

Abstract
HIV-1 Nef and the unrelated murine leukemia virus glycoGag strongly enhance the infectivity of HIV-1 virions produced in certain cell types in a clathrin-dependent manner. Here we show that Nef and glycoGag prevent the incorporation of the multipass transmembrane proteins SERINC3 and SERINC5 into HIV-1 virions to an extent that correlates with infectivity enhancement. Silencing of SERINC3 together with SERINC5 precisely phenocopied the effects of Nef and glycoGag on HIV-1 infectivities. The infectivity of nef-deficient virions increased more than 100-fold when produced in double-knockout human CD4+ T cells that lack both SERINC3 and SERINC5, and re-expression experiments confirmed that the absence of SERINC3 and SERINC5 accounted for the infectivity enhancement. Furthermore, SERINC3 and SERINC5 together restricted HIV-1 replication, and this restriction was evaded by Nef. SERINC3 and SERINC5 are highly expressed in primary human HIV-1 target cells, and inhibiting their downregulation by Nef is a potential strategy to combat HIV/AIDS.
Nef is an accessory protein encoded by HIV-1 and other primate lentiviruses. In vivo, Nef is a major pathogenicity determinant that is required for high virus loads1–3. Although not essential for virus replication in cell culture, Nef enhances virus spreading in primary CD4+ T cells, particularly when such cells are infected prior to mitogenic stimulation4–6. Nef robustly down-regulates the viral entry receptor CD4 from the surface of virus-producing cells by inducing its clathrin-dependent endocytosis and subsequent lysosomal degradation7–10. A recent study suggests that an important physiological function of CD4 down-regulation by Nef is to prevent the CD4-induced exposure of epitopes in HIV-1 envelope (Env) that make infected cells susceptible to antibody-dependent cell-mediated cytotoxicity11. Apart from CD4, Nef down-regulates multiple other cell surface proteins12. The selective down-modulation of HLA-A and HLA-B but not of HLA-C by Nef serves to protect infected cells both from cytotoxic T cells and from natural killer cells13–15. The Nef proteins of most primate lentiviruses also down-modulate the T cell receptor complex, which is thought to protect infected T cells from activation-induced cell death in non-pathogenic natural SIV infections16. This function of Nef was lost in HIV-1 and closely related viruses, which may contribute to the pathogenicity of HIV-1 in humans16.
One of the most conserved yet poorly understood functions of Nef is the enhancement of progeny virion infectivity17, 18. Although Nef exerts its effect on HIV-1 infectivity in virus producer cells, it does not detectably affect virus morphogenesis or maturation19–22. Nevertheless, progeny virions produced in the absence of Nef do not efficiently reverse transcribe their genome in target cells19, 20. It has been reported that high levels of cell surface CD4 inhibit the release or infectivity of HIV-1 progeny virions, and that Nef relieves these effects23, 24. However, the enhancement of HIV-1 infectivity depends on residues within Nef that are dispensable for its ability to down-regulate CD425. Furthermore, Nef enhances HIV-1 progeny virion infectivity even when CD4 is not expressed or cannot be down-regulated17, 19, 20. Finally, the glycosylated Gag (glycoGag) protein of Moloney murine leukemia virus (MLV) closely mimics the effect of Nef on HIV-1 infectivity, even though glycoGag does not down-regulate CD426. MLV glycoGag is an accessory protein whose translation begins at an inefficient CTG start codon upstream and in-frame with the gag gene27. The resulting product is a type II transmembrane protein with an N-terminal cytosolic non-Gag portion and an extracellular Gag domain28. The potent Nef-like activity of glycoGag on HIV-1 infectivity resides entirely in its cytosolic domain, which is unrelated to Nef29. Nevertheless, the effects of Nef and glycoGag on HIV-1 infectivity appear mechanistically related. Both are similarly dependent on the producer cell type26, are similarly determined by variable regions of HIV-1 Env30, and exhibit a similar reliance on clathrin-mediated endocytosis29, 31, 32. However, the molecular basis for these similarities remains unknown.
Nef inhibits the incorporation of SERINCs
Because of the essential role of the endocytic machinery in the enhancement of HIV-1 infectivity by Nef or glycoGag, we examined the possibility that both proteins down-regulate a restriction factor that gets incorporated into assembling virions in their absence. To identify factors whose incorporation is prevented by both Nef and glycoGag, we conducted a proteomic analysis of OptiPrep gradient-purified virions produced by T lymphoid cells infected with wild type (WT; Nef+) or Nef− HIV-1NL43, or with a version that encodes a fully active minimal glycoGag (termed glycoMA30) instead of Nef (Extended Data Fig. 1a). The only host protein that could reproducibly be identified in Nef− virion samples in independent experiments but was not identified in any Nef+ or glycoMA virion sample was serine incorporator 3 (SERINC3), a member of a family of putative carrier proteins with at least 10 transmembrane domains33 (Extended Data Fig. 1b). In one experiment, STOM and PFKP were also identified in Nef− but not in Nef+ or glycoMA virion samples (Extended Data Fig. 1b). However, in another experiment STOM was identified in all virions samples, and PFKP was not identified in any sample. Thus, STOM and PFKP were not further pursued.
Immunoblotting of virion samples confirmed that the incorporation of HA-tagged SERINC3 is strongly inhibited by the Nef proteins of several laboratory-adapted and primary HIV-1 isolates from different clades (Fig. 1a) and by glycoMA (Extended Data Fig. 2a). Furthermore, the effects of glycoMA truncation mutants on the incorporation of SERINC3-HA (Extended Data Fig. 2a) correlated closely with their abilities to enhance HIV-1 infectivity29. Two of the Nef proteins tested did not inhibit the incorporation of SERINC3-HA (Fig. 1a), and one of these (Nef90CF056) also had no effect on HIV-1 infectivity (Fig. 1c). Because the other (NefSF2) did enhance HIV-1 infectivity (Fig. 1c), we examined its effect on the incorporation of other human SERINC family members. Although NefSF2 did not affect the incorporation of SERINC3-HA (Fig. 1a), it strongly inhibited the incorporation of SERINC5-HA (Fig. 1b). Among the primary Nefs examined, those that were most active in enhancing HIV-1 infectivity (Nef97ZA012 and Nef93BR020) strongly inhibited the incorporation both of SERINC3 and of SERINC5, the less active Nef94UG114 was a less effective inhibitor particularly of SERINC5 incorporation, and the inactive Nef90CF056 inhibited neither SERINC3 nor SERINC5 incorporation (Fig. 1a–c). Like the most active Nefs, WT glycoMA, which enhances HIV-1 infectivity at least as potently30, also strongly inhibited the incorporation both of SERINC3 and of SERINC5 (Extended Data Fig. 2a, b). Further, the effects of glycoMA truncation mutants on SERINC5 incorporation (Extended Data Fig. 2b), like those on SERINC3 incorporation (Extended Data Fig. 2a), correlated with their effects on HIV-1 infectivity enhancement29.
Figure 1. Inhibition of incorporation of SERINC proteins into HIV-1 virions by Nef correlates with infectivity enhancement.
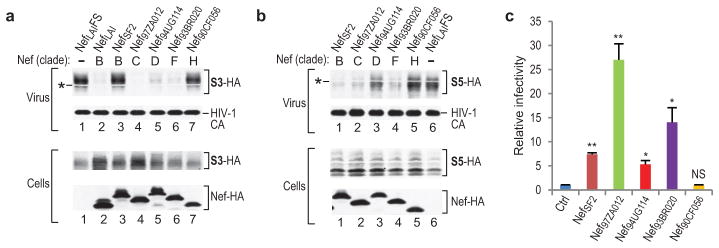
a, b, Western blots showing the effects of Nef proteins from various HIV-1 clades on the incorporation of SERINC3-HA (S3-HA) (a) or SERINC5-HA (S5-HA) (b) into Nef− HIV-1 virions. The white bands marked by asterisks are caused by co-migrating HIV-1 Pr55gag. The experiment shown in (a) was performed twice. Supplementary Information contains full scans for (a, b). c, Ability of Nef proteins from different HIV-1 clades to enhance HIV-1 infectivity. Env−/Nef− HIV-1HXB2 particles trans-complemented with EnvHXB2 were produced in JurkatTAg cells in the absence or presence of the indicated Nef proteins, and infectivities normalized for p24 antigen were determined using TZM-bl indicator target cells (n =3). Bars, mean with s.d. *P < 0.05, **P < 0.01, NS, not significant (P > 0.05), two-tailed unpaired t-test with Welch’s correction in case of unequal variance (F-test, α = 0.025).
Subcellular localization of SERINC5
SERINC5-mCherry clearly localized to the plasma membrane and to filopodia-like protrusions when expressed alone, but accumulated in perinuclear vesicles when co-expressed with Nef or glycoGag (Extended Data Fig. 3a, and data not shown). Furthermore, SERINC5(iHA), which harbors an internal HA tag next to a conserved consensus glycosylation site within a proposed extracellular loop34, could be readily detected on the surface of transfected JurkatTAg T lymphoid cells by flow cytometry, and its surface expression was greatly reduced when either NefSF2 or glycoGag were co-expressed (Extended Data Fig. 3b). We infer that Nef and glycoGag decrease the virion-association of SERINC5 by decreasing its cell surface levels.
Effects of exogenous SERINCs on HIV infectivity
HIV-1 virions produced in Jurkat T lymphoid cells are more dependent on Nef for optimal infectivity than virions produced in 293T cells26, which in turn are more dependent on Nef than virions produced in exceptionally permissive MT4 cells35. Interestingly, the relative requirement for Nef correlates with SERINC5 mRNA levels, which are high in Jurkat cells, lower in 293T cells, and lower yet in MT4 cells (Extended Data Fig. 4a, b). SERINC5 mRNA levels in unstimulated or stimulated human PBMC were even higher than in Jurkat cells, and were not further increased by treatment with interferon-α (INF-α) (Extended Data Fig. 4b, c).
In single cycle replication assays, exogenous SERINC5 reduced the specific infectivity of Nef− HIV-1 virions produced in 293T cells for TZM-bl indicator target cells >100-fold, even when as little as 100 ng of the relatively weak pBJ5-based SERINC5 expression vector was used (Fig. 2a). Under the same conditions, exogenous SERINC3 reduced progeny virus infectivity only 2- to 3-fold (Fig. 2a). However, while endogenous SERINC5 mRNA levels in 293T cells are low, these cells have relatively high endogenous SERINC3 mRNA levels (Extended Data Fig. 4b). Even at 500 ng, the vectors expressing SERINC3 or SERINC5 did not affect virus particle production, Gag processing, or HIV-1 Env incorporation (Fig. 2b). However, late reverse transcriptase (RT) products in target cells exposed to Nef− HIV-1 virions produced in 293T cells transfected with 500 ng of the SERINC5 expression vector were reduced >100-fold (Fig. 2c).
Figure 2. Effects of exogenous SERINCs on HIV-1 infectivity.

a, Overexpression of SERINC5 in virus producer cells dramatically reduces Nef− HIV-1 progeny virus single-round infectivity. The effects of exogenous SERINC3 (S3) and SERINC5 (S5) were measured using TZM-bl indicator cells (n = 3). b, Western blots showing that virus production, Gag processing, and gp41 (Env) incorporation were unaffected. c, Nef− HIV-1 progeny virions produced in the presence of exogenous SERINC5 are defective in the synthesis of late RT products (n = 2). d, Effects of exogenous SERINCs on the single-round infectivity of VSV G-pseudotyped Nef− HIV-1 virions measured as in Fig. 2a (n = 3). e, VSV G reduces the association of SERINC5-HA (S5-HA) with Env− HIV-1 virions. The HIV-1 proviral plasmid in lane 1 has a disrupted gag gene. This experiment was repeated twice. Supplementary Information contains full scans for (b, e). f, NefSF2 and glycoGag expressed in trans in virus producer cells counteract the effect of exogenous SERINC5 on Nef− HIV-1 progeny virion infectivity (n = 3). Bars, mean with s.d. *P < 0.05, **P < 0.01, NS, not significant (P > 0.05), two-tailed unpaired t-test with Welch’s correction in case of unequal variance.
To examine whether SERINC5 affects HIV-1 virion fusion with target cells, we co-expressed a chimeric β-lactamase-Vpr (BlaM-Vpr) protein that is taken up into virions36. Fusion was then quantified based on the cleavage of a fluorescent substrate following the transfer of BlaM-Vpr from virions into target cells. We initially used 1 μg of the SERINC5 expression vector to compensate for potential competition by the strong promoter driving BlaM-Vpr expression, and found that the ability of Nef− HIV-1 progeny virions to fuse with target cells was largely abolished (Extended Data Fig. 5). However, 100 ng of the SERINC5 expression vector, which reduced the infectivity of Nef− HIV-1 virions ~20-fold when co-transfected with the BlaM-Vpr expression vector, caused only an ~4-fold reduction in the ability to fuse with target cells (Extended Data Fig. 5).
The effects on HIV-1 infectivity were specific, because 500 ng of the vectors expressing SERINC3 or SERINC5 had at most modest effects on the infectivities of Env− HIV-1 particles pseudotyped with the vesicular stomatitis virus G protein (VSV G) (Fig. 2d), which do not require Nef or glycoGag for optimal infectivity26, 37, 38. Surprisingly, the incorporation of HA-tagged SERINC5 into Env− HIV-1 particles was reduced in the presence of VSV G (Fig. 2e). Thus, reduced incorporation may have contributed to the relative resistance of VSV G-pseudotyped HIV-1 to exogenous SERINC5. Crucially, the effect of exogenous SERINC5 on Nef− HIV-1 was counteracted by NefSF2 or glycoGag expressed in trans (Fig. 2f). Indeed, exogenous SERINC5 had no effect whatsoever in the presence of glycoGag (Fig. 2f). In two independent experiments performed with cells from different donors, exogenous SERINC5 also greatly reduced the infectivity of Nef− HIV-1 virions produced in 293T cells for primary human target cells (Extended Data Fig. 6).
Effects of SERINC depletion on HIV infectivity
JurkatTAg T lymphoid cells express both SERINC3 and SERINC5 at relatively high levels (Extended Data Fig. 4b). Short interfering RNAs (siRNAs) that knocked down HA-tagged SERINC3 or SERINC5 (Fig. 3a) enhanced the specific infectivity of Nef−, EnvHXB2-pseudotyped HIV-1 particles produced in JurkatTAg cells by >4- or >8-fold, respectively (Fig. 3b). In five independent experiments, both siRNAs together enhanced the specific infectivity of Nef− progeny virions 18- to 45-fold (Fig. 3b, d, and data not shown). The siRNAs against SERINC3 and SERINC5 together also significantly enhanced the infectivity of Nef−, Env89.6-bearing HIV-1 virions produced by infected primary macrophages (Fig. 3c). However, they had little effect on the already high specific infectivity of Nef− progeny virions produced in JurkatTAg cells that was observed when Nef97ZA012 or glycoGag were expressed in trans (Fig. 3d).
Figure 3. Effects of depleting SERINCs in virus producer cells.

a, Depletion of HA-tagged SERINCs in JurkatTAg cells by specific siRNAs. b, Single-round infectivities of Nef− HIV-1 virions produced in JurkatTAg cells (n = 3) subjected to non-targeting siRNA or to siRNAs targeting SERINC3 (si_S3), SERINC5 (si_S5), or both (si_S3+5). c, Single-round infectivities of Nef− HIV-1 virions produced in primary MDM subjected to siRNAs (n = 3). d, Simultaneous depletion of SERINC3 and SERINC5 has negligible effects on Nef− HIV-1 progeny virion infectivity when Nef or glycoGag are provided in trans (n = 3). e, The effects of depleting SERINC3 together with SERINC5 on virus infectivity are governed by the same determinants in gp120 V1/V2 that govern Nef-responsiveness (n = 3). f, Western blots showing that the combined siRNAs targeting SERINC3 and SERINC5 did not affect particle production, Gag processing, or Env incorporation. Supplementary Information contains full scans for (a, f). Bars, mean with s.d. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, NS, not significant (P > 0.05) two-tailed unpaired t-test with Welch’s correction in case of unequal variance. The experiments shown in (a) and (b) were performed twice.
The Env proteins of the R5-tropic primary HIV-1 isolates SF162 and JRFL differ considerably in their responsiveness to Nef or glycoGag, which is determined by variable regions 1 and 2 (V1/V2) of gp12030. Remarkably, the siRNAs against SERINC3 and SERINC5 together precisely phenocopied the differential effects of Nef97ZA012 on the specific infectivities of Nef− HIV-1 progeny particles bearing EnvSF162 or EnvJRFL (Fig. 3e). Furthermore, responsiveness to Nef97ZA012 and to the siRNAs targeting SERINC3 and SERINC5 could be switched simultaneously by exchanging the V1/V2 regions of EnvSF162 and EnvJRFL (Fig. 3e). Nef− HIV-1 virions bearing Envs that differed profoundly in their responsiveness to Nef or to SERINC depletion incorporated comparable amounts of SERINC5-HA (Extended Data Fig. 7). Furthermore, Nef97ZA012 largely prevented the incorporation of SERINC5-HA in the presence of both Envs (Extended Data Fig. 7). Importantly, the simultaneous depletion of SERINC3 and SERINC5 in virus producer cells affected neither particle morphogenesis nor Env incorporation (Fig. 3f). Collectively, these data imply that SERINC3 and SERINC5 together account for the effects of Nef and glycoGag on HIV-1 infectivity.
HIV infectivity and replication in SERINC KO cells
Next, we knocked out the SERINC3 and SERINC5 genes in JurkatTAg cells using the CRISPR/Cas9 system39 (Extended Data Fig. 8). Nef−, EnvHXB2-pseudotyped HIV-1 particles produced in JurkatTAg clones lacking either SERINC3 or SERINC5 were ~5-fold or 13- to 20-fold more infectious, respectively, than particles produced in the parental cells (Fig. 4a). Strikingly, the specific infectivity of particles produced in double-knockout (KO) cells lacking SERINC3 and SERINC5 was >100-fold higher (Fig. 4a). Furthermore, the markedly increased specific infectivity of viral particles produced in double-KO cells could be confirmed by visualizing GFP-positive cells following exposure to recombinant HIV-1 expressing GFP (Fig. 4b). Nef and glycoGag potently enhanced the specific infectivity of particles produced in parental JurkatTAg cells as expected, but had no significant effects on the already highly infectious particles produced in double-KO cells (Fig. 4c). The introduction of expression cassettes for SERINC3, for SERINC5, and for both, into the double-KO cells via retroviral transduction led to 3.6-fold, 5.7-fold, and 32-fold reductions, respectively, in the specific infectivities of Nef− HIV-1 particles produced in these cells (Fig. 4d). These data confirm that SERINC3 and SERINC5 synergistically restrict HIV-1 infectivity in the absence of Nef.
Figure 4. Effects of SERINC knockout and reconstitution on HIV-1 infectivity.

a, Single-round infectivities of Nef− HIV-1 progeny virions produced in parental or in KO JurkatTAg cells lacking SERINC3, SERINC5, or both (n = 3). b, TZM-bl cells were incubated with equal amounts of single-cycle Nef− HIV-1-GFP produced in parental or double-KO cells lacking SERIN3 and SERINC5. Infected TZM-bl cells expressing GFP were detected by fluorescence microscopy. c, Effects of Nef and glycoGag provided in trans on the single-round infectivities of Nef− HIV-1 virions produced in parental or double-KO cells (n = 3). d, Effects of introducing expression cassettes for SERINC3, SERINC5, or both into the double-KO cells on Nef− HIV-1 progeny virus infectivities (n = 3). Bars, mean with s.d. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, NS, not significant (P > 0.05) two-tailed unpaired t-test with Welch’s correction in case of unequal variance. The experiments shown in (a) and (b) were repeated three times.
The effects of Nef on HIV-1 replication in cell lines have generally been more modest than in primary lymphocytes4, 6. However, apart from Jurkat cells, T cell lines often express only low levels of SERINC5 mRNA (Extended Data Fig. 4 and data not shown). We observed that at low input virus concentrations NefNL43 and Nef97ZA012 robustly enhanced HIV-1 spreading in highly permissive Jurkat E6.1 cells (Fig. 5a). NefNL43 and Nef97ZA012 also enhanced HIV-1 replication in JurkatTAg cells, as judged from Gag protein expression levels in the infected cells and from the release of p24 antigen over time (Fig. 5b, c). In marked contrast, the Nef+ and Nef− viruses replicated with similar kinetics in double-KO JurkatTAg cells lacking SERINC3 and SERINC5, which were generally more permissive than the parental cells (Fig. 5b, c). Crucially, the requirement for Nef was restored in double-KO cells reconstituted with SERINC3 and SERINC5 expression cassettes (Fig. 5b, c). While the levels of SERINC3 and SERINC5 in the reconstituted cells were somewhat higher than in the parental cells (Extended Data Fig. 9), they were comparable to those in human PBMC (Extended Data Fig. 4).
Figure 5. Nef counteracts inhibition of HIV-1 replication by SERINC3 and SERINC5.
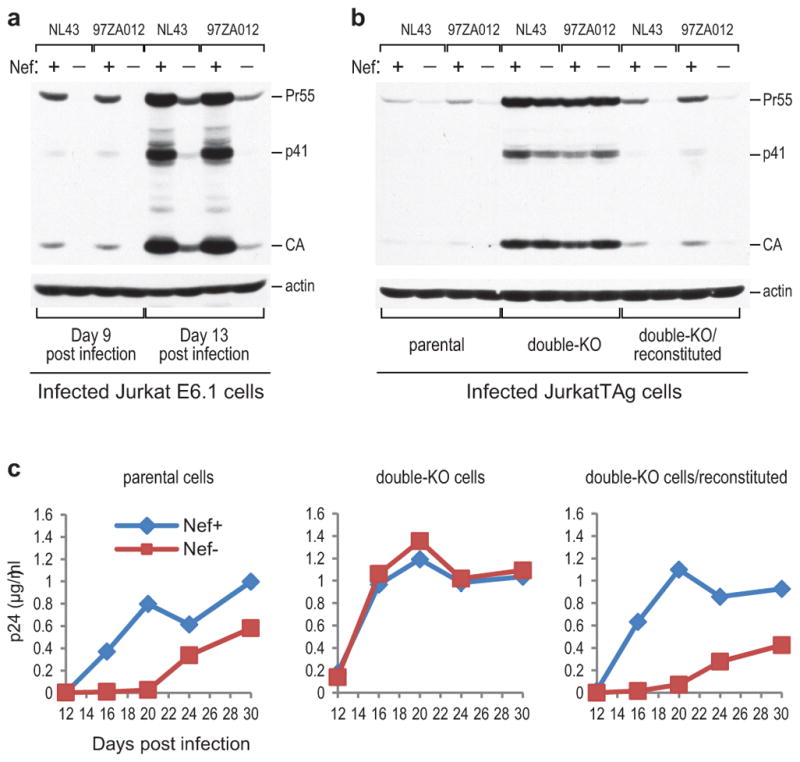
a, Effect of Nef on HIV-1 spreading in Jurkat E6. 1 cells infected at a low input virus concentration (100 pg p24/ml). Gag protein expression in the cultures 9 and 13 days post infection was examined by Western blotting as a measure of virus replication. b, c, Effects of Nef on virus spreading in parental JurkatTAg cells, double-KO cells lacking SERINC3 and SERINC5, and SERINC3 +SERINC5-reconstituted double-KO cells. The spreading of HIV-1NL43-based viruses encoding either WT or disrupted versions of NefNL43 or Nef97ZA012 was examined by Western blotting of cell lysates with anti-CA antibody 9 days post infection with 20 ng p24/ml (b), or by monitoring p24 accumulation in the supernatants after infection with 4 ng p24/ml (c). Relatively high input virus concentrations were used to compensate for low CD4 levels on JurkatTAg cells. The experiment shown in (b) was repeated twice. Supplementary Information contains full scans for (a, b).
Although endogenous CD4 levels in JurkatTAg cells are low, similar results were obtained with more permissive CD4high versions generated by retroviral transduction. In the presence of extra CD4, Nef again clearly enhanced virus replication in parental JurkatTAg cells, but was entirely dispensable in double-KO cells lacking SERINC3 and SERINC5 (Extended Data Fig. 10). Furthermore, the role of Nef in virus replication was restored upon reconstitution of SERINC3 and SERINC5 expression in the double-KO cells (Extended Data Fig. 10). These results demonstrate that SERINC3 and SERINC5 together restrict HIV-1 replication, and that Nef antagonizes this restriction.
Discussion
Our findings reveal that HIV-1 Nef and MLV glycoGag efficiently down-regulate SERINC3 and SERINC5 form the cell surface, which prevents their incorporation into HIV-1 virions and consequently counteracts their inhibitory effect on HIV-1 infectivity. Importantly, these findings offer an explanation for why the enhancement of HIV-1 infectivity by Nef and glycoGag is highly dependent on dynamin 2, clathrin, and the AP-2 clathrin adapter complex29, 31. SERINC family members are present in all eukaryotes, but their functions remain largely unknown. SERINCs reportedly enhance the incorporation of serine into phosphatidylserine and sphingolipids33. In principle, this activity could affect the lipid composition of the viral envelope, which is considered crucial for virion infectivity40. Our data demonstrate that SERINC3 and SERINC5 together account for most if not all of the effects of Nef on HIV-1 infectivity and on HIV-1 replication in JurkatTAg cells. Notably, Nef enhances HIV-1 infectivity and stimulates HIV-1 replication in human peripheral blood mononuclear cells (PBMC)4–6, 18, 41, whose SERINC3 and SERINC5 mRNA levels exceed those of Jurkat cells (Extended Data Fig. 4).
The ability of virions produced in the absence of Nef to reverse transcribe their genome in target cells is impaired17, 19, 20. Consistent with these observations, we find that SERINC5 in virus producer cells strongly inhibits the ability of Nef− HIV-1 virions to complete reverse transcription. We also find that SERINC5 can in principle abolish the ability of progeny HIV-1 virions to fuse with target cells. However, lower levels of SERINC5 inhibited the fusion step to a lesser extent than the ability of progeny virions to productively infect target cells. Although there is controversy regarding the effect of Nef on HIV-1 entry35, 42, 43, a two-fold reduction in the ability of Nef− HIV-1 virions to fuse with target cells was noted in one study35. In all of these studies, virus was produced in 293T cells, whose endogenous SERINC5 mRNA levels are low (Extended Data Fig. 4). It is conceivable that relatively low levels of virion-associated SERINC5 impair primarily fusion pore enlargement, which poses a higher energy barrier to overcome than pore formation44. This would be expected to impair passage of the viral core but not necessarily of the much smaller BlaM-Vpr fusion indicator into target cells. Consistent with a role in entry, Nef enhances the cytoplasmic delivery of viral cores45. Further, the requirement for Nef is determined by HIV-1 Env30.
Interestingly, the Env proteins of HIV-1NL43 and HIV-1SF162, which are highly responsive to Nef and glycoGag, require a higher number of Env trimers to complete entry than the poorly Nef/glycoGag-responsive EnvJRFL30, 46. Furthermore, the naturally occurring N160K mutation in the V2 loop of HIV-1 Env, which results in the loss of a glycosylation site, can increase both the responsiveness to Nef and glycoGag and the stoichiometry of entry30, 46. Mechanistically, a link between Nef/glycoGag responsiveness and the stoichiometry of entry could be due to an inhibitory effect of virion-associated SERINCs on the clustering of Env trimers. Notably, such clusters have been visualized on the surface of mature HIV-1 virions and, most prominently, at virus-cell contact zones47, 48. Alternatively, SERINCs embedded in the virion membrane could increase the energy barrier for fusion pore expansion. In both scenarios, differences in Nef/glycoGag-responsiveness among HIV-1 Envs, as well as differences in SERINC-sensitivity, may ultimately be due to differences in the amount of energy that these Envs provide towards fusion. Regardless of the mechanism, our observation that viruses as distant as HIV-1 and MLV have evolved to counteract SERINC3 and SERINC5 raises the possibility that these proteins have a broader role in innate antiviral immunity.
METHODS
Cells
JurkatTAg, 293T, MT4, A549, and U2-OS cells were gifts from G. Crabtree, D. Baltimore, W. Haseltine, M. Bujny, and A. Brass, respectively. Jurkat E6.1 and MOLT-3 cells were obtained from the ATCC. TZM-bl cells were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. PBMC were isolated from the blood of healthy donors by Ficoll-Hypaque density gradient centrifugation. The MycoSensor PCR assay system (Agilent) was used to check all cell lines for mycoplasma. The cell lines were not authenticated for this study.
HIV-1 proviral constructs
NL4-3/Nefstop, the nef-deficient variant of the prototypic HIV-1NL4-3 used in this study, has nef codons 31–33 replaced by three consecutive premature termination codons. NL-nef97ZA012 is a version of HIV-1NL4-3 that has the nef gene precisely replaced by that of p97ZA012.1 (GenBank Accession # AF286227), a near full-length molecular clone of a primary subtype C HIV-1 isolate49. NL-nef97ZA012fs is a nef-deficient variant of NL-nef97ZA012 owing to a frameshift at a unique XhoI site in nef. HXB/89.6ecto-GFP is a macrophage-tropic variant of the infectious HIV-1 molecular clone HXBH10 that encodes GFP within the nef region, and that has a KpnI-BamHI fragment (nt 6351-8475 of K03455) encoding the Env ectodomain replaced by the corresponding fragment from p89.6, a biologically active molecular clone of the primary HIV-189.6 isolate50. The proviral plasmids NL4-3/glycoMA, HXB/Env−/Nef+, HXB/Env−/Nef−, and HXBH10-gag− have been described29, 51, 52.
Expression plasmids
The pBJ5-based Nef expression vectors used, the nef-deficient control vector pNefLAIFS, and the pBJ5-based expression vectors for glycoGag-HA, for WT glycoMA-HA, and for its truncation mutants have been described29, 31 The latter plasmids were used as templates to amplify fragments encoding C-terminally FLAG-tagged versions of WT glycoMA and of its mutants, which were also cloned into the mammalian expression vector pBJ5. The HIV-1 Env expression vectors used have also been described30. The coding sequences for SERINC3 and SERINC5 without or with a C-terminal HA-tag were amplified from BC006088 (SERINC3) and from BC101281 and AW005635 (SERINC5) (GE Healthcare). The primers included a Kozak sequence and XhoI and NotI cloning sites for insertion into pBJ5. The vectors expressing SERINC5(iHA) and SERINC5-mCherry are also pBJ5-based. SERINC5(iHA) has an HA tag inserted between residues 290 and 291 of SERINC5. SERINC5-mCherry has Thr-Gly-Ala-Gly linker inserted between SERINC5 and mCherry.
Retroviral vectors
The human SERINC3 and SERINC5 genes preceded by a Kozak sequence were inserted into pMSCVhyg and pMSCVpuro, respectively (Clontech). The human CD4 gene was inserted into the retroviral vector pCXbsr53.
Protein identification
For the identification of virus-associated host proteins, virions released by chronically infected T-lymphoid MOLT-3 cells were pelleted through sucrose, resuspended in PBS, and further purified in OptiPrep velocity gradients as described 54. OptiPrep gradient fractions were collected from the top and diluted with PBS. Viral particles were harvested from the fractions by ultracentrifugation and lysed in SDS-PAGE loading buffer [60 mM Tris-HCl, pH 6.8, 1% SDS, 10% (w/v) glycerol, 0.005% bromophenol blue, 5% (v/v) 2-mercaptoethanol]. Virus-containing fractions were then identified by Western blotting with antibody 183-H12-5C against HIV-1 capsid (CA)55.
For mass spectrometry, virion-associated proteins were briefly run into an SDS-PAGE gel to allow removal of SDS. After in-gel digestion with trypsin, peptides were separated on a NanoAcquity (Waters) UPLC and analyzed with a Q Exactive hybrid mass spectrometer (Thermo). The run conditions followed the “sensitive” settings recommended for optimizing the Q Exactive for low abundance proteins56. Raw data files were peak processed with Proteome Discoverer (version 1.3, Thermo) prior to searching with Mascot Server (version 2.4) against the SwissProt database. Search results were then loaded into the Scaffold Viewer (Proteome Software, Inc.).
Viral particle analysis
To examine the incorporation of SERINCs, 293T cells were co-transfected with NL4-3/Nefstop, vectors expressing HA-tagged SERINCs, and vectors expressing various epitope-tagged Nef or glycoGag proteins, or the appropriate control vectors. To examine whether VSV G affects SERINC5 incorporation, 293T cells were co-transfected with 1 μg HXBH10-gag− (a control HIV-1 proviral construct unable to express Gag) or HXB/Env−/Nef−, 100 ng of a plasmid expressing VSV G or control vector, and 500 ng of a plasmid expressing SERINC5-HA. Virions released into the medium were pelleted through sucrose, and virus- and cell-associated proteins were detected by Western blotting as described previously57. Samples used for the detection of SERINCs were maximally heated to 37°C, because SERINCs are highly aggregation-prone at higher temperatures (data not shown). In some cases, 25 mM TCEP was used as the reducing agent. The antibodies used were 183-H12-5C against HIV-1 CA, HA.11 (Covance) against the HA epitope, M2 against the FLAG epitope (Sigma-Aldrich), and AC-40 (Sigma-Aldrich) against actin. To examine Env incorporation, virions produced by transiently transfected 293T or JurkatTAg cells were pelleted through 20% sucrose cushions by ultracentrifugation, and examined by Western blotting using the anti-gp41 MAb Chessie 858, an anti-gp120 polyclonal antibody (20-HG81; Fitzgerald), and the anti-CA MAb 183-H12-5C.
SERINC overexpression experiments
Pseudovirions capable of a single round of replication were produced by transfecting 293T cells in triplicate using a calcium phosphate precipitation method. The cells were co-transfected with 1 μg HXB/Env−/Nef−, 100 ng of a plasmid expressing EnvHXB2 or VSV G, and 100 or 500 ng of plasmids expressing SERINC3 or SERINC5, or with equimolar amounts of the empty vector. To examine the effects of Nef or glycoGag, 293T cells were co-transfected with 1 μg HXB/Env−/Nef−, a plasmid expressing EnvHXB2 (100 ng), a plasmid expressing SERINC5 (100 ng) or the empty vector, and plasmids expressing NefSF2 (2 μg) or glycoGag (200 ng) or the empty vector. Supernatants containing progeny virions were harvested two days post transfection, clarified by low-speed centrifugation, filtered through 0.45 μm pore filters, and then used immediately to infect TZM-bl indicator cells in T25 flasks. Aliquots of the filtered virus stocks were frozen for HIV-1 CA (p24) antigen quantitation by a standard ELISA. Three to five days post infection, the indicator cells were lysed in reporter lysis buffer (Promega), and β-galactosidase activity was determined as a measure of infection using a kit (E2000; Promega) according to the manufacturer’s instructions.
To examine the effects of exogenous SERINC5 on the single-cycle infectivity of nef-deficient HIV-1 for primary target cells, viral stocks were obtained by co-transfecting 293T cells with HXB/Env−/Nef−, a plasmid expressing EnvHXB2, a plasmid expressing SERINC5 or the empty vector, and an HIV-1-based lentiviral vector expressing GFP. Filtered viral stocks normalized for p24 antigen were used to infect human PBMC, and infected cells expressing GFP were quantified by flow cytometry.
SERINC depletion experiments
To obtain pseudovirions capable of a single round of replication, Lipofectamine 2000 (Invitrogen) was used to transfect JurkatTAg cells in triplicate with 1 μg HXB/Env−/Nef−, 100 ng of an HIV-1 Env expression plasmid, and small interfering RNAs (siRNAs; 40 nM each). Additionally, 500 ng of a plasmid expressing Nef97ZA012, or 200 ng of a plasmid expressing glycoGag, or the empty pBJ5 expression plasmid, were co-transfected in some experiments. The siRNAs targeting SERINC3 [Hs_TDE1_2; target sequence: CACGGTGACTCGCCTCATTTA] or SERINC5 [Hs_C5orf12_3; target sequence: CACCGTCTACATCTACTCCTA], and AllStars negative control siRNA were purchased from Qiagen. As a control for experiments in which the siRNAs targeting SERINC3 and SERINC5 were cotransfected, the concentration of the control siRNA was doubled. The infectivities of JurkatTAg-derived virus stocks normalized for p24 antigen content were determined as above using TZM-bl indicator cells.
To examine the effects of SERINCs on the infectivity of HIV-1 progeny virions produced in primary cells, monocyte-derived macrophages (MDM) were infected with replication-competent, dual-tropic HXB/89.6ecto-GFP. On day 5 post infection, Lipofectamine 2000 was used to simultaneously transfect the MDM with the siRNAs targeting SERINC3 and SERINC5 (240 nM each), or with the negative control siRNA. The cells were washed 5 hours later to remove the transfection agent. Virus-containing culture medium was harvested on day 3 post transfection, and infectivities normalized for p24 antigen were determined using TZM-bl indicator cells. Indinavir (2 μM) was added to the TZM-bl cells together with virus to limit replication to a single cycle, and AMD3100 (5 μM) and maraviroc (50 nM) were added the next day to prevent Env-induced cell-cell fusion.
Generation and use of KO cells
Expression plasmids for single guide RNAs (sgRNAs) targeting exons within the SERINC3 and SERINC5 genes were transiently transfected into JurkatTAg cells by nucleofection, along with a plasmid expressing Cas9. The sites targeted by the sgRNAs are depicted in Extended Data Fig. 8. Whereas the two sgRNAs targeting the SERINC3 gene were expressed individually, the two sgRNAs targeting the SERINC5 gene were expressed together. To obtain double-KO cells, JurkatTAg S3−/− (2) cells were co-transfected with the two sgRNAs targeting the SERINC5 gene and the Cas9 expression plasmid. Nine days post transfection, gene editing in the bulk cultures was confirmed by PCR amplification of the targeted regions of the genome, followed by digestion of the PCR products with appropriate restriction enzymes (NcoI and BtsCI for target sites A and B within the SERINC3 gene, respectively; BsoBI for target site B within the SERINC5 gene). Clones were then obtained by limiting dilution in 96-well flat-bottomed culture plates. Whenever possible, the clones were pre-screened by PCR amplification of the targeted regions of the genome and restriction analysis. Furthermore, the PCR products were cloned into pCR-Blunt II-TOPO (Invitrogen/Life Technologies), and up to 10 independent clones were sequenced in each case. The primer pairs used for PCR amplification of the sgRNA target sites were: 5′-CCATAGTCAGTCTTGCAGTTG-3′ and 5′-GTACGTAGTATCTAGCATAGTGC-3′ (SERINC3 target site A), 5′-CTTCTAGGCTAATGTTGTCC-3′ and 5′-GTGAGTTGCAGGTACTAAGTC-3′ (SERINC3 target site B), 5′-CACACGATCCATTTCCACAG-3′ and 5′-CGCATCATGGTACCAGGTG-3′ (SERINC5 target site A), and 5′-GATCATTGGCAGGTAAGAGC-3′ and 5′-CACACCGCAAACACAAGC-3′ (SERINC5 target site B). Deletions between SERINC5 target sites A and B were identified using primers 5′-CACACGATCCATTTCCACAG-3′ and 5′-CACACCGCAAACACAAGC-3′ for PCR amplification and direct sequencing of the products. An inversion between SERINC5 target sites A and B was characterized using primer pair 5′-CACACGATCCATTTCCACAG-3′ and 5′-GATCATTGGCAGGTAAGAGC-3′, and primer pair 5′-CGCATCATGGTACCAGGTG-3′ and 5′-CACACCGCAAACACAAGC-3′. Ectopic SERINC expression cassettes were introduced into the double-KO cells by retroviral transduction with MSCVhygSERINC3 and/or MSCVpuroSERINC5, followed by selection with hygromycin and/or puromycin. SERINC3 expression was examined by Western blotting with a rabbit anti-TDE1 (SERINC3) antibody (GTX115512; GeneTex).
To determine the effects of SERINCs on HIV-1 infectivity in the absence of Nef, parental, KO, double-KO, and gene-reconstituted double-KO JurkatTAg cells were co-transfected in triplicate with a plasmid expressing EnvHXB2 and HXB/Env−/Nef−. To determine the effects of Nef and glycoGag in cells lacking SERINCs, parental and double-KO JurkatTAg cells were co-transfected with a plasmid expressing EnvHXB2 and either HXB/Env−/Nef−, HXB/Env−/Nef+, or HXB/Env−/Nef− together with a plasmid expressing glycoGag. Progeny virus infectivities normalized for p24 antigen were determined using TZM-bl indicator cells. Alternatively, the HIV-1 vector HIVec2.GFP was co-transfected together with HXB/Env−/Nef− and the Env expression plasmid. Following exposure to equal amounts of virus, infected TZM-bl cells were then identified based on GFP expression.
For virus replication studies, replication-competent HIV-1 was produced by transiently transfecting 293T cells with NL4-3, NL4-3/Nefstop, NL-nef97ZA012, or NL-nef97ZA012fs. Virus-containing supernatants were passed through 0.45 μm filters, normalized for p24 antigen, and used to infect parental, double-KO, and gene-reconstituted double-KO JurkatTAg cells, or CD4high versions obtained by retroviral transduction with pCXbsrCD4 and selection with blasticidin.
Analysis of mRNA expression
Total cellular RNA was extracted from cell lines and PBMC using an RNeasy mini kit (Qiagen) and treated with RNase-free DNase (Qiagen). The A260/A280 ratio was > 2.0 for all samples analyzed. Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) was performed in triplicate for each biological sample using a LightCycler 96 real-time PCR system (Roche) and a Kapa SYBR FAST One-Step qRT-PCR Universal kit (Kapa Biosystems) according to the manufacturer’s instructions. Threshold cycle values were normalized for those obtained for GAPDH, and relative expression levels were calculated using the 2−ΔΔCT method59. The primer pairs used were: SERINC3, 5′-AATTCAGGAACACCAGCCTC-3′ and 5′-GGTTGGGATTGCAGGAACGA-3′; SERINC5, 5′-ATCGAGTTCTGACGCTCTGC-3′ and 5′-GCTCTTCAGTGTCCTCTCCAC-3′; GAPDH, 5′-TGCACCACCAACTGCTTAGC-3′ and 5′-GGCATGGACTGTGGTCATGAG-3′.
Analysis of late RT products
Virions were produced by co-transfecting 293T cells with NL4-3/Nefstop (1.5 μg) and the pBJ5-based vector expressing SERINC5 (500 ng) or an equimolar amount of empty pBJ5. Cell-free virions were treated with RNase-free DNase I (Roche), and used to infect A549/CD4/CXCR4 cells in duplicate in T25 flasks for 14 hrs in the absence or presence of a cocktail of RT inhibitors. Genomic DNA was extracted with DNAzol (Life Technologies), and 100 ng of each template DNA was used for quantitative PCR using a LightCycler 96 real-time qPCR system (Roche) and a Kapa SYBR FAST Universal qPCR kit (Kapa Biosystems) according to the manufacturer’s instructions. The primers used to quantify late RT products were J1 fwd (5′-ACAAGCTAGTACCAGTTGAGCCAGATAAG-3′) and J2 rev (5′-GCCGTGCGCGCTTCAGCAAGC-3′). The J1 fwd primer exploits differences between the 5′ and 3′LTRs of pNL4-3 to help distinguish between late RT products and contaminating plasmid DNA. Standard curves were obtained from 10-fold serial dilutions of DNA extracted from cells infected with virions produced in the absence of exogenous SERINC5. Quantitative PCR results were normalized for input virus based on p24 antigen quantifications. A549/CD4/CXCR4 target cells were generated by transduction with retroviral vectors expressing CD4 (pMSCVpuroCD4) and CXCR4 (pCXbsrCXCR4).
Virion fusion assay
Virions containing BLAM-Vpr were produced by transfecting 293T cells with HXB/Env−/Nef− (2.5 μg), a vector expressing the Env protein of HIV-1HXB2 or a frameshifted version unable to express Env (200 ng), the BlaM-Vpr expression vector pMM310 (1μg), and a pBJ5-based vector expressing SERINC5 (1 μg or 100 ng) or an equimolar amount of empty pBJ5 (0.7 μg or 70 ng). Cell-free virions were normalized for p24 antigen and incubated with 2 × 105 TZM-bl or A549/CD4/CXCR4 cells in 6-well plates for 4 hrs at 37°C. After washing with PBS, 1 ml CCF4-AM dye solution in phenol-free DMEM/2% FBS was added to the cells. The CCF4-AM dye solution was prepared using a LiveBLAzer FRET-B/G loading kit (Life Technologies) according to the alternative protocol recommended by the manufacturer. After incubation for 12–14 hrs at 11°C in an ECHOterm chilling incubator (Torrey Pines Scientific), the cells were washed 3 x with PBS, detached with Versene (Life Technologies), fixed in 2% paraformaldehyde/PBS, and analyzed on a Becton Dickinson LSR II flow cytometer. Samples were excited with a 405 nm violet laser, and fluorescence emission was measured in the Pacific Blue channel (450/50 nm filter) and in the AmCyan channel (525/20 nm filter).
Extended Data
Extended Data Figure 1. Identification of SERINC3 as a candidate target of Nef and glycoGag.

a, Anti-HIV-1 CA immunoblot of Nef+, Nef−, and glycoMA+ HIV-1 virions harvested from the indicated fractions of OptiPrep gradients. b, Proteins identified by mass spectrometry in Nef− but not in Nef+ or glycoMA+ virion lysates. The data are from two independent experiments.
Extended Data Figure 2. MLV glycoGag inhibits the incorporation of SERINC3 and SERINC5 into HIV-1 virions.
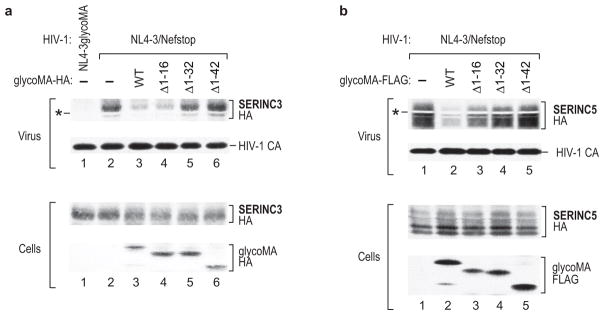
a, b, Western blots showing the effects of WT or mutant glycoMA on the incorporation of SERINC3-HA (a) or SERINC5-HA (b) into Nef− HIV-1 virions. The NL4-3/glycoMA proviral construct expresses untagged glycoMA in cis. In all other cases, HA-tagged (a) or FLAG-tagged (b) glycoMA proteins were expressed in trans. The white bands marked by asterisks are caused by co-migrating HIV-1 Pr55gag. Both experiments were performed twice.
Extended Data Figure 3. Nef and glycoGag down-regulate SERINC5 from the cell surface.

a, SERINC5 re-localizes from the plasma membrane to perinuclear vesicles in the presence of glycoGag. HeLa or U2-OS cells transiently expressing SERINC5-mCherry alone or together with glycoGag were examined by live-cell fluorescence microscopy. b, Nef and glycoGag both down-regulate SERINC5. JurkatTAg cells transiently expressing SERINC5(iHA), either alone or together with NefSF2 or glycoGag, were surface-stained with anti-HA antibody and analyzed by flow cytometry. Per cent fractions of cells expressing SERINC5(iHA) on the surface are indicated. This experiment was performed twice.
Extended Data Figure 4. SERINC mRNA expression levels.

a, Expression of SERINC family members in uninfected and HIV-infected Jurkat E6.1 cells. RNA was extracted at the peak of infection with WT (Nef+) or Nef− HIV-1NL43, and gene expression was quantified by RNA-seq as reads per kilobase of coding sequence per million reads (RPKM) (n = 1). The HIV-1 budding factor TSG101 and the housekeeping gene HPRT1 are included for comparison. b, Levels of SERINC3 and SERINC5 mRNA (arbitrary units) in cell lines and primary cells, as measured by qRT-PCR (n = 3). PBMC were left unstimulated or stimulated with 0.5 μg/ml phytohemagglutinin (PHA) and 20 U/ml IL-2 for 2 days. c, SERINC5 mRNA expression is not induced by INF-α. PBMC were left untreated or treated with 1000 U/ml human INF-α 2a (PBL Assay Science) for 14 hrs (n = 2). Bars, mean with s.d. NS, not significant (P > 0.05) two-tailed unpaired t-test.
Extended Data Figure 5. Exogenous SERINC5 inhibits the fusion of progeny virions with target cells.

TZM-bl or A549/CD4/CXCR4 cells were exposed to equal amounts of virus containing BlaM-Vpr, and fusion was analyzed by measuring the Env-dependent increase in blue fluorescence using multiparameter flow cytometry. Virions were produced in 293T cells transfected with an Env− HIV-1 provirus, a vector expressing EnvHXB2 (Env+) or a frameshift mutant (Env−), a vector expressing BlaM-Vpr, and a vector expressing SERINC5 (1 μg or 100 ng) or an equimolar amount of the empty vector (0.7 μg or 70 ng). The percentage of cells displaying increased blue fluorescence is indicated.
Extended Data Figure 6. Exogenous SERINC5 reduces the infectivity of Nef− HIV-1 progeny virions for primary target cells.

In two independent experiments, PHA-stimulated PBMC from different donors were infected with equal amounts of single-cycle GFP-HIV-1 virions produced in 293T cells in the absence or presence of exogenous SERINC5. Per cent fractions of infected (GFP-positive) cells are indicated.
Extended Data Figure 7. SERINC5 incorporation into HIV-1 virions that differ in Nef responsiveness.
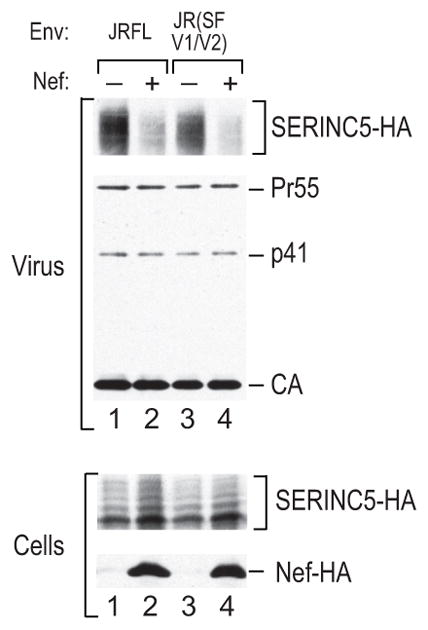
Recombinant virions were produced in 293T cells co-transfected with the HXB/Env−/Nef− provirus and vectors expressing the poorly Nef-responsive EnvJRFL or the highly Nef-responsive JR (SF V1/V2) Env chimera, along with a vector expressing SERINC5-HA. Empty pBJ5 vector or a version expressing HA-tagged Nef97ZA012 was also co-transfected. SERINC5-HA in purified virions was detected by Western blotting. This experiment was performed twice.
Extended Data Figure 8. Characterization of JurkatTAg KO cells.

a, Mutant SERINC3 alleles identified in SERINC3 KO clones. b, Mutant SERINC5 alleles identified in SERINC5 KO and SERINC3/5 double-KO clones. The sgRNA target sites are highlighted, and the predicted Cas9 target sites are indicated by arrowheads. Inserted nucleotides are in red. One of the two mutated SERINC5 alleles in JurkatTAg S3−/− S5−/− (1) cells has an inversion between sgRNA target sites A and B. JurkatTAg S5−/− (2) cells harbor 3 mutated SERINC5 alleles. All mutations cause frameshifts and/or large deletions of coding sequence. No WT alleles were detected in any of the KO clones.
Extended Data Figure 9. SERINC3 and SERINC5 expression levels in reconstituted double-KO cells.
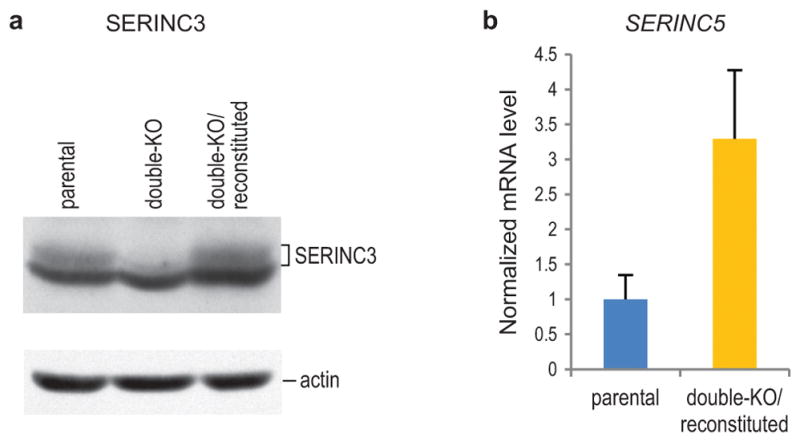
a, SERINC3 protein levels in parental, double-KO, and reconstituted double-KO JurkatTAg cells were compared by Western blotting. SERINC3 migrated close to a prominent background band that was also recognized by the anti-SERINC3 antibody. b, SERINC5 mRNA levels in parental and reconstituted double-KO JurkatTAg cells were compared by qRT-PCR (n = 3).
Extended Data Figure 10. Effects of SERINC knockout and reconstitution on HIV-1 replication.
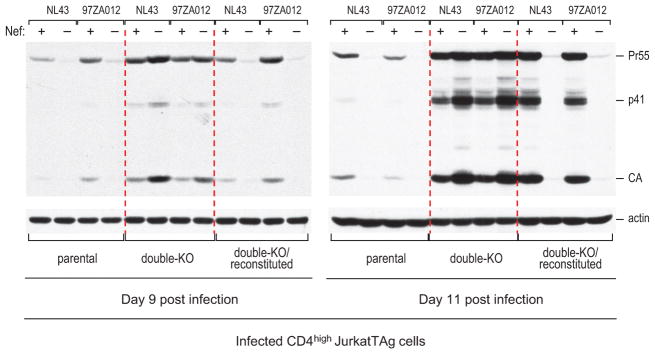
Parental, double-KO, and SERINC3 + SERINC5-reconstituted double-KO CD4high JurkatTAg cells were analyzed by immunoblotting with anti-HIV CA at day 9 and day 11 post infection with equal amounts (2 ng p24/ml) of HIV-1NL43 encoding either WT or disrupted versions of NefNL43 or Nef97ZA012.
Supplementary Material
Acknowledgments
We thank J. Leszyk and S. Shaffer for protein microsequencing, BGI Americas for RNA-seq, R. Maehr for sgRNA and Cas9 expression plasmids, J. Sodroski for HIVec2.GFP, T. Akagi for pCXbsr, and the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH for p89.6, indinavir, maraviroc, the MAbs 183-H12-5C and Chessie 8, and TZM-bl cells. This work was supported by NIAID/NIH grant R01AI029873 and by NIDA/NIH grant DP1DA038034.
Footnotes
Author Contributions Y.U, Y.W. and H.G.G. designed the experiments and analyzed the data. Y.U. carried out the analysis of virions and the SERINC overexpression and depletion experiments. Y.W. generated and characterized the SERINC KO cells, carried out all experiments involving KO cells and primary cells, and performed the qRT-PCR experiments and the BlaM-Vpr-based fusion assays. H.G.G. wrote the manuscript.
The authors declare no competing financial interests.
References
- 1.Kestler HW, 3rd, et al. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell. 1991;65:651–62. doi: 10.1016/0092-8674(91)90097-i. [DOI] [PubMed] [Google Scholar]
- 2.Deacon NJ, et al. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science. 1995;270:988–91. doi: 10.1126/science.270.5238.988. [DOI] [PubMed] [Google Scholar]
- 3.Zou W, et al. Nef functions in BLT mice to enhance HIV-1 replication and deplete CD4+CD8+ thymocytes. Retrovirology. 2012;9:44. doi: 10.1186/1742-4690-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim S, Ikeuchi K, Byrn R, Groopman J, Baltimore D. Lack of a negative influence on viral growth by the nef gene of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1989;86:9544–8. doi: 10.1073/pnas.86.23.9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller MD, Warmerdam MT, Gaston I, Greene WC, Feinberg MB. The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J Exp Med. 1994;179:101–13. doi: 10.1084/jem.179.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spina CA, Kwoh TJ, Chowers MY, Guatelli JC, Richman DD. The importance of nef in the induction of human immunodeficiency virus type 1 replication from primary quiescent CD4 lymphocytes. J Exp Med. 1994;179:115–23. doi: 10.1084/jem.179.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia JV, Miller AD. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature. 1991;350:508–11. doi: 10.1038/350508a0. [DOI] [PubMed] [Google Scholar]
- 8.Aiken C, Konner J, Landau NR, Lenburg ME, Trono D. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell. 1994;76:853–64. doi: 10.1016/0092-8674(94)90360-3. [DOI] [PubMed] [Google Scholar]
- 9.Rhee SS, Marsh JW. Human immunodeficiency virus type 1 Nef-induced down-modulation of CD4 is due to rapid internalization and degradation of surface CD4. J Virol. 1994;68:5156–63. doi: 10.1128/jvi.68.8.5156-5163.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaudhuri R, Lindwasser OW, Smith WJ, Hurley JH, Bonifacino JS. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J Virol. 2007;81:3877–90. doi: 10.1128/JVI.02725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veillette M, et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J Virol. 2014;88:2633–44. doi: 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haller C, et al. HIV-1 Nef and Vpu are functionally redundant broad-spectrum modulators of cell surface receptors, including tetraspanins. J Virol. 2014;88:14241–57. doi: 10.1128/JVI.02333-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. 1996;2:338–42. doi: 10.1038/nm0396-338. [DOI] [PubMed] [Google Scholar]
- 14.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. 1998;391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 15.Cohen GB, et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity. 1999;10:661–71. doi: 10.1016/s1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- 16.Schindler M, et al. Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell. 2006;125:1055–67. doi: 10.1016/j.cell.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 17.Chowers MY, Pandori MW, Spina CA, Richman DD, Guatelli JC. The growth advantage conferred by HIV-1 nef is determined at the level of viral DNA formation and is independent of CD4 downregulation. Virology. 1995;212:451–7. doi: 10.1006/viro.1995.1502. [DOI] [PubMed] [Google Scholar]
- 18.Munch J, et al. Nef-mediated enhancement of virion infectivity and stimulation of viral replication are fundamental properties of primate lentiviruses. J Virol. 2007;81:13852–64. doi: 10.1128/JVI.00904-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aiken C, Trono D. Nef stimulates human immunodeficiency virus type 1 proviral DNA synthesis. J Virol. 1995;69:5048–56. doi: 10.1128/jvi.69.8.5048-5056.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz O, Marechal V, Danos O, Heard JM. Human immunodeficiency virus type 1 Nef increases the efficiency of reverse transcription in the infected cell. J Virol. 1995;69:4053–9. doi: 10.1128/jvi.69.7.4053-4059.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller MD, Warmerdam MT, Page KA, Feinberg MB, Greene WC. Expression of the human immunodeficiency virus type 1 (HIV-1) nef gene during HIV-1 production increases progeny particle infectivity independently of gp160 or viral entry. J Virol. 1995;69:579–84. doi: 10.1128/jvi.69.1.579-584.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forshey BM, Aiken C. Disassembly of human immunodeficiency virus type 1 cores in vitro reveals association of Nef with the subviral ribonucleoprotein complex. J Virol. 2003;77:4409–14. doi: 10.1128/JVI.77.7.4409-4414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ross TM, Oran AE, Cullen BR. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr Biol. 1999;9:613–21. doi: 10.1016/s0960-9822(99)80283-8. [DOI] [PubMed] [Google Scholar]
- 24.Lama J, Mangasarian A, Trono D. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr Biol. 1999;9:622–31. doi: 10.1016/s0960-9822(99)80284-x. [DOI] [PubMed] [Google Scholar]
- 25.Goldsmith MA, Warmerdam MT, Atchison RE, Miller MD, Greene WC. Dissociation of the CD4 downregulation and viral infectivity enhancement functions of human immunodeficiency virus type 1 Nef. J Virol. 1995;69:4112–21. doi: 10.1128/jvi.69.7.4112-4121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pizzato M. MLV glycosylated-Gag is an infectivity factor that rescues Nef-deficient HIV-1. Proc Natl Acad Sci U S A. 2010;107:9364–9. doi: 10.1073/pnas.1001554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prats AC, De Billy G, Wang P, Darlix JL. CUG initiation codon used for the synthesis of a cell surface antigen coded by the murine leukemia virus. J Mol Biol. 1989;205:363–72. doi: 10.1016/0022-2836(89)90347-1. [DOI] [PubMed] [Google Scholar]
- 28.Pillemer EA, Kooistra DA, Witte ON, Weissman IL. Monoclonal antibody to the amino-terminal L sequence of murine leukemia virus glycosylated gag polyproteins demonstrates their unusual orientation in the cell membrane. J Virol. 1986;57:413–21. doi: 10.1128/jvi.57.2.413-421.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Usami Y, Popov S, Gottlinger HG. The Nef-like effect of murine leukemia virus glycosylated gag on HIV-1 infectivity is mediated by its cytoplasmic domain and depends on the AP-2 adaptor complex. J Virol. 2014;88:3443–54. doi: 10.1128/JVI.01933-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Usami Y, Gottlinger H. HIV-1 Nef Responsiveness Is Determined by Env Variable Regions Involved in Trimer Association and Correlates with Neutralization Sensitivity. Cell Rep. 2013;5:802–12. doi: 10.1016/j.celrep.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pizzato M, et al. Dynamin 2 is required for the enhancement of HIV-1 infectivity by Nef. Proc Natl Acad Sci U S A. 2007;104:6812–7. doi: 10.1073/pnas.0607622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Craig HM, Pandori MW, Guatelli JC. Interaction of HIV-1 Nef with the cellular dileucine-based sorting pathway is required for CD4 down-regulation and optimal viral infectivity. Proc Natl Acad Sci U S A. 1998;95:11229–34. doi: 10.1073/pnas.95.19.11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inuzuka M, Hayakawa M, Ingi T. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J Biol Chem. 2005;280:35776–83. doi: 10.1074/jbc.M505712200. [DOI] [PubMed] [Google Scholar]
- 34.Grossman TR, Luque JM, Nelson N. Identification of a ubiquitous family of membrane proteins and their expression in mouse brain. J Exp Biol. 2000;203:447–57. doi: 10.1242/jeb.203.3.447. [DOI] [PubMed] [Google Scholar]
- 35.Day JR, Munk C, Guatelli JC. The membrane-proximal tyrosine-based sorting signal of human immunodeficiency virus type 1 gp41 is required for optimal viral infectivity. J Virol. 2004;78:1069–79. doi: 10.1128/JVI.78.3.1069-1079.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol. 2002;20:1151–4. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- 37.Aiken C. Pseudotyping human immunodeficiency virus type 1 (HIV-1) by the glycoprotein of vesicular stomatitis virus targets HIV-1 entry to an endocytic pathway and suppresses both the requirement for Nef and the sensitivity to cyclosporin A. J Virol. 1997;71:5871–7. doi: 10.1128/jvi.71.8.5871-5877.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo T, Douglas JL, Livingston RL, Garcia JV. Infectivity enhancement by HIV-1 Nef is dependent on the pathway of virus entry: implications for HIV-based gene transfer systems. Virology. 1998;241:224–33. doi: 10.1006/viro.1997.8966. [DOI] [PubMed] [Google Scholar]
- 39.Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10:957–63. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waheed AA, Freed EO. The Role of Lipids in Retrovirus Replication. Viruses. 2010;2:1146–1180. doi: 10.3390/v2051146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lundquist CA, Tobiume M, Zhou J, Unutmaz D, Aiken C. Nef-mediated downregulation of CD4 enhances human immunodeficiency virus type 1 replication in primary T lymphocytes. J Virol. 2002;76:4625–33. doi: 10.1128/JVI.76.9.4625-4633.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tobiume M, Lineberger JE, Lundquist CA, Miller MD, Aiken C. Nef does not affect the efficiency of human immunodeficiency virus type 1 fusion with target cells. J Virol. 2003;77:10645–50. doi: 10.1128/JVI.77.19.10645-10650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cavrois M, Neidleman J, Yonemoto W, Fenard D, Greene WC. HIV-1 virion fusion assay: uncoating not required and no effect of Nef on fusion. Virology. 2004;328:36–44. doi: 10.1016/j.virol.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 44.Cohen FS, Melikyan GB. The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J Membr Biol. 2004;199:1–14. doi: 10.1007/s00232-004-0669-8. [DOI] [PubMed] [Google Scholar]
- 45.Schaeffer E, Geleziunas R, Greene WC. Human immunodeficiency virus type 1 Nef functions at the level of virus entry by enhancing cytoplasmic delivery of virions. J Virol. 2001;75:2993–3000. doi: 10.1128/JVI.75.6.2993-3000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brandenberg OF, Magnus C, Rusert P, Regoes RR, Trkola A. Different infectivity of HIV-1 strains is linked to number of envelope trimers required for entry. PLoS Pathog. 2015;11:e1004595. doi: 10.1371/journal.ppat.1004595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sougrat R, et al. Electron tomography of the contact between T cells and SIV/HIV-1: implications for viral entry. PLoS Pathog. 2007;3:e63. doi: 10.1371/journal.ppat.0030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chojnacki J, et al. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science. 2012;338:524–8. doi: 10.1126/science.1226359. [DOI] [PubMed] [Google Scholar]
- 49.Rodenburg CM, et al. Near full-length clones and reference sequences for subtype C isolates of HIV type 1 from three different continents. AIDS Res Hum Retroviruses. 2001;17:161–8. doi: 10.1089/08892220150217247. [DOI] [PubMed] [Google Scholar]
- 50.Collman R, et al. An infectious molecular clone of an unusual macrophage-tropic and highly cytopathic strain of human immunodeficiency virus type 1. J Virol. 1992;66:7517–21. doi: 10.1128/jvi.66.12.7517-7521.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dorfman T, Popova E, Pizzato M, Gottlinger HG. Nef enhances human immunodeficiency virus type 1 infectivity in the absence of matrix. J Virol. 2002;76:6857–62. doi: 10.1128/JVI.76.13.6857-6862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dorfman T, Mammano F, Haseltine WA, Gottlinger HG. Role of the matrix protein in the virion association of the human immunodeficiency virus type 1 envelope glycoprotein. J Virol. 1994;68:1689–96. doi: 10.1128/jvi.68.3.1689-1696.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Akagi T, Shishido T, Murata K, Hanafusa H. v-Crk activates the phosphoinositide 3-kinase/AKT pathway in transformation. Proc Natl Acad Sci U S A. 2000;97:7290–5. doi: 10.1073/pnas.140210297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dettenhofer M, Yu XF. Highly purified human immunodeficiency virus type 1 reveals a virtual absence of Vif in virions. J Virol. 1999;73:1460–7. doi: 10.1128/jvi.73.2.1460-1467.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chesebro B, Wehrly K, Nishio J, Perryman S. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol. 1992;66:6547–54. doi: 10.1128/jvi.66.11.6547-6554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelstrup CD, Young C, Lavallee R, Nielsen ML, Olsen JV. Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. J Proteome Res. 2012;11:3487–97. doi: 10.1021/pr3000249. [DOI] [PubMed] [Google Scholar]
- 57.Accola MA, Strack B, Gottlinger HG. Efficient particle production by minimal gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J Virol. 2000;74:5395–402. doi: 10.1128/jvi.74.12.5395-5402.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abacioglu YH, et al. Epitope mapping and topology of baculovirus-expressed HIV-1 gp160 determined with a panel of murine monoclonal antibodies. AIDS Res Hum Retroviruses. 1994;10:371–81. doi: 10.1089/aid.1994.10.371. [DOI] [PubMed] [Google Scholar]
- 59.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.