 kcal/mol, pairing the two mutations changes GB1 only by +5.2 kcal/mol, which is ~70% of the energy sum. The rings of γW55 and αY93 (the −OH has little effect) interact favorably, by −2.1 kcal/mol. This means that removing either one of the rings interferes with the other’s ability to stabilize ACh. The special pair residues αY93 and γW55 appear to act in concert at the αγ site.
To summarize, the agonist sites operate independently but can be different with regard to agonist energy (αγ>αδ≈αε). The Core binding site apparatus has two working parts, the aromatic triad (that joins loops C and B) and the special pair (that joins loop A and strand β2 of the complimentary subunit). These two components do not operate completely independently, as mutations of special pair residue γW55 and triad residue αY190 (but not αY198 or αW149) are coupled energetically. In adult AChRs, interactions of the QA of ACh with the triad, in particular αY190, are the primary sources of agonist energy. The aromatic groups of the special pair do not provide much binding energy at adult sites, but they swing into action in a big way at the fetal αγ site, in particular γW55.
There are several other interesting residues that are near the agonist site, just outside the Core. Two notable ones are αG153 (in loop B) and ε/δP121 (in the complementary β6 strand). Neither of these affect the coupling constant (with an alanine substitution) and both happen to be exceptions to the relationship GB1=0.59lnKd [15]. Mutations of αG153 increase Kd and Jd to similar extents, so that their ratio (the coupling constant) remains unchanged [23]. The increase in Kd caused by the αG153K mutation, which is present in some neuronal AChRs, is particularly large for nicotine. Most mutations of the triad and pair do not alter the allosteric constant substantially, but those of αG153 (and αW149) are exceptions. With regard to ε/δP121, most mutations here have little effect on either the allosteric or coupling constant, except for L (which causes CMS; [24]) and R. Either of these substitutions knocks-out the local site by lowering its affinity [25]. An unusual aspect of ε/δP121 mutations is that the change in the coupling constant is larger than expected given the change in Kd. αG153 and ε/δP121 are interesting parts of the binding site apparatus, even if they are not part of the Core.
The Core aromatics are the same at αγ vs. αδ, yet the special pair behaves differently at each site. Apparently, the complementary, non-α side of the binding pocket influences the action of the special pair (and to a lesser extent, αW149). The non-α surface is a β-sheet comprised of the β5-5′ linker, strand β5′, loop E, strand β6, loop D and strand β2 (Fig. 2). It will be interesting to learn exactly how structural differences between complementary subunits influence action of the special pair.
Simulations
There are no high-resolution structures of liganded nicotinic AChR transmitter binding sites, and none of agonist sites in low- vs. high-affinity conformations. AChBP does not appear to undergo an affinity switch, but this protein has a KdACh that is similar to that of the resting mouse muscle αγ site (4 vs 8 μM). To gain some sense of the structural correlates of the different GB1 changes consequent to Core mutations, members of the lab built homology models of the AChR extracellular domain based on AChBP, docked ACh and carried out short (20 ns) molecular dynamic (MD) simulations to estimate binding energies [26]. These calculations provide enthalpy estimates, but these can be compared to in vivo free energies because for some agonists there is only a small entropic component to affinity [27].
The simulated energies and those measured experimentally from single-channels agree. The simulated ACh energy was ~40% larger at αγ compared to αδ≈αε (Fig. 5). Snapshots of simulated structures suggest that the αγ site is more compact than αδ, with the tryptophans more orthogonal and αW149 and the special pair closer to the QA. The two loop C tyrosine ‘pincers’ have a similar configuration at all 3 kinds of agonist site, consistent with their similar in vivo energy contributions. Simulated and experimental affinity estimates also were in agreement for another agonist (TMA) at WT sites, and for ACh in receptors having an alanine substitution of a Core aromatic residue, including the substantial structural and massive energy differences between W55 at αγ vs. αδ. These results indicate that with regard to affinity-estimation, AChBP is a good model for the muscle AChR agonist sites (the full receptor structure is not necessary) and that using standard force fields and brief MD simulations are adequate.
Figure 5. MD simulations.

a. In silico and in vivo ACh binding energies match. The histogram entries are energies calculated from the MD trajectories and the white circles are the corresponding free energies estimated from single-channel currents. b. Representative snapshots from the MD trajectories. The different positions of aromatic triad and special pair residues in αγ (green) vs. αδ (gray) are consistent with the in vivo energy differences (Fig. 4). Blue ball, quaternary ammonium (QA) group of ACh.
Simulations, in combination with energy measurements from electrophysiology, may prove to be useful in addressing some unanswered questions. We are beginning to understand the components of affinity at the resting binding sites, but it is also important to understand those at activated, high-affinity sites. Resting and active agonist binding energies are correlated, so it is likely that there are common rearrangements. It may be possible to use simulations and experiments in combination to identify structural elements in the complementary subunit that influence the action of the special pair, and to explore the reasons for the effects of the −OH group of αY190, the two loop B glycines and other α subunit amino acids outside the Core that influence its behavior. The excellent agreement between the energy estimates from MD simulations and single-molecule kinetic analyses provides optimism that comparisons between simulations and single-channel experiments can be applied to probe different ligands, neuronal AChR agonist sites and beyond. Matching energies from simulations and experiments, to deconstruct the agonist site apparatus and identify sources of affinity, promises to be useful approach for engineering agonists, receptors and synapses.
Acknowledgments
I thank J Howe, TK Nayak, P Purohit and S Gupta for comments on the manuscript. Supported by NIH NS 064969.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Franke C, Parnas H, Hovav G, Dudel J. A molecular scheme for the reaction between acetylcholine and nicotinic channels. Biophysical journal. 1993;64:339–56. doi: 10.1016/S0006-3495(93)81374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papke RL. Merging old and new perspectives on nicotinic acetylcholine receptors. Biochemical pharmacology. 2014;89:1–11. doi: 10.1016/j.bcp.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson MB. Kinetics of unliganded acetylcholine receptor channel gating. Biophysical journal. 1986;49:663–72. doi: 10.1016/S0006-3495(86)83693-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Purohit P, Gupta S, Jadey S, Auerbach A. Functional anatomy of an allosteric protein. Nat Commun. 2013;4:2984. doi: 10.1038/ncomms3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jadey SV, Purohit P, Bruhova I, Gregg TM, Auerbach A. Design and control of acetylcholine receptor conformational change. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4328–33. doi: 10.1073/pnas.1016617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou M, Engel AG, Auerbach A. Serum choline activates mutant acetylcholine receptors that cause slow channel congenital myasthenic syndromes. PNAS. 1999;96:10466–71. doi: 10.1073/pnas.96.18.10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jadey S, Auerbach A. An integrated catch-and-hold mechanism activates nicotinic acetylcholine receptors. The Journal of general physiology. 2012;140:17–28. doi: 10.1085/jgp.201210801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Purohit P, Auerbach A. Unliganded gating of acetylcholine receptor channels. Proceedings of the National Academy of Sciences. 2009;106:115–20. doi: 10.1073/pnas.0809272106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Purohit P, Bruhova I, Auerbach A. Sources of energy for gating by neurotransmitters in acetylcholine receptor channels. Proceedings of the National Academy of Sciences. 2012;109:9384–9. doi: 10.1073/pnas.1203633109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nayak TK, Auerbach A. Asymmetric transmitter binding sites of fetal muscle acetylcholine receptors shape their synaptic response. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:13654–9. doi: 10.1073/pnas.1308247110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grosman C, Zhou M, Auerbach A. Mapping the conformational wave of acetylcholine receptor channel gating. Nature. 2000;403:773. doi: 10.1038/35001586. [DOI] [PubMed] [Google Scholar]
- 12.Auerbach A. A statistical analysis of acetylcholine receptor activation in Xenopus myocytes: stepwise versus concerted models of gating. J Physiol. 1993;461:339–78. doi: 10.1113/jphysiol.1993.sp019517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lape R, Colquhoun D, Sivilotti LG. On the nature of partial agonism in the nicotinic receptor superfamily. Nature. 2008;454:722–7. doi: 10.1038/nature07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakmann B, Patlak J, Neher E. Single acetylcholine-activated channels show burst-kinetics in presence of desensitizing concentrations of agonist. Nature. 1980;286:71–3. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- 15.Purohit P, Bruhova I, Gupta S, Auerbach A. Catch-and-hold activation of muscle acetylcholine receptors having transmitter binding site mutations. Biophys J. 2014;107:88–99. doi: 10.1016/j.bpj.2014.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jha A, Auerbach A. Acetylcholine receptor channels activated by a single agonist molecule. Biophysical journal. 2010;98:1840–6. doi: 10.1016/j.bpj.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nayak TK, Bruhova I, Chakraborty S, Gupta S, Zheng W, Auerbach A. Functional differences between neurotransmitter binding sites of muscle acetylcholine receptors. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:17660–5. doi: 10.1073/pnas.1414378111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purohit P, Auerbach A. Glycine Hinges with Opposing Actions at the Acetylcholine Receptor-Channel Transmitter Binding Site. Molecular Pharmacology. 2011;79:351–9. doi: 10.1124/mol.110.068767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olsen JA, Balle T, Gajhede M, Ahring PK, Kastrup JS. Molecular recognition of the neurotransmitter acetylcholine by an acetylcholine binding protein reveals determinants of binding to nicotinic acetylcholine receptors. PLoS One. 2014;9:e91232. doi: 10.1371/journal.pone.0091232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA, Dougherty DA. From ab initio quantum mechanics to molecular neurobiology: a cation-π binding site in the nicotinic receptor. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:12088–93. doi: 10.1073/pnas.95.21.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purohit P, Auerbach A. Loop C and the mechanism of acetylcholine receptor-channel gating. The Journal of general physiology. 2013;141:467–78. doi: 10.1085/jgp.201210946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grosman C, Auerbach A. The dissociation of acetylcholine from open nicotinic receptor channels. Proceedings of the National Academy of Sciences. 2001;98:14102–7. doi: 10.1073/pnas.251402498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jadey S, Purohit P, Auerbach A. Action of nicotine and analogs on acetylcholine receptors having mutations of transmitter-binding site residue alphaG153. The Journal of general physiology. 2013;141:95–104. doi: 10.1085/jgp.201210896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohno K, Wang HL, Milone M, Bren N, Brengman JM, Nakano S, et al. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron. 1996;17:157–70. doi: 10.1016/s0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- 25.Gupta S, Purohit P, Auerbach A. Function of interfacial prolines at the transmitter-binding sites of the neuromuscular acetylcholine receptor. The Journal of biological chemistry. 2013;288:12667–79. doi: 10.1074/jbc.M112.443911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nayak TK, Bruhova I, Chakraborty S, Gupta S, Zheng W, Auerbach A. Functional differences between neurotransmitter binding sites of muscle acetylcholine receptors. Proceedings of the National Academy of Sciences of the United States of America; 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta S, Auerbach A. Temperature Dependence of Acetylcholine Receptor Channels Activated by Different Agonists. Biophysical journal. 2011;100:895–903. doi: 10.1016/j.bpj.2010.12.3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elenes S, Auerbach A. Desensitization of diliganded mouse muscle nicotinic acetylcholine receptor channels. The Journal of physiology. 2002;541:367–83. doi: 10.1113/jphysiol.2001.016022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Purohit P, Auerbach A. Unliganded gating of acetylcholine receptor channels. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:115–20. doi: 10.1073/pnas.0809272106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41:907–14. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]