

Abstract
Activation of the transcription factor Nrf2 has been posited to be a promising therapeutic strategy in a number of inflammatory and oxidative stress diseases due to its regulation of detoxifying enzymes. In this work, we have developed a comprehensive structure-activity relationship around a known, naphthalene-based non-electrophilic activator of Nrf2, and we report highly potent non-electrophilic activators of Nrf2. Computational docking analysis of a subset of the compound series demonstrates the importance of water molecule displacement for affinity, and the X-ray structure of di-amide 12e supports the computational analysis. One of the best compounds, acid 16b, has an IC50 of 61 nM in a fluorescence anisotropy assay and a Kd of 120 nM in a surface plasmon resonance assay. Additionally, we demonstrate that the ethyl ester of 16b is an efficacious inducer of Nrf2 target genes, exhibiting ex vivo efficacy similar to the well-known electrophilic activator, sulforaphane.
Graphical Abstract
1. Introduction
During periods of oxidative or electrophilic stress, one of the body’s main defenses is induction of cytoprotective proteins, including detoxification enzymes, such as those that reduce quinones (e.g., NAD(P)H quinone oxidoreductase 1, NQO1) [1], those that degrade heme (heme oxygenase 1, HMOX1) [2], and those involved in glutathione synthesis and transfer (e.g., glutamate-cysteine ligase catalytic subunit, glutamate-cysteine ligase regulatory subunit, glutathione S-transferase, GST) [3]. These genes are regulated by the transcription factor, Nrf2 (nuclear factor-erythroid2-related factor 2), which belongs to a cap ‘n’ collar family of basic leucine zipper transcription factors that comprise seven highly conserved domains (Neh1 to Neh7) [4]. In the absence of electrophilic or oxidative stressors, Nrf2 is negatively regulated by Keap1 (Kelch like ECH associated protein 1), a 69 kDa sensor protein that contains 27 cysteine residues [5]. Keap1 is a tightly-associated dimer that acts as an adaptor protein that simultaneously binds both Nrf2, through Keap1’s Kelch domain, and the E3 ubiquitin ligase Cul3 [6], through Keap1’s BTB domain. Cul3 polyubiquitinates Nrf2, and Nrf2 is subsequently degraded through the ubiquitin/proteasome system [4]. Nrf2 is activated by inhibiting its degradation (see below). Nrf2 is then translocated to the nucleus, where it heterodimerizes with the small MAF (sMAF) transcription factors and binds to antioxidant response elements in the promoter regions of many detoxification genes, such as NQO1, GST, HMOX1 and glutathione peroxidase [7].
As shown in Figure 1, there are two prevailing mechanisms that explain how Nrf2 degradation is inhibited: 1) the Cul3 dissociation model [8,9] and 2) the hinge and latch model [10]. In the Cul3 dissociation model, Keap1 recognizes and binds Nrf2 through the ETGE and DLG motifs of the Neh2 region of Nrf2. Keap1 brings Nrf2 into close proximity with a ubiquitin-conjugating enzyme (Cul3), which then transfers ubiquitin to target lysine residues on Nrf2. If electrophilic inhibitors bind to Keap1 so that Cul3 is dissociated from Keap1, Nrf2 is not degraded, but it is also not released from Keap1 [8,9,11]. Newly formed Nrf2 is then available for regulating expression of cytoprotective enzymes. The alternative hinge and latch model is governed by modifications affecting Nrf2 [10,12–14]. In this model, upon electrophilic modification of key cysteine residues in the IVR region of Keap1, the protein undergoes a slight conformational modification and releases the DLG motif, while maintaining binding to the ETGE motif [12,13]. Once the DLG motif is released, Nrf2 swings out from its ideal position, becoming inaccessible for ubiquitination [13]. With this change, Nrf2 does not get degraded, and nascent Nrf2 production gives rise to Nrf2 accumulation and activation. A recent structural study [15] lends support for the Cul3-dissociation model, but there is still an active dialogue in the literature as to which model dominates in Nrf2 activation or whether both are operative [8–10,16–18].
Figure 1.

A model of Nrf2 activation by both electrophilic and non-electrophilic inhibition of Keap1-Nrf2 complex
Given its central importance in regulating cytoprotective enzymes, Nrf2 activation has been proposed as a promising pharmacotherapeutic strategy in a number of inflammatory and oxidative stress disorders, including chronic kidney disease [19], multiple sclerosis [20], pulmonary fibrosis [21], cancer chemoprevention [22], and chronic obstructive pulmonary disorder [23]. As alluded to above, electrophilic Nrf2 activators are known (Chart 1). Two of the more well-known electrophilic Nrf2 activators are sulforaphane (1), an isothiocyanate derived from cruciferous vegetables like broccoli; and dimethyl fumarate, a new therapeutic for multiple sclerosis (i.e., Tecfidera® (2); see Chart 1). The therapeutic benefit of each of these electrophilic molecules is thought to arise, in part, from Nrf2 activation. These compounds covalently react with the sensor cysteines of Keap1, particularly Cys 151 [24–28], which results in Nrf2 activation (see above). These covalent activators are obviously efficacious, but, because they are electrophiles, they may not be selective for Keap1. This point is exemplified by a proteomics study done with the oleanic triterpenoid bardoxolone imidazole (3) (See Chart 1) [29]. That study found that this potent electrophilic activator of Nrf2 interacts with at least 577 different proteins in whole cells [29]. It acts as a Michael acceptor for reactive Cys residues on Keap1 en route to activating Nrf2. A related compound, bardoxolone methyl (4), proceeded as far as a phase III clinical trial in patients with type 2 diabetes and chronic kidney disease before adverse cardiovascular events derailed its development [30]. Although their cause is unknown, these adverse events may be attributable to off-target toxicity.
Chart 1.
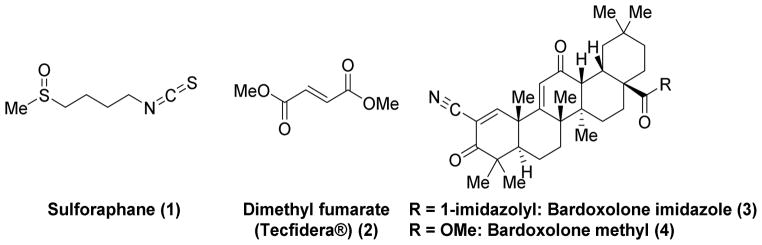
Known electrophilic Nrf2 activators that react with cysteine residues in the BTB and IVR domains of Keap1.
Recently, there has been great interest in developing reversible covalent drugs to activate Nrf2 [31,32]; however, we and others have taken a different approach and have begun to develop non-covalent compounds that might be more selective Nrf2 activators [31,33–37]. Non-covalent compounds may serve two important functions: first, as tool compounds that can help to disentangle the rather complicated pharmacology of Nrf2 activation, and, second, as lead compounds for eventual therapeutic development.
In developing non-covalent activators of Nrf2, a sensible approach is to inhibit the interaction of Nrf2 with its negative regulator, Keap1. In this case, an inhibitor would occupy the site on Keap1’s Kelch domain where the ETGE motif of Nrf2 is bound, which is distal from the binding site of known electrophilic activators [38]; thus, if a molecule binds in the ETGE pocket, Nrf2 is displaced and activated [29,34].
Several hit compounds have been identified through high-throughput screening (Chart 2). These hits include tetrahydroisoquinoline 5 (IC50 = 1.0 μM) [35], carbazone 6 (IC50 = 9.8 μM) [36], naphthalene 7 (IC50 = 2.7 μM) [33], naphthalene 8 (IC50 = 29 nM [34]; Kd = 9.9 nM), thiopyrimidine 9 (IC50 = 118 M) [39,40] and urea 10 (affinity unknown) [33]. Limited SAR and molecular modeling studies with some of these scaffolds have been carried out in hopes of uncovering potent non-electrophilic inhibitors of the Keap1/Nrf2 complex [7,34,35]. These studies have revealed that many of these non-electrophilic activators behave similarly to the ETGE motif; significant interactions seem to occur between acidic portions of the inhibitors and Arg 415 in the binding pocket, with other contributions stemming from hydrogen bonding and π-stacking [34]. These studies represent a powerful starting point in developing more comprehensive SARs of non-electrophilic activators of Nrf2. In this work, we have expanded the SAR around 7 and 8. We detail here a more complete understanding of the structural requirements for binding of non-electrophilic molecules to Keap1 via computational modeling and X-ray crystallography, and we report a compound with greater ligand efficiency, equipotent in vitro activity, and enhanced ex vivo activity.
Chart 2.

Known Non-electrophilic Nrf2 Activators that Inhibit the Keap1/Nrf2 Interaction in the Kelch domain.
2. Methods and materials
2.1. Kelch domain expression and purification [24]
The gene for the Kelch domain (amino acids 321–609) of human Keap1 was codon-optimized for expression in E. coli, synthesized and cloned into pET15b (Bio Basic Inc.). For crystallographic work, the codon-optimized gene was PCR amplified and subcloned via ligation-free cloning into the vector pEV-L8 to incorporate a TEV-cleavable, N-terminal (His)8-tag. For expression and purification of both Kelch proteins from the vectors described above, BL21 (DE3) cells (Novagen) were transformed with each construct via electroporation. These cells were plated on LB agarose plates containing 50 μg/mL carbenicillin. Single colonies were picked and then used to inoculate small starter cultures (LB medium containing 50 μg/mL carbenicillin). Cells were allowed to grow overnight at 37 °C and were then used to inoculate 1 L of fresh media (LB:carbenicillin) and grown at 37 °C until OD600 of 0.6. At this point, protein expression was induced by the addition of 1 mM isopropyl-β-D-galactopyranoside for 4 hours at 37 °C. After expression, cells were harvested by centrifugation at 8000 g at 4 °C for 25 min. Cell pellets were frozen at −80 °C. The frozen cell pellets were resuspended in Buffer A (50 mM Tris, pH 7.5, 500 mM NaCl, 10 mM imidazole, 3 mM DTT) containing one EDTA-free protease inhibitor pellet (Roche), 7.5 mM MgSO4 and minimal amounts of DNaseI and lysozyme and the cells were lysed via sonication. The lysed cells were pelleted by centrifugation at 28,960 g for 25 min at 4 °C. The supernatant was applied to a 5 mL HisTrap affinity column charged with Ni2+ (GE Healthcare Life Sciences) equilibrated with Buffer A. The protein was eluted in a gradient of 0–60% Buffer B (50 mM Tris, pH 7.5, 500 mM NaCl, 500 mM imidazole, 3 mM DTT) in 5 mL fractions. The final purity of the proteins was analyzed by SDS/PAGE.
To generate purified Kelch protein for crystallization experiments, pooled fractions from the HisTrap column were dialyzed overnight at 4 °C against Buffer A in the presence of TEV protease (1:5(w/w)). Separation of the cleaved tag was achieved by reverse HisTrap chromatography following the same procedure described above. Pooled fractions were concentrated and dialyzed against Buffer C (20 mM Tris, pH 7.5, 5 mM DTT). The concentrated protein was loaded onto a size-exclusion Superdex75 HR 26/60 column (Amersham Biosciences) equilibrated with Buffer C. Protein was used fresh for co-crystallization with the compounds and aliquots were flash frozen on liquid nitrogen and stored at −80 °C for further experimentation. Protein purity at all steps was analyzed by SDS-PAGE.
2.2 Co-crystallization and X-ray structure determination of the Kelch-compound 12e complex
Co-crystallization of the Kelch-12e complex was carried out by sitting drop vapor diffusion using a 1:1 (Kelch:reservoir) drop ratio. The Kelch-12e complex was generated by adding Kelch at 5 mg/mL (160 μM) to a reservoir solution containing 2-fold molar excess of compound 12e (320μM), 0.2 M potassium chloride and 20% (w/v) PEG 3350. Crystals were grown at 4 °C and reached an average size of 200 μm x 150 μm x 20 μm in 11 days. Crystals were soaked briefly in reservoir solution supplemented with 20% PEG 400 and then flash-cooled in liquid nitrogen for X-ray data collection. A complete X-ray dataset was collected at 100 K and at a X-ray wavelength of 1.547 Å at distance of 200 mm from our Raxis 4++ detector. The X-ray dataset was processed and scaled using HKL2000 to a resolution of 2.47 Å. The Kelch-12e complex crystallized as a monomer in the asymmetric unit and in space group P2 with unit cell dimensions of a=38.47 Å b=51.52 Å c=67.24 Å and β =101.27°. Molecular replacement using the Kelch:compound 7 structure (4IQK) as a search model was conducted to obtain initial phases. The structure was then refined using the program Phenix. The final coordinates for the Kelch:12e complex have been deposited under PDB code 4XMB.
2.3 Cell culture and treatment
Mouse alveolar epithelial cells (MLE-12) [41,42] (kindly provided by Dr. Jeff Whitsett, University of Cincinnati) were grown in DMEM (Dubecco’s modified Eagle’s medium) with 10% FBS (fetal bovine serum). Cells were cultured at 37 °C under 5% CO2 in a humidified incubator. Cells were treated with the indicated compounds at 10 μM concentration for 6 h in the presence of complete medium.
2.4 Immunoblot analyses
Total protein was extracted in lysis buffer consisting of 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM Na3VO4, 5 mM β-glycerophosphate, and 1 μg/ml leupeptin. Comparable amount of total protein (~40 μg) from each sample were separated on a 10% SDS-PAGE and the membranes were probed with antibodies specific for Nrf2 (Santa Cruz Biotech, Santa Cruz, CA), HMOX1 (Santa Cruz Technologies, Santa Cruz, CA), NQO1 (Abcam Company Cambridge, MA). β-actin (Sigma, St. Louis, MO) antibody was used as the loading control. The blots were developed using an HyGlo-Chemi antibody detection kit (Denville Scientific Inc, Metuchen, NJ) and images were visualized on ChemiDoc (Biorad) and bands were quantified using image J software.
2.5 Fluorescence anisotropy assays
Fluorescence anisotropy assays were performed on a Biotek Microplate Reader. The plates used for the fluorescence anisotropy assay were nonbinding Corning 3686 96-well black, half area, flat bottom plate polystyrene plates. The assay solution was 40 μL per well consisting of 10 μL of modified HEPES buffer (10 mM) pH 7.4 containing 50 mM EDTA, 150 mM NaCl, and 0.005% Tween-20 (final concentrations), 10 μL of 100 nM Keap1 Kelch domain protein (final concentraton) [43], 10 μL of ligand sample of varying concentrations, and 10 μL of FITC-9mer Nrf2 peptide (10 nM final concentration) [43]. The DMSO concentration in the final assay solution was not more than 5%. The experiments were performed in triplicate with initial concentration of the ligand set at 25 μM and were further diluted to 20, 15, 10, 5, 3, 1, 0.3, 0.1, 0.03, 0.01, and 0.003 μM. The plate was protected from light, centrifuged for 2 min at 2250 rpm, rocked for 1 h, and centrifuged again for 2 min at 2250 rpm to remove any bubbles formed during the rocking stage. The fluorescence anisotropy measurement was performed at λex = 480 ± 20 nm and λem = 520 ± 20 nm emission filters. Fluorescence anisotropy was determined by measuring the parallel (F||) and perpendicular (F⊥) fluorescence intensity with respect to the linearly polarized excitation light. The IC50 of the ligand was calculated by plotting the normalized fluorescence anisotropy values vs. the log concentration of the ligand, and the relationships were analyzed by Prism 6.1 software.
2.6 Determination of dissociation equilibrium constant (Kd) by Surface Plasmon Resonance (SPR)
Purified Keap1 protein was diluted to 80 μg/mL with 10 mM sodium acetate (pH 5.0) and injected for 7 minutes at a 10 μL/min flow rate on a CM5 sensor chip for immobilization at 25 °C with running buffer PBS-P (10 mM phosphate pH 7.4, 2.7 mM KCl, 137 mM NaCl, and 0.05% surfactant P-20) using a Biacore T200 instrument. Blank surface was used as a control on flow channels 1 and 3. Keap1 was immobilized to flow channels 2 and 4 after sensor surface activation by a mixture of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrocholoride (EDC)/ N-hydroxysuccinimide (NHS) followed by ethanolamine blocking on unoccupied surface area. Keap1 immobilization levels of flow channels 2 and 4 were ~6,560 RU and ~5,700 RU, respectively. Compound solutions with a series of increasing concentrations were applied to all four channels at a 30 μL/min flow rate with assay buffer (10 mM phosphate, pH 7.4, 2.7 mM KCl, 137 mM NaCl, 0.05% surfactant P-20, 2 mM TCEP, and 2% DMSO), and real-time response units (RU) were monitored. Sensorgrams were analyzed using the Biacore T200 evaluation software 2.0. Data were referenced with blank channel RU values, and the Kd values were determined by fitting the reference subtracted data to a single rectangular hyperbolic curve equation (Eq. 1) where y is the RU, ymax is the maximum RU, and x is the compound concentration. Kinetic fittings were done by 1 to 1 binding equation embedded in the Biacore T200 evaluation software 2.0.
| (1) |
2.7 Ensemble Docking
An ensemble containing four structures of the Keap domain was generated based on the crystal structure 4IQK containing different patterns of waters 866 and 807. To construct the ensemble the waters were 1) unaltered, 2) water 866 (water 1) was removed, 3) water 807 (water 2) was removed, or 4) both waters were removed. Docking was performed using the Virtual Screening Workflow implementation of the Glide docking package from Schrödinger (Small-Molecule Drug Discovery Suite 2014-4: Glide, version 6.5, Schrödinger, LLC, New York, NY, 2014) [44–46]. Docking was performed using the Standard Precision (SP) settings, and up to two poses per compound state were allowed.
2.8 Hydration Site Analysis
The docking results obtained in the previous section were used as input for the hydration site analysis program, WATsite [47], which is freely available from http://people.pharmacy.purdue.edu/~mlill/software/. All settings of WATsite used were standard, with the exception that the production molecular dynamics simulation was extended from 1 ns (default) to 5 ns and the number of frames used was 5000.
2.9 Chemistry
1H and 13C NMR spectra were recorded on a Bruker 400 MHz spectrometer using tetramethylsilane (TMS) as an internal standard. Peak positions are given in parts per million (δ). Purity of each of the final, tested compounds was determined by HPLC on a Shimadzu LC-20AB (Solvent system; 55% MeCN/40% H2O, isocratic; Column: Shimadzu C18, 50 μm, 50 x 4.6 mm) and was ≥95% (UV, 254 nm). The HRMS spectra were recorded on a Shimadzu LCMS-IT-TOF, and the molecular weight of the compounds was within 0.05% of calculated values. Flash chromatography was performed using silica gel (230–400 mesh). All reactions were monitored by thin-layer chromatography (TLC) on silica gel GHLF plates (250 μm, Macherey-Nagel, Inc., Bethlehem, PA). Compounds 7, 8, and 12a were synthesized according to previously reported literature [33,34,39]. It should be noted that aminonaphthalenes are generally carcinogenic in nature. Caution should be used while handling them.
2.9.1. General method for synthesis of 11a–j, and 20 (Method A)
These previously unreported compounds were synthesized according to a known, similar procedure [48]. A solution of 1,4-diaminonaphthalene dihydrochloride (1 equivalent) and substituted-benzenesulfonyl chloride (2.2 equivalent) in pyridine (3–5 mL) was allowed stir at room temperature for 24 h. On completion, the reaction mixture was diluted with EtOAc (40 mL), washed with H2O (2 x 50 mL) and HCl (2N, 2 x 50 mL), dried (Na2SO4) and evaporated to yield crude products, which were recrystallized or washed with appropriate solvents to yield pure compounds.
2.9.1.1. Synthesis of N,N′-(naphthalene-1,4-diyl)dibenzenesulfonamide (11a)
The crude product was recrystallized from MeOH to yield 0.14 g (51% yield) of 11a as pink needles. 1H NMR (400 MHz, DMSO-d6, δ) 10.20 (s, 2H), 7.92-7.89 (m, 2H), 7.63-7.34 (m, 10H), 7.24 (d, J = 8.0 Hz, 2H), 7.03 (s, 2H); 13C NMR (100 MHz, DMSO-d6, δ) 139.8, 132.8, 131.1, 130.1, 129.2, 126.7, 126.2, 123.3, 123.0; HRMS-ESI (−) (m/z): [M-H]− calcd for C20H19N2O6S2, 437.0655; found, 437.0635.
2.9.1.2. Synthesis of N,N′-(naphthalene-1,4-diyl)bis(3-methoxybenzenesulfonamide) (11b)
The crude product was recrystallized from CH2Cl2/acetone to yield 0.08 g (25% yield) of 11b as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ) 10.32 (s, 2H), 7.95-7.93 (m, 2H), 7.38 (t, J = 8.0 Hz, 2H), 7.20-7.06 (m, 9H), 3.67 (s, 6H); 13C NMR (100 MHz, DMSO-d6, δ) 159.3, 141.1, 131.1, 130.1, 130.4, 126.3, 123.1, 123.4, 118.8, 118.9, 111.5, 55.5; HRMS-ESI (−) (m/z): [M-H]− calcd for C24H21N2O6S2, 497.0887; found, 497.0864.
2.9.1.3. Synthesis of N,N′-(naphthalene-1,4-diyl)bis(4-chlorobenzenesulfonamide) (11c)
The crude product was washed with Et2O to afford 0.12 g (37% yield) of 11c as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ) 10.33 (s, 2H), 7.92 (br s, 2H), 7.69-7.41 (m, 10H), 7.06 (m, 2H); 13C NMR (100 MHz, DMSO-d6, δ) 138.6, 137.7, 130.9, 130.2, 129.3, 128.7, 126.4, 123.3; HRMS-ESI (−) (m/z): [M-H]− calcd for C22H15N2O4S2Cl2, 504.9874; found, 504.9874.
2.9.1.4. Synthesis of 3,3′-(((4-(hydrosulfonylamino)naphthalen-1-yl)amino)sulfonyl)dibenzoic acid (11d)
The crude methyl ester was washed with Et2O to afford 0.21 g (58% yield) of the methyl ester of 11d as a purple solid. 1H NMR (400 MHz, DMSO-d6, δ): 10.41 (s, 2H), 8.28 (s, 2H), 8.13 (d, J = 8.0 Hz, 2H), 7.89 (dd, J = 6.4, 3.2 Hz, 2H), 7.80 (d, J = 8.0 Hz, 2H), 7.61 (t, 2H), 7.37 (dd, J = 6.4, 3.0 Hz, 2H), 6.98 (s, 2H), 3.85 (s, 6H). The methyl ester was further hydrolyzed using NaOH (15%, 5 mL) in MeOH (15 mL) at reflux for 3 h. On completion, the organic solvent was evaporated, and the residue was suspended in H2O (40 mL), acidified with HCl (2N, to pH 1–2), and extracted with EtOAc (3 x 30 mL). The combined organic portion was washed with H2O (3 x 50 mL) and brine (50 mL), dried (Na2SO4) and evaporated to yield crude product which, upon recrystallization from EtOAc/MeOH, yielded 0.01 g (51% yield) of 11d as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ): 13.46 (br s, 2H), 10.36 (s, 2H), 8.30 (s, 2H), 8.11 (d, J = 7.6 Hz, 2H), 7.89 (dd, J = 6.4, 3.2 Hz, 2H), 7.74 (d, J = 7.6 Hz, 2H), 7.57 (t, 2H), 7.37 (dd, J = 6.0, 2.8 Hz, 2H), 6.99 (s, 2H); 13C NMR (100 MHz, DMSO-d6, δ): 165.9, 140.3, 133.3, 131.8, 130.9, 130.7, 130.2, 129.8, 127.4, 126.4, 123.5; HRMS-ESI (−) (m/z): [M-H]− calcd for C24H17N2O8S2, 525.0432; found, 525.0456.
2.9.1.5. Synthesis of N,N′-(naphthalene-1,4-diyl)bis(4-trifluoromethylbenzenesulfonamide) (11e)
The crude product was washed with Et2O to afford 0.06 g (16% yield) of 11e as buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ) 10.49 (s, 2H), 7.83-7.81 (m, 10H), 7.22 (s, 2H), 7.09 (s, 2H); 13C NMR (100 MHz, DMSO-d6, δ) 143.9, 132.9 (q, JCF = 32 Hz), 131.2, 130.6, 128.1, 126.8 (d, JCF = 3 Hz), 126.7, 124.2, 123.8 (d, JCF = 270 Hz), 123.5; HRMS-ESI (−) (m/z): [M-H]− calcd for C24H15N2O6F6S2, 573.0383; found, 573.0393.
2.9.1.6. Synthesis of N,N′-(naphthalene-1,4-diyl)bis(3-trifluoromethylbenzenesulfonamide) (11f)
The crude product was recrystallized from i-PrOH to afford 0.10 g (50% yield) of 11f as a grey-colored solid; 1H NMR (400 MHz, DMSO-d6, δ): 10.48 (s, 2H), 7.98 (d, J = 8.0 Hz, 2H), 7.90 (d, J = 8.0 Hz, 2H), 7.86-7.83 (m, 4H), 7.46–7.71 (m, 2H), 7.36-7.34 (m, 2H), 7.06 (s, 2H); 13C NMR (100 MHz, DMSO-d6, δ): 140.8, 130.9, 130.9, 130.6, 130.1, 129.8 (d, JCF = 33 Hz), 129.6 (d, JCF = 5 Hz) 126.4, 123.7, 123.3 (d, JCF = 4 Hz), 123.3 (d, JCF = 271 Hz), 123.2; HRMS-ESI (−) (m/z): [M-H]− calcd for C24H15F6N2O4S2, 573.0383; found, 573.0370.
2.9.1.7. Synthesis of N,N′-(naphthalene-1,4-diyl)bis(3,4-dimethoxybenzenesulfonamide) (11g)
The crude product was recrystallized from i-PrOH to afford 0.15 g (65% yield) of 11g as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ): 10.03 (s, 2H), 8.00-7.93 (m, 2H), 7.42-7.40 (m, 2H), 7.18 (d, J = 8.0 Hz, 2H), 7.10-7.05 (m, 4H), 6.98 (d, J = 8.0 Hz, 2H), 3.77 (s, 6H), 3.61 (s, 6H); 13C NMR (100 MHz, DMSO-d6, δ): 152.1, 148.5, 131.4, 131.2, 130.2, 126.2, 123.5, 122.9, 120.5, 110.9, 109.4, 55.8, 55.6; HRMS-ESI (−) (m/z): [M-H]− calcd for C26H25N2O8S2, 557.1052; found, 557.1051.
2.9.1.8. Synthesis of N,N′-(naphthalene-1,4-diyl)bis(4-methylbenzenesulfonamide) (11h)
The crude product was recrystallized from i-PrOH to afford 0.092 g (53% yield) of 11h as a white-colored solid; 1H NMR (400 MHz, DMSO-d6) d 10.12 (s, 7H), 7.95 (dd, J = 3.3, 6.4 Hz, 2H), 7.51 (d, J = 8.2 Hz, 4H), 7.39 (dd, J = 3.4, 6.9 Hz, 2H), 7.27 (d, J = 8.1 Hz, 4H), 6.99 (s, 2H), 2.32 (s, 6H); 13C NMR (100 MHz, DMSO-d6, δ): 143.5, 137.5, 131.4, 130.5, 129.9, 127.2, 126.6, 123.8, 123.1, 21.4; HRMS-ESI (+) (m/z): [M+H]+ calcd for C24H23N2O4S2, 467.1099; found, 467.1105.
2.9.1.9. Synthesis of N,N′-(naphthalene-1,4-diyl)bis(4-(N,N-dimethylamino)benzenesulfonamide) (11i)
The crude product was washed with H2O (50 mL) and Et2O (20 mL) to obtain a grey solid which was recrystallized from MeOH to afford 0.13 g (65% yield) of 11i as a grey-colored solid; 1H NMR (400 MHz, DMSO-d6, δ): d 9.76 (s, 2H), 8.03-8.02 (m, 2H), 7.42--7.40 (m, 6H), 7.01 (s, 2H), 6.64(d, J = 8.4 Hz, 4H), 2.94 (s, 12H); 13C NMR (100 MHz, DMSO-d6, δ): 152.9, 131.6, 130.3, 128.8, 126.3, 125.4, 123.9, 122.4, 111.1.
2.9.1.10. Synthesis of N,N′-(naphthalene-1,4-diyl)bis(4-fluorobenzenesulfonamide) (11j)
The crude product was recrystallized from i-PrOH/Et2O to afford 0.12 g (57% yield) of 11i as pink-colored needles; 1H NMR (400 MHz, CDCl3, δ): 7.80-7.78 (dd, J = 2.8, 3.2 Hz, 1H), 7.75-7.72 (m, 2H), 7.48-7.46 (dd, J = 2.8, 3.2 Hz, 1H), 7.07 (t, J = 8.4 Hz, 2H); 13C NMR (100 MHz, DMSO-d6, δ): 164.7 (d, JCF = 250 Hz), 136.5 (d, JCF = 3 Hz), 131.4, 130.6, 130.2 (d, JCF = 10 Hz), 126.7, 123.7, 123.7, 116.7 (d, JCF = 23 Hz).
2.9.2. General method for synthesis of 12a–f, and 21 (Method B)
These previously unreported compounds were synthesized according a published procedure [34]. Potassium carbonate (3 equivalents) and suitable bromo-substituted electrophiles (2.5 equivalents) were added to a solution of 7 (1 equivalents) in anhydrous DMF (2 mL), and the reaction was stirred at room temperature until complete as monitored by TLC. On completion the reaction was quenched with H2O (25 mL) and acidified with HCl (2N, to pH 5). The precipitate was collected by filtration and purified by column chromatography or recrystallized with appropriate solvents to yield pure compounds.
2.9.2.1. Synthesis of diethyl 2,2′-(naphthalene-1,4-diyl)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetate (12a)
The crude product was recrystallized from EtOH to yield 0.26 g (65% yield) of 12a as a brown-colored solid; 1H NMR (400 MHz, DMSO-d6, δ): 8.34 (dd, J = 6.3, 3.3 Hz, 1H), 8.20 (dd, J = 6.4, 3.4 Hz, 1H), 7.63-7.57 (m, 6H), 7.16-7.06 (m, 5H), 6.84 (s, 1H), 4.59-4.40 (m, 4H), 4.07-3.96 (m, 4H), 3.89 (s, 3H), 3.84 (s, 3H), 1.05 (m, 6H); 13C NMR (100 MHz, DMSO-d6, δ): 168.9, 163.4, 137.5, 103.6, 103.4, 127.1, 114.8, 61.3, 56.2, 25.9, 14.3; HRMS-ESI (+) (m/z): [M+Na]+ calcd for C34H32N2O10S2Na, 693.1547; found, 693.1588.
2.9.2.2. Synthesis of N,N′-(naphthalene-1,4-diyl)bis(N-allyl-4-methoxybenzenesulfonamide) (12b)
The crude product was purified by column chromatography (silica gel; hexanes/EtOAc; 1:0 to 3:1) to afford 0.09 g (90% yield) of 12b as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.19-8.14 (m, 2H), 7.63 (d, J = 8.0 Hz, 4H), 7.53 (d, J = 8.0 Hz, 2H), 7.19 (d, J = 12 Hz, 2H), 7.08 (d, J = 8.0 Hz, 2H), 6.78 (s, 1H), 6.64 (s, 1H), 5.70-5.61 (m, 2H), 5.06-4.83 (m, 4H), 4.49-4.40 (m, 2H), 4.06-3.85 (m, 8H); 13C NMR (100 MHz, DMSO-d6, δ): 162.9, 135.9, 133.8, 133.7, 132.6, 132.4, 130.1, 129.9, 128.9, 128.7, 126.9, 125.6, 125.2, 124.2, 119.7, 119.5, 114.5, 55.8, 54.6, 54.4; HRMS-ESI (−) (m/z): [M-H]− calcd for C30H29N2O6S2, 577.1473; found, 577.1202.
2.9.2.3. Synthesis of N,N′-(naphthalene-1,4-diyl)bis(4-methoxy-N-methylbenzenesulfonamide) (12c)
The crude product was recrystallized from EtOH to yield 0.07 g (89% yield) of 12c as a buff-colored solid; 1H NMR (400 MHz, DMSO-d6, δ): 8.24 (s, 1H), 8.19 (s, 1H), 7.69 (dd, J = 6.4, 3.2 Hz, 2H), 7.63 (d, J = 8.8 Hz, 4H), 7.20 (d, J = 8.0 Hz, 2H), 7.14 (d, J = 8.0 Hz, 2H), 6.87 (s, 1H), 6.65 (s, 1H), 3.90 (s, 3H), 3.86 (s, 3H), 3.21 (s, 3H), 3.16 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ): 162.9, 138.4, 138.3, 130.2, 130.0, 127.6, 127.3, 124.1, 114.6, 114.6 55.8; HRMS-ESI (+) (m/z): [M+Na]+ calcd for C26H26N2O6S2Na, 549.1130; found, 549.1115.
2.9.2.4. Synthesis of N,N′-(naphthalene-1,4-diyl)bis(N-ethyl-4-methoxybenzenesulfonamide) (12d)
The crude product was purified by column chromatography (silica gel; hexanes/EtOAc; 1:0 to 1:1) to afford 0.06 g (77% yield) of 12d as a buff-colored solid; 1H NMR (400 MHz, DMSO-d6, δ): 8.25 (dd, J = 7.6, 4.0 Hz, 1H), 8.16 (dd, J = 6.4, 3.2 Hz, 1H), 7.69-7.60 (m, 6H), 7.20 (d, J = 8.0 Hz, 2H), 7.12 (d, J = 8.0 Hz, 2H), 6.92 (s, 1H), 6.68 (s, 1H), 3.98-3.81 (m, 2H), 3.87 (s, 3H), 3.82 (s, 3H), 3.45 (app. sextet, J = 7.2 Hz, 1H), 0.96 (t, J = 7.0 Hz, 3H), 0.88 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6, δ): 162.9, 136.3, 134.3, 129.8, 126.5, 125.1, 124.7, 113.8, 54.9, 54.8, 46.6, 13.1, 12.9; HRMS-ESI (+) (m/z): [M+Na]+ calcd for C28H30N2O6S2Na, 577.1437; found, 577.1405.
2.9.2.5. Synthesis of 2,2′-(naphthalene-1,4-diylbis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetamide (12e)
The crude product was recrystallized from i-PrOH to afford 0.04 g (65% yield) of 12e as a brown-colored solid; 1H NMR (400 MHz, DMSO-d6, δ): 8.19-7.99 (m, 2H), 7.64-7.41 (m, 6H), 7.13-6.93 (m, 6H), 4.48-4.32 (m, 4H), 3.92-3.82 (m, 6H); 13C NMR (100 MHz, DMSO-d6, δ): 169.1, 163.2, 162.9, 134.9, 133.7, 133.1, 132.0, 130.4, 130.3, 129.9, 129.4, 126.9, 126.8, 126.6, 125.1, 123.7, 121.7, 114.7, 114.6, 56.2, 56.1, 54.4; HRMS-ESI (+) (m/z): [M+Na]+ calcd for C28H28N4O8S2Na, 635.1241; found, 635.1294.
2.9.2.6. Synthesis of N,N′-bis-propargyl-(naphthalene-1,4-diyl)bis(4-methoxybenzenesulfonamide) (12f)
Yield: 50 mg (62% yield) of 12f as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.21 (dd, J = 6.6, 3.0 Hz, 1H), 8.17-8.11 (m, 1H), 7.70-7.62 (m, 6H), 7.17 (d, J = 8.9 Hz, 2H), 7.11 (d, J = 8.8 Hz, 2H), 7.01 (s, 1H), 6.82 (s, 1H), 4.60-4.46 (m, 4H), 3.90 (s, 3H), 3.85 (s, 3H), 3.25 (br s, 1H), 3.16 (s, 1H); 13C NMR (400MHz, DMSO-d6, δ):163.10, 163.05, 136.60, 133.55, 133.39, 130.30, 130.03, 129.55, 128.93, 127.28, 126.03, 125.66, 124.09, 114.55, 114.48, 78.41, 76.83, 76.65, 5.86, 41.69, 41.48; HRMS-ESI (+) (m/z): [M+Na]+ calcd for C30H26N2O6S2Na, 597.1158; found, 597.1130.
2.9.3. Synthesis of 4-methoxy-N-(4-nitronaphthalen-1-yl)benzenesulfonamide
This previously unreported compound was synthesized according to a similar procedure [48]. In a round-bottomed flask, 4-nitronaphthalen-1-amine (synthesized according to a reported procedure) [49] (5.0 g, 26.6 mmol) and 4-methoxybenzenesulfonyl chloride (6.6 g, 31.9 mmol) was dissolved in THF (30 mL) and pyridine (9 mL). The stirring mixture was brought to reflux and stirred for 48 hours. Upon reaction completion, monitored by TLC (1:1 Hex:EtOAc), the reaction mixture was concentrated under vacuum, taken up in EtOAc (50 mL), and washed with HCl (2N, 50 mL) and H2O (2 x 50 mL). The organic fraction was dried over sodium sulfate, filtered, and dried under vacuum. The resulting solid was washed with ether (2 x 25 mL) and recrystallized from toluene to yield 7.19 g (76% yield) of 4-methoxy-N-(4-nitronaphthalen-1-yl)benzenesulfonamide as an off white solid; 1H NMR (400 MHz, DMSO-d6, δ): 10.82 (br s, 1H), 8.38 (d, J = 8.7 Hz, 1H), 8.28 (t, J = 8.4 Hz, 2H), 7.70-7.83 (m, 3H), 7.66 (t, J = 7.2 Hz, 1H), 7.43 (d, J = 8.5 Hz, 1H), 7.05 (d, J = 8.9 Hz, 2H), 3.77 (s, 3H).
2.9.4. Synthesis of N-(4-aminonaphthalen-1-yl)-4-methoxybenzenesulfonamide hydrochloride
This previously unreported compound was synthesized according to a known, similar procedure [50]. Pd/C (10%, 0.05 g) was added to a suspension of previously prepared N-(4-nitronaphthalen-1-yl)-4-methoxybenzenesulfonamide (0.50 g, 1.4 mmol) in EtOH (30 mL) and HCl/EtOH (1.25 M, 5.0 mL), and the reaction was hydrogenated using a Parr shaker apparatus (35 psi) for 5 h. On completion the reaction mixture was filtered over Celite®, and the filter bed was washed with EtOH (20 mL). The combined organic portions were evaporated and dried to yield 0.50 g (98% yield) of N-(4-aminonaphthalen-1-yl)-4-methoxybenzenesulfonamide hydrochloride as a buff-colored solid which was utilized for the synthesis of 14a, 14b and 14d without further purification or characterization due to the relative instability of the aniline salt.
2.9.5. Synthesis of N-(4-aminonaphthalen-1-yl)benzenesulfonamide Hydrochloride
This previously unreported compound was synthesized according to a known, similar procedure [50]. Pd/C (10%, 0.45 g) was added to a suspension of N-(4-nitronaphthalen-1-yl)-4-benzenesulfonamide (0.40 g, 1.2 mmol) in EtOH (15 mL) and HCl/EtOH (1.25 M, 5 mL), and the reaction mixture was hydrogenated using a Parr shaker apparatus (40 psi) for 5 h. On completion the reaction was filtered over Celite®, and the filter bed was washed with EtOH (20 mL). The combined organic portions were evaporated and dried to yield 0.40 g (100% yield) of N-(4-aminonaphthalen-1-yl)benzenesulfonamide hydrochloride as a buff-colored solid which was utilized for the synthesis of 14c without further purification or characterization due to the instability of the aniline salt.
2.9.6. Synthesis of 4-methoxy-N-(4-(phenylsulfonamido)naphthalen-1-yl)benzenesulfonamide (14a)
This previously unreported compound was synthesized according to a known, similar procedure [48]. N-(4-Aminonaphthalen-1-yl)-4-methoxybenzenesulfonamide hydrochloride (0.10 g, 0.31 mmol) was added to a solution of anhydrous THF (10 mL), pyridine (0.040 mL, 0.46 mmol) and benzenesulfonyl chloride (0.070 g, 0.37 mmol), and the reaction mixture was allowed to stir at room temperature for 24 h. On completion, the organic solvent was evaporated, and the residue was dissolved in CH2Cl2 (40 mL), washed with H2O (2 x 30 mL) and HCl (2N, 2 x 20 mL), dried (Na2SO4) and evaporated to yield crude product, which was washed with hot acetone and Et2O to afford 0.14 g (89% yield) of 14a as a purple solid. 1H NMR (400 MHz, DMSO-d6, δ): 10.19 (s, 1H), 10.04 (s, 1H), 7.97-7.89 (m, 2H), 7.63-7.34 (m, 9H), 7.02 (s, 2H), 6.98 (d, J = 8.8 Hz, 2H), 3.77 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ): 162.4, 139.8, 132.8, 131.4, 130.8, 130.1, 129.0, 126.7, 126.2, 123.3, 123.1, 122.7, 114.3, 55.7; HRMS-ESI (−) (m/z): [M-H] calcd for C23H19N2O5S2, 467.0741. Found, 467.0755.
2.9.7. Synthesis of 4-methoxy-N-(4-((4-(trifluoromethyl)phenyl)sulfonamido)naphthalen-1-yl)benzenesulfonamide (14b)
This previously unreported compound was synthesized according to a known, similar procedure [48]. N-(4-Aminonaphthalen-1-yl)-4-methoxybenzenesulfonamide hydrochloride (0.20 g, 0.55 mmol) was added to a solution of anhydrous THF (10 mL), pyridine (1 mL) and p-trifluoromethylbenzenesulfonyl chloride (0.16 g, 0.70 mmol), and the reaction was allowed to stir at room temperature for 24 h. On completion, the organic solvent was evaporated and the residue was dissolved in CH2Cl2 (40 mL), washed with H2O (2 x 30 mL) and HCl (2N, 2 x 20 mL), dried (Na2SO4) and evaporated to yield crude product, which was recrystallized from MeOH to yield 0.15 g (55% yield) of 14b as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ): 10.45 (s, 1H), 10.45 (s, 1H), 7.96 (d, J = 8.0 Hz, 2H), 7.87-7.79 (m, 5H), 7.56 (d, J = 8.0 Hz, 2H), 7.40-7.33 (m, 2H), 7.07-7.02 (m, 2H), 6.98 (d, J = 8.0 Hz, 2H), 3.76 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ): 162.4, 143.7, 132.5 (q, JCF = 32 Hz), 131.8, 131.5, 130.3, 130.2, 130.01, 128.9, 127.7, 126.4 (d, JCF = 3 Hz), 126.3, 126.2, 123.8, 123.5, 123.4 (q, JCF = 271 Hz), 123.1, 122.6, 55.6; HRMS-ESI (−) (m/z): [M-H]− calcd for C23H19N2O5S2, 535.0615; found, 535.0636.
2.9.8. Synthesis of N-(4-(phenylsulfonamido)naphthalen-1-yl)-4-(trifluoromethyl)benzenesulfonamide (14c)
This previously unreported compound was synthesized according to a similar procedure [48]. N-(4-Aminonaphthalen-1-yl)benzenesulfonamide hydrochloride (0.20 g, 0.59 mmol) was added to a solution of anhydrous THF (10 mL) and pyridine (1 mL) and p-trifluoromethylbenzenesulfonyl chloride (0.22 g, 0.89 mmol), and the reaction was allowed to stir at room temperature for 20 h. On completion, the organic solvent was evaporated and the residue was dissolved in CH2Cl2 (40 mL), washed with H2O (2 x 30 mL) and HCl (2N, 2 x 20 mL), dried (Na2SO4) and evaporated to yield crude product, which was recrystallized from EtOH to afford 0.10 g (37% yield) of 14c as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ): 10.47 (s, 1H), 10.25 (s, 1H), 7.92-7.80 (m, 6H), 7.63 (d, J = 8.0 Hz, 2H), 7.58-7.54 (m, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.36-7.32 (m, 2H), 7.05 (s, 2H); 13C NMR (100 MHz, DMSO-d6, δ): 144.1, 140.2, 133.2, 132.9 (q, JCF = 32 Hz), 131.9, 130.8, 130.6, 130.5, 129.6, 128.1, 127.1, 126.9 (d, JCF = 3 Hz), 126.7, 126.7, 124.1, 123.8 (d, JCF = 270 Hz), 123.8, 123.5, 123.4; HRMS-ESI (+) (m/z): [M-H]− calcd for C23H16F3N2O4S2, 507.0628; found, 507.0631.
2.9.9. Synthesis of 3-methoxy-N-(4-((4-methoxyphenyl)sulfonamido)naphthalen-1-yl)benzenesulfonamide (14d)
This previously unreported compound was synthesized according to a known, similar procedure [48]. N-(4-Aminonaphthalen-1-yl)-4-methoxybenzenesulfonamide hydrochloride (0.10 g, 0.31 mmol) was added to a solution of pyridine (2 mL) and 3-methoxybenzenesulfonyl chloride (0.070 g, 0.35 mmol), and the reaction mixture was allowed to stir at room temperature for 24 h. On completion, the organic solvent was evaporated, and the residue was dissolved in CH2Cl2 (40 mL), washed with H2O (2 x 30 mL) and HCl (2N, 2 x 20 mL), dried (Na2SO4) and evaporated to yield crude product, which was recrystallized with EtOH to afford 0.10 g (63% yield) of 14d as light brown-colored needles. 1H NMR (400 MHz, DMSO-d6, δ): 10.19 (s, 1H), 10.05 (s, 1H), 7.98-7.92 (m, 2H), 7.57-7.55 (AA′XX′, J = 8.8 Hz, 2H), 7.41-7.37 (m, 3H), 7.21-7.10 (m, 3H), 7.04 (s, 2H), 6.99-6.97 (AA′XX′, J = 8.8 Hz, 2H), 3.78 (s, 3H), 3.67 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ): 162.7, 159.6, 141.5, 131.9, 131.7, 131.2, 130.7, 130.5, 130.4, 129.3, 126.6, 123.8, 123.7, 123.4, 123.1, 119.2, 119.2, 114.6, 111.9, 56.1, 55.9.
2.9.10. Synthesis of ethyl N-((4-methoxyphenyl)sulfonyl)-N-(4-nitronaphthalen-1-yl)glycinate (15)
This previously unreported compound was synthesized according to a similar procedure [34]. In a round-bottomed flask, 4-methoxy-N-(4-nitronaphthalen-1-yl)benzenesulfonamide (1.0 g, 2.8 mmol) and potassium carbonate (1.16 g, 8.4 mmol) was dissolved in N-methylpyrrolidinone (4 mL) and stirred for 30 minutes at 85 °C. Ethyl bromoacetate (1.17 g, 6.98 mmol) was added to the stirring mixture and stirred for an additional 1 hour. Upon reaction completion, as determined by TLC, the reaction mixture was poured into stirring ice water (75 mL). The aqueous mixture was extracted with EtOAc (3 x 50 mL), and the combined organic phases were washed with HCl (2N, 30 mL), H2O (2 x 50 mL), and saturated brine (50 mL). The organic fraction was dried over sodium sulfate, filtered, and evaporated under vacuum to yield 1.21 g (98% yield) 15 as a yellow-colored solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.39 (d, J = 8.3 Hz, 1H), 8.23 (d, J = 8.2 Hz, 1H), 8.26 (d, J = 8.6 Hz, 1H), 7.83 (t, J = 7.2 Hz, 1H), 7.76 (t, J = 7.3 Hz, 1H), 7.68-7.59 (m, J = 8.9 Hz, 2H), 7.34 (d, J = 8.2 Hz, 1H), 7.15-7.08 (m, J = 8.9 Hz, 2H), 4.58 (s, 2H), 4.18-4.10 (m, 2H), 3.86 (s, 3H), 1.11-1.03 (m, 3H).
2.9.11. Synthesis of ethyl N-(4-((4-methoxyphenyl)sulfonamido)naphthalen-1-yl)-N-((4-methoxyphenyl) sulfonyl)glycinate (16a)
This previously unreported compound was synthesized according to a known procedure [48]. In a round bottomed flask, previously prepared 15 (1.21 g, 2.72 mmol) and tin chloride dihydrate were dissolved in EtOH (35 mL). The solution was stirred at reflux for 3 hours. Upon completion, as determined by TLC, the reaction mixture was concentrated under vacuum. The mixture was basified with NaHCO3 to pH 9 and extracted with EtOAc (200 mL). The organic phase was then washed with H2O (3 x 100 mL) and brine (100 mL), dried over sodium sulfate, and filtered. 2M HCl in EtOH (10 mL) was then added to the organic layer, the precipitate was collected and washed with EtOAc to yield 0.9 g (70% yield) of aniline salt as a fluffy yellow-colored solid, of which 0.5 g (1.1 mmol), and 4-methoxybenzenesulfonyl chloride (0.298 mg, 1.44 mmol) were dissolved in pyridine (3 mL). The solution was stirred at room temperature for 24 hours. Upon completion, determined by TLC, the reaction mixture was diluted with EtOAc (100 mL) and washed with HCl (2N, 2 x 75 mL), H2O (2 x 75 mL), and brine (75 mL). The organic section was dried over sodium sulfate, filtered, and evaporated to yield 0.59 g (90% yield) of 16a as an off-white solid; 1H NMR (400 MHz, DMSO-d6, δ): 10.21 (s, 1H), 8.15 (d, J = 8.3 Hz, 1H), 8.09 (d, J = 7.8 Hz, 1H), 7.65 (d, J = 8.8 Hz, 2H), 7.55 (d, J = 8.7, 2H), 7.53-7.46 (m, 2H), 7.10-7.01 (m, 4H), 7.02-6.96 (m, 2H), 4.46 (s, 2H), 3.99 (q, J = 7.5 Hz, 2H), 3.87 (s, 3H), 3.80 (s, 3H), 1.04 (t, J = 7.1 Hz, 3H); 13C NMR (400 MHz, DMSO-d6, δ): 168.6, 163.0, 162.5, 134.4, 133.5, 132.7, 131.6, 130.0, 129.9, 129.2, 129.0, 126.7, 126.6, 126.4, 124.6, 123.3, 121.3, 114.4, 60.9, 59.8, 55.8, 55.7, 53.3, 20.8, 14.1, 13.9; HRMS-ESI (+) (m/z): [M+H]+ calcd for C28H29N2O8S2, 585.1365; found, 585.1360
2.9.12. Synthesis of N-(4-((4-methoxyphenyl)sulfonamido)naphthalen-1-yl)-N-((4-methoxyphenyl)sulfonyl) glycine (16b)
This previously unreported compound was synthesized according to a known similar procedure [34]. In a round-bottomed flask ethyl 16a (0.04 g, 0.06 mmol) and NaOH (0.24 g, 5.99 mmol) were dissolved in MeOH (2 mL) and H2O (2 mL). The solution was stirred at reflux for 3 hours. Upon completion, as determined by TLC, the reaction mixture was acidified with HCl (2 N, to pH 3) and extracted with EtOAc (25 mL). The organic phase was washed with H2O (3 x 30 mL), dried over Na2SO4, filtered, and evaporated to yield 0.03 g (90% yield) of 16b as a white-colored solid; 1H NMR (400 MHz, DMSO-d6, δ): 10.21 (s, 1H), 8.13 (d, J = 9.4 Hz, 1H), 8.06 (d, J = 9.6 Hz, 1H), 7.64 (d, J = 8.9 Hz, 2H), 7.52 (d, J = 8.8 Hz, 2H), 7.50-7.45 (m, 2H), 7.04 (dd, J = 9.0, 2.8 Hz, 4H), 6.99 (d, J = 3.1 Hz, 2H), 4.35 (s, 2H), 3.84 (s, 3H), 3.79 (s, 3H); 13C NMR (400 MHz, DMSO-d6, δ): 170.0, 162.9, 162.5, 134.5, 133.4, 132.6, 131.6, 129.9, 129.9, 129.4, 129.0, 126.8, 126.6, 126.3, 124.6, 123.3, 121.4, 114.4, 114.3, 59.8, 55.8, 55.7, 53.2, 20.8, 14.2; HRMS-ESI (+) (m/z): [M+H]+ calcd for C26H25N2O8S2, 557.1052; found, 557.1055.
2.9.13. Synthesis of 4-methoxy-N-(naphthalen-1-yl)benzenesulfonamide (17)
This previously unreported compound was synthesized according to a known, similar procedure [48]. A solution of 1-aminonaphthalene hydrochloride (0.50 g, 2.2 mmol) and p-methoxybenzenesulfonyl chloride (0.55 g, 2.7 mmol) in pyridine (5 mL) was allowed to stir at room temperature for 19 h. On completion, the reaction mixture was quenched with H2O (30 mL), acidified with HCl (2N, to pH 6) and the precipitate was collected by filtration. The collected precipitate was washed with H2O (2 x 50 mL), dried and the crude product was recrystallized from i-PrOH to afford 0.50 g (73% yield) of 17 as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ): 10.01 (s, 1H), 8.05 (d, J = 8.3 Hz, 1H), 7.88 (d, J = 7.3 Hz, 1H), 7.77 (d, J = 8.3 Hz, 1H), 7.61 (d, J = 8.8 Hz, 2H), 7.50-7.38 (m, 3H), 7.15 (d, J = 7.1 Hz, 1H), 7.01 (d, J = 8.8 Hz, 2H), 3.78 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ): 162.4, 133.9, 132.6, 129.4, 128.9, 128.0, 126.6, 126.2, 126.1, 125.6, 123.2, 122.9, 114.3, 55.7; HRMS-ESI (−) (m/z): [M-H]− calcd for C17H14NO3S, 312.0700; found, 312.0655.
2.9.14. Synthesis of N-((4-methoxyphenyl)sulfonyl)-N-(naphthalen-1-yl)glycine (18)
This previously unreported compound was synthesized according to a published procedure [34]. Potassium carbonate (0.01 g, 0.72 mmol) and ethyl bromoacetate (0.09 g, 0.58 mmol) were added to a solution of 17 (0.15 g, 0.48 mmol) in anhydrous DMF (2 mL), and the reaction mixture was stirred at room temperature overnight. On completion the reaction was quenched with H2O (50 mL) and extracted with EtOAc (2 x 20 mL). The combined organic portion was washed with H2O (5 x 50 mL), brine (30 mL), dried (Na2SO4) and evaporated to yield the crude which was suspended in NaOH (2.5 g, 37 mmol), MeOH (12 mL) and H2O (12 mL) and allowed to reflux for 4 h. On completion the reaction mixture was cooled, diluted with H2O (20 mL) and extracted with EtOAc (2 x 25 mL). The aqueous portion was acidified with HCl (2N, to pH 2) and extracted with EtOAc (2 x 50 mL). The combined organic portion was washed with H2O (3 x 50 mL) and brine (50 mL), dried (Na2SO4) and evaporated to yield the crude product which, upon recrystallization from i-PrOH/Hexanes, yielded 0.13 g (71% yield) of 18 as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ): 12.80 (br s, 1H), 8.17-8.15 (m, 1H), 7.96-7.93 (m, 2H), 7.60 (d, J = 9.1 Hz, 2H), 7.55-7.51 (m, 2H), 7.43 (t, J = 7.8 Hz, 1H), 7.18 (d, J = 7.3 Hz, 1H), 7.09 (d, J = 9.1 Hz, 2H) 4.49-4.36 (m, 2H), 3.85 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ): 170.4, 163.2, 136.9, 134.6, 132.2, 130.3, 129.4, 128.4, 128.3, 126.9, 125.8, 124.7, 114.8, 56.2, 53.6; HRMS-ESI (−) (m/z): [M-H]− calcd for C19H16NO5S, 370.0755; found, 370.0711.
2.9.15. Synthesis of N,N′-(naphthalene-1,3-diyl)bis(4-methoxybenzenesulfonamide) (19)
This previously unreported compound was synthesized according to a known, similar procedure [51]. A solution of 1,3-dinitronaphthalene (0.15 g, 0.70 mmol) and tin chloride dihydrate (2.3 g, 10 mmol) in EtOAc (15 mL) was heated at reflux for 30 min. On completion the reaction was cooled to room temperature and quenched in ice, basified with sat. NaHCO3 (to pH 7–8), and extracted with EtOAc (3 x 25 mL). The combined organic portion was washed with H2O (3 x 30 mL), brine (30 mL), dried (Na2S2O4) and evaporated to yield crude 0.10 g (100% yield) of naphthalene-1,3-diamine, which was utilized in the synthesis of 19 without further purification or characterization due to the instability of the free base [52]. Naphthalene-1,3-diamine (0.10 g, 0.70 mmol) was dissolved in anhydrous CH2Cl2 (10 mL) and pyridine (0.20 mL, 2.1 mmol) at 0 °C. p-Methoxybenzenesulfonyl chloride (0.40 g, 2.1 mmol) was added and the reaction was allowed to stir at room temperature for 24 h. On completion, the precipitate was collected by filtration, washed with CH2Cl2 (20 mL), washed with H2O (3 x 40 mL), dried (Na2SO4), and evaporated to yield a brown oil which was purified by column chromatography (silica gel; hexanes/EtOAc; 4:1 to 2:1) to afford 0.30 g (96% yield) of 19 as a yellow-colored solid. 1H NMR (400 MHz, DMSO-d6, δ) 10.40 (s, 1H), 10.12 (s, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.68-7.56 (m, 5H), 7.39-7.25 (m, 4H), 6.99 (t, J = 8.0 Hz, 4H), 3.76 (s, 3H), 3.74 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ) 162.5, 162.4, 135.3, 133.9, 133.7, 131.5, 130.3, 128.9, 127.4, 126.9, 124.8, 122.8, 115.9, 114.4, 111.3, 113.7, 55.7, 55.6; HRMS-ESI (−) (m/z): [M-H]− calcd for C24H21N2O6S2, 497.0887; found, 497.0864.
2.9.16. Synthesis of N,N′-(naphthalene-2,3-diyl)bis(4-methoxybenzenesulfonamide) (20): (Method A)
2,3-Diaminonaphthalene (0.40 g, 2.5 mmol) was used as the amine source. Crude product was recrystallized from EtOAc/MeOH to afford 1.3 g (92% yield) of 20 as yellow flakes. 1H NMR (400 MHz, DMSO-d6, δ): 9.40 (s, 2H), 7.67 (d, J = 8.8 Hz, 6H), 7.50 (s, 2H), 7.37 (dd, J = 6.4, 3.2 Hz, 2H), 7.03 (d, J = 8.8 Hz, 4H), 3.77 (s, 6H); 13C NMR (100 MHz, DMSO-d6, δ): 162.8, 130.3, 129.4, 128.7, 127.1, 126.3, 120.7, 114.5, 55.7; HRMS-ESI (+) (m/z): [M+H]+ calcd for C24H23N2O6S2, 499.0992; found, 499.0973.
2.9.17. Synthesis of 2,2′-(naphthalene-1,3-diylbis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetamide (21): (Method B)
Crude product was recrystallized with EtOH/iPrOH yielded 0.09 g (71% yield) of 21 as buff-colored needles; 1H NMR (400 MHz, DMSO-d6, δ): 8.32-8.29 (m, 1H), 7.84-7.81 (m, 1H), 7.78 (s, 1H), 7.56-7.52 (m, 6H), 7.36 (br s, 1H), 7.27 (br s, 1H), 7.17 (br s, 1H), 7.06 (dd, J = 1.6, 8.8 Hz, 4H), 6.97 (d, J = 2 Hz, 2H), 4.27-4.18 (m, 2H), 4.02-3.95 (m 2H), 3.86 (s, 3H), 3.84 (S, 3H); 13C NMR (100 MHz, DMSO-d6, δ): 169.1, 168.8, 163.2, 137.9, 137.3, 133.9, 131.7, 130.5, 130.1, 129.5, 129.4, 128.2, 127.4, 127.3, 126.6, 125.3, 114.9, 114.8, 56.5, 56.1, 54.4, 53.3.
2.9.18. Synthesis of 2,2′-(naphthalene-1,3-diylbis(((4-methoxyphenyl)sulfonyl)azanediyl))acetic acid (22)
This previously unreported compound was synthesized according to a known, similar procedure [34]. Potassium carbonate (0.1 g, 0.7 mmol) and ethylbromoacetate (0.09 g, 0.5 mmol) were added to a solution of 19 (0.12 g, 0.2 mmol) in anhydrous DMF (2 mL), and the reaction mixture was stirred at room temperature overnight. On completion the reaction was quenched with H2O (30 mL), and the yellow-colored residue was collected by filtration to yield 0.14 g (90% yield) of yellow-colored intermediate. Further, the intermediate (0.08 g, 0.1 mmol) was suspended in NaOH (15%, 3 mL) and MeOH (10 mL) and the reaction mixture was allowed to reflux overnight. On completion the reaction mixture was cooled and MeOH was removed by evaporation. The solid was dissolved with H2O (20 mL) and the aqueous portion was acidified with HCl (2N, to pH 2). The solid was collected by filtration which, upon recrystallization from MeOH, yielded 0.05 g (68% yield) of 22 as a brown-colored solid; 1H NMR (400 MHz, DMSO-d6, δ): 8.26 (d, J = 8.8 Hz, 1H), 7.89 (d, J = 6.8 Hz, 1H), 7.80 (s, 1H), 7.69-7.52 (m, 6H), 7.08-7.01 (m, 4H), 6.88 (s, 1H), 3.86 (s, 3H), 4.44-4.09 (m, 4H), 3.83 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ): 170.1, 169.9, 163.3, 163.2, 137.9, 137.1, 134.1, 131.5, 130.4, 130.1, 129.9, 129.4, 128.4, 127.6, 127.5, 127.3, 126.6, 124.9, 114.8, 114.8, 56.1, 53.5, 52.2.
2.9.19. Synthesis of 2-(naphthalene-2,3-diylbis(((4-methoxyphenyl)sulfonyl)azanediyl))acetamide (23)
This previously unreported compound was synthesized according to a known, similar procedure [34]. Potassium carbonate (0.08 g, 0.6 mmol) and bromoacetamide (0.06 g, 0.4 mmol) were added to a solution of 20 (0.1 g, 0.20 mmol) in anhydrous DMF (2 mL), and the reaction mixture was stirred at room temperature overnight. On completion the reaction was quenched with H2O (70 mL) and extracted with EtOAc (2 x 30 mL). The combined organic portions were washed with H2O (4 x 100 mL), brine (50 mL), dried (Na2SO4) and evaporated to yield the crude product which was purified using preparative TLC (Hexanes:EtOAc 1:2) to afford 0.06 g (50% yield) of 22 as a buff-colored solid; 1H NMR (400 MHz, DMSO-d6, δ): (m, 2H), 7.64-7.41 (m, 6H), 7.13-6.93 (m, 6H), 4.48-4.32 (m, 4H), 3.92-3.82 (m, 6H); 13C NMR (100 MHz, DMSO-d6, δ): 169.1, 163.2, 162.9, 134.9, 133.7, 133.1, 132.0, 130.4, 130.3, 129.9, 129.4, 126.9, 126.8, 126.6, 125.1, 123.7, 121.7, 114.7, 114.6, 56.2, 56.1, 54.4.
2.9.20. Synthesis of N,N′-(1,4-Phenylene)bis(4-methoxybenzenesulfonamide) (24)
This previously unreported compound was synthesized according to a known, similar procedure [51]. 1,4-Phenylenediamine (0.5 g, 4.6 mmol) was added to a solution of anhydrous CH2Cl2 (10 mL) and pyridine (1.2 mL, 13.9 mmol) at 0 °C. p-Methoxybenzenesulfonyl chloride (2.4 g, 11.6 mmol) was added, and the reaction was allowed to stir at room temperature for 24 h. On completion, the precipitate was collected by filtration, washed with CH2Cl2 (30 mL), and recrystallized from EtOH to yield 1.6 g (76%) of 24 as pink-colored flakes. 1H NMR (400 MHz, DMSO-d6, δ): 9.93 (s, 2H), 7.58 (t, J = 8.8, 1.6 Hz, 4H), 7.01 (d, J = 8.8 Hz, 4H), 6.90 (s, 4H), 3.79 (s, 6H); 13C NMR (100 MHz, DMSO-d6, δ): 162.4, 134.1, 131.1, 128.9, 121.5, 114.3, 55.7; HRMS-ESI (−) (m/z): [M-H]− calcd for C20H20N2O6S2, 447.0696; found, 447.0690.
2.9.21. Synthesis of N,N′-(Naphthalene-1,4-diyl)bis(4-methoxybenzamide) (25)
This previously unreported compound was synthesized according to a known, similar procedure [48]. 1,4-Diaminonaphthalene (0.10 g, 0.63 mmol) was added to a solution of anhydrous CHCl3 (10 mL) and pyridine (0.15 mL, 1.9 mmol) at 0 °C. p-Methoxybenzoyl chloride (0.21 mL, 1.6 mmol) was added in a dropwise manner and the reaction was allowed to stir at room temperature for 24 h. On completion the reaction mixture was diluted with H2O (25 mL), and the precipitate was collected by filtration, washed with EtOH (10 mL), and recrystallized with EtOH to yield 0.15 g (56% yield) of 22 as pink-colored solid. 1H NMR (400 MHz, DMSO-d6, δ) 10.31 (s, 2H), 8.11 (d, J = 8.0 Hz, 2H), 8.02-8.01 (m, 2H), 7.60-7.57 (m, 4H), 7.11 (d, J = 8.0 Hz, 4H), 3.87 (s, 6H); 13C NMR (100 MHz, DMSO-d6, δ) 165.7, 162.1, 132.4, 130.0, 129.8, 126.6, 125.9, 123.8, 113.7, 55.5; HRMS-ESI (+) (m/z): [M+H]+ calcd for C26H23N2O4, 427.1649; found, 427.1652.
3. Results
3.1. Chemistry
7 and 8 were synthesized as described previously [34]. Briefly, 1,4-diaminonaphthalene was subjected to sulfonylation with p-methoxybenzenesulfonyl chloride in pyridine to yield 7. This route was also used to synthesize various substituted analogs (i.e., 11a–j; Scheme 1). 7 was further alkylated with ethyl bromoacetate in the presence of a base to obtain the di-ethyl carboxylate (i.e., 12a), which yielded the di-acid analog 8 after saponification.
Scheme 1. Synthesis of symmetrically substituted N,N′-dialkylated sulfonamidesa.

Reagents: (a) pyridine, rt; (b) R′Br, K2CO3, DMF, rt; (c) NaOH, MeOH, H2O, reflux.
As reported previously by Jiang, et al. [34], the 1H NMR spectrum of 8 showed a doubling of signals; for instance, there were two singlets of 3H each at 3.83 and 3.88 ppm, corresponding to the methyl ethers, and a multiplet of 4H centered around 4.39 ppm, corresponding to the methylene groups α to the ethyl esters. This effect was not seen with the unsubstituted sulfonamides (e.g., 7) and was surprising, given the elements of symmetry suggested by the planar structure. The same effect was observed in the 1D/2D spectra of other di-substituted sulfonamides collected at room temperature. In DMSO, 8 and congeners may adopt one or more solution conformations where the flanking p-methoxybenzenesulfonamide substituents are oriented in opposite directions, based on 2D ROESY data collected at room temperature. We believe this phenomenon is due to restricted rotation about the Nsulfonamide-Cnaphthalene bond that arises with increasing steric bulk on the substituted sulfonamide nitrogen. The coalescence temperature was found to be 313 K, which translates into a low activation barrier for this motion that can be easily overcome upon binding. When NMR studies were performed at temperatures higher than 313 K (e.g., 353 K) each set of peaks appeared as one signal (see supplementary information). On the other hand, crystals of this molecule obtained from slow evaporation of methanol showed the “expected” form with a mirror plane of symmetry (see supplementary information). The methodology used to synthesize 7 and 8 was also used to prepare various di-substituted analogs using the appropriate electrophiles (i.e., 12b–f). 1,4-diaminobenzene was used to synthesize 24. Compound 25 was synthesized from p-anisoyl chloride and 1,4-diaminonaphthalene. Analogs bearing unsymmetrical substitutions were synthesized as shown in Scheme 2. 1-Nitronaphthalene was aminated in the presence of hydroxylamine and a base to yield 4-amino-1-nitronaphthalene [49], which upon sulfonylation yielded 13. Further reduction of the nitro group afforded a mono-sulfonylated amine, which was further sulfonylated using various benzenesulfonyl chlorides to yield unsymmetrical sulfonamides (i.e., 14a–d). In an alternate route, sulfonamide 13 was alkylated before reduction and sulfonylation to give mono-alkylated analogs 16a–b. 1-Aminonaphthalene was sulfonylated to yield 17, which upon alkylation and saponification yielded 18. Similar to the synthesis of 7, 1,3- and 2,3-diaminonaphthalene analogs were used as starting materials to synthesize 19 and 20. Further alkylation of 19 and 20 with 2-bromoacetamide yielded 21 and 23, respectively. Similar to the synthesis of 8, alkylation of 19 with ethyl bromoacetate, followed by saponification, yielded 22.
Scheme 2. Synthesis of unsymmetrical and mono-alkylated p-substituted sulfonamidesa.

aReagents: (a) NH2OH·HCl, NaOH, MeOH, H2O, reflux; (b) pyridine, rt; (c) R′Br, K2CO3, DMF/NMP, rt/reflux; (d) 10% Pd/C, HCl/EtOH, EtOH, H2 (35 psi), rt; (e) R″-Ph-SO2Cl, pyridine, rt; (f) SnCl2·H2O, MeOH, reflux; (g) NaOH, MeOH, H2O, reflux.
3.2. In vitro assays
To test the potency of the analogs in vitro, a fluorescence anisotropy assay was carried out according to a previously reported procedure [43]. Briefly, inhibition of the Keap1-Nrf2 interaction with small molecules was assayed using both the Kelch domain of Keap1 and an N-terminally fluorescein-labeled 9-mer peptide containing the ETGE motif derived from the Neh2 domain of Nrf2 [43]. The experiments were performed in triplicate. Sigmoidal concentration-response curves were fitted to the data using Graphpad Prism 6.1 software (see Supplementary information and Tables 1 and 2).
Table 1.
Inhibition of association of Nrf2 peptide with Keap1 (Kelch domain) using fluorescence anisotropy assay and dissociation equilibrium constant (Kd) by surface plasmon resonance (SPR) assay.
| ||||||
|---|---|---|---|---|---|---|
| Cmpd. | R | R1 | R2 | R3 | IC50 (nM) (95% CL)b | Kd (nM) ± SDc |
| 7 | p-OCH3 | p-OCH3 | H | H | 940 (850–1300) | 1700 ± 120 |
| 8 | p-OCH3 | p-OCH3 | CH2COOH | CH2COOH | 24 (19–31) | 20 ± 3 |
| 11a | H | H | H | H | > 25000 | NDa |
| 11b | m-OCH3 | m-OCH3 | H | H | 630 (400–1000) | 2300 ± 110 |
| 11c | p-Cl | p-Cl | H | H | > 25000 | NDa |
| 11d | m-COOH | m-COOH | H | H | > 25000 | NDa |
| 11e | p-CF3 | p-CF3 | H | H | > 25000 | NDa |
| 11f | m-CF3 | m-CF3 | H | H | > 25000 | NDa |
| 11g | m,p-di OCH3 | m,p-di OCH3 | H | H | 1100 (800–1500) | NDa |
| 11h | p-CH3 | p-CH3 | H | H | 890 (480–1650) | NDa |
| 11i | p-N(CH3)2 | p-N(CH3)2 | H | H | 190 (84–444) | NDa |
| 11j | p-F | p-F | H | H | 660 (172–2565) | NDa |
| 12a | p-OCH3 | p-OCH3 | CH2COOEt | CH2COOEt | > 25000 | > 250000 |
| 12b | p-OCH3 | p-OCH3 | CH2CH=CH2 | CH2CH=CH2 | > 25000 | NDa |
| 12c | p-OCH3 | p-OCH3 | CH3 | CH3 | > 25000 | NDa |
| 12d | p-OCH3 | p-OCH3 | Et | Et | > 25000 | NDa |
| 12e | p-OCH3 | p-OCH3 | CH2CONH2 | CH2CONH2 | 63 (52–85) | 44 ± 8 |
| 12f | p-OCH3 | p-OCH3 | CH2C≡CH | CH2C≡CH | > 25000 | NDa |
| 14a | H | p-OCH3 | H | H | > 25000 | NDa |
| 14b | p-OCH3 | p-CF3 | H | H | > 25000 | NDa |
| 14c | H | p-CF3 | H | H | > 25000 | NDa |
| 14d | p-OCH3 | m-OCH3 | H | H | 410 (150–1100) | NDa |
| 16a | p-OCH3 | p-OCH3 | H | CH2COOEt | 85 (45–160) | 400 ± 84 |
| 16b | p-OCH3 | p-OCH3 | H | CH2COOH | 61 (46–82) | 110 ± 11 |
ND: Not determined;
95% confidence level, as calculated by Graphpad Prism 6.1; fluorescence anisotropy experiments were performed in triplicate;
standard deviation from three independent experiments.
Table 2.
Inhibition of association of Nrf2 peptide with Keap1 (Kelch domain) using fluorescence anisotropy assay.
| |||||
|---|---|---|---|---|---|
| Cmpd. | Substitution pattern | R1 | R2 | X | IC50 (nM) (95% CL)a |
| 17 | 1- | -H | _ b | -SO2- | >25000 |
| 18 | 1- | -CH2COOH | _ b | -SO2- | >25000 |
| 19 | 1,3- | -H | -H | -SO2- | > 25000 |
| 20 | 2,3- | -H | -H | -SO2- | > 25000 |
| 21 | 1,3- | -CH2CONH2 | -CH2CONH2 | -SO2- | > 25000 |
| 22 | 1,3- | -CH2COOH | -CH2COOH | -SO2- | 5500 (1400–22000) |
| 23 | 2,3- | -H | -CH2CONH2 | -SO2- | > 25000 |
| 24 | 1,4- | -H | -H | -CO- | > 25000 |
| 25 | 1,4- | -H | -H | -SO2- | >25000 |
95% confidence level, as calculated by Graphpad Prism 6.1; fluorescence anisotropy experiments were performed in triplicate;
Compounds 17 and 18 lack the second arylsulfonamide substituent.
A subset of the compounds with demonstrated ability to bind to the Kelch domain via the FP assays was tested in a surface plasmon resonance (SPR) assay. Briefly, the Kelch domain of Keap1 was immobilized on a CM5 sensor chip. Compound solutions of increasing concentrations were applied to the protein-bound chip, and response units were measured (see Supplementary Information and Table 1). The experiments were conducted three separate times. The Nrf2 peptide (FITC-LDEETGEFL-NH2) alone was tested by SPR as a control. The determined Kd value was 68.9 nM, which is slightly weaker than the reported Kd (25 nM). Approximately 60% of captured Keap1 was active based on the comparison between calculated RUmax and achieved RUmax:
3.3. Docking and Hydration Site Analysis
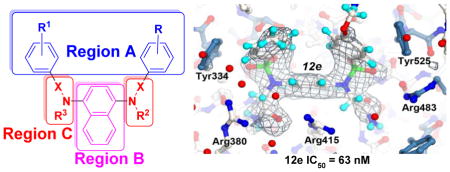
Inspection of the previously determined X-ray structure of 7 bound to the Kelch domain of Keap 1 (4IQK) reveals an interesting binding mode wherein the ligand is flanked by two co-crystallized water molecules (water 1 and water 2, shown in red spheres in Figure 2A) that are separated by Arg 415. We hypothesized that substitutions at the R2 and R3 positions in Region C (Figure 2B; see Discussion section) may potentially utilize the pockets that these waters occupy, displacing the water molecules in the process. To investigate this hypothesis and further elucidate structure-activity relationships and the role of these water molecules in ligand binding, ensemble docking and hydration site analyses were performed on a subset of six ligands (7, 8, 12a, 12e, 16a, 16b) with substitutions at the R2 and/or R3 positions. The top-ranked docking pose for each molecule along with the results of the WATsite analysis are shown in Figure 3.
Figure 2.
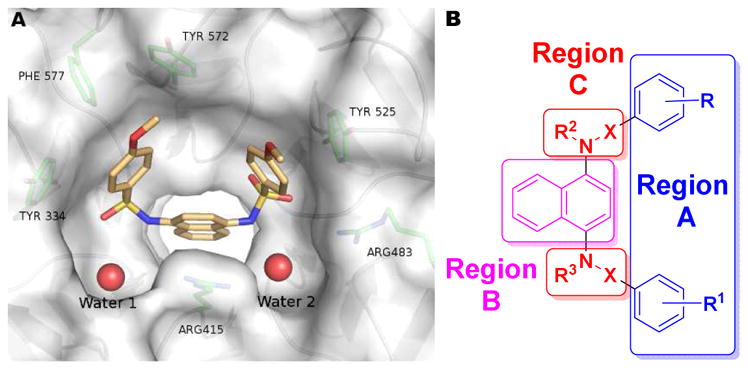
A) Co-crystal structure of 7 with Keap1 Kelch domain (4IQK). B) Schematic of the regions of modification on 7.
Figure 3.
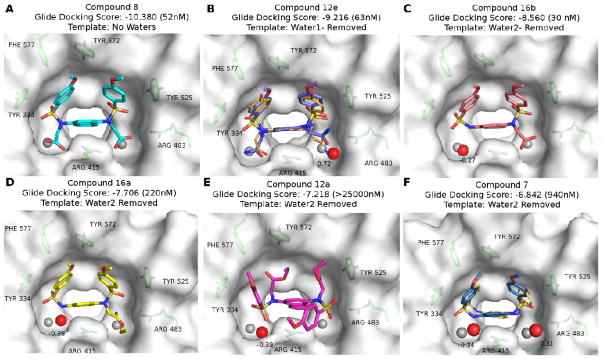
Docking and WATsite results for compounds (A) 8, (B) 12e, (C) 16b, (D) 16a, (E) 12a and (F) 7. For each molecule, the Glide docking score is shown, and the measured IC50 is noted in parentheses. The template selected by the ensemble docking procedure is shown below the docking score for each compound. The locations of the waters (shown as red spheres in Figure 2A) in the crystal structure 4IQK are shown as grey spheres in each frame. High occupancy (>80%) hydration sites predicted by WATsite that were within 2.0 Å of crystal waters are shown as red spheres. The ΔG of the hydration sites are shown in kcal/mol next to each site; a negative value indicates a favorable hydration site, and a positive value indicates an unfavorable site. The crystal structures of compound 12e (PDB: 4XMB; Panel B) and 7 (PDB: 4IQK; panel F) are shown in smaller orange sticks.
3.4. X-ray structure of the Kelch-compound 12e complex
To help support the computational docking analysis, we co-crystallized Keap1 Kelch domain as a complex with compound 12e and determined the X-ray structure of the complex to 2.47 Å (Rfree = 22.8%, Rwork = 17.3%). Compound 12e was chosen due to its nanomolar potency and computational prediction that one water (water 1) would be displaced (shown in Figure 3B). The X-ray structure of 12e and its associated electron density in the Kelch binding site are shown in Figure 4.
Figure 4.
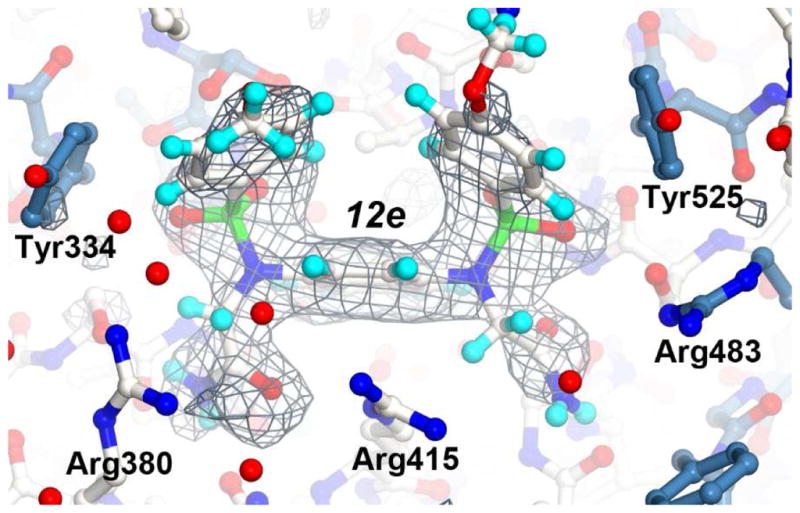
X-ray structure of the Kelch domain from human Keap1 in complex with compound 12e. The structure is shown as a ball-and-stick representation with atoms colored as follows: oxygen (red), sulfur (green), nitrogen (bright blue) and hydrogen (light blue). Carbon atoms for the 12e and residues in loop regions colored white. Carbon atoms of residues in beta sheets are colored steel blue. A final simulated annealing Fo-Fc electron density omit map, with 12e removed from the map calculations, is shown as a grey mesh contoured at 2.7 σ. The figure was generated with CueMOl2.
As predicted, the X-ray structure confirms that water 1 was displaced by one of the acetamide functional groups and that water 2 was shifted forward in comparison to its location in the co-crystal structure of 7 (shown in grey spheres in Figure 3B). To accommodate the di-acetamide functional groups, the guanidinium side chain portion of Arg 380 flipped approximately 180° to pack against one of the di-acetamides (Figure 4B). The result of Arg 380 adopting this position is a significant structural shift of Asn 381 to Thr 388 into a position that alters crystal packing and hence crystallization of the Kelch-12e complex in the P21 space group.
3.5 Immunoblot analysis
Six non-electrophilic compounds that were either active in the primary fluorescence anisotropy assay or that were esters of active compounds were tested for their ability to regulate Nrf2 and its target genes in MLE12 (mouse lung alveolar epithelial cell line) cells.42,43 Western blots were used to demonstrate the ability of the compounds to transactivate Nrf2 target genes heme oxygenase 1 (HMOX1) and NAD(P)H quinone oxidoreductase 1 (NQO1) (Figure 5). Nrf2 protein levels were also measured to demonstrate stabilization by the activators, and compared to the electrophilic positive control sulforaphane 1. The experiments were carried out three separate times.
Figure 5.

The effects of non-electrophilic activators on the expression levels of Nrf2 and its targets in lung alveolar epithelial cells. (A) MLE 12 cells were treated with DMSO (vehicle) or indicated compound (10 μM) for 6 hours, cell lysates were prepared, equal amount of protein was immunoblotted using antibodies specific for Nrf2, NQO1 or HMOX1. β-actin was used as loading control. (B) Quantification of target gene band intensity using β-actin as reference. DMSO-treated sample value was used as one arbitrary unit. Error bars represent the standard deviation from three independent experiments.
4. Discussion
In 2013, Marcotte, et al. reported one of the first non-electrophilic Nrf2 activators, 7, with an IC50 of 2.7 μM. In 2014, Jiang, et al. reported 8, based on modification of 7, as one of the first sub-nanomolar non-electrophilic Nrf2 activators, with an IC50 in a fluorescence anisotropy assay of 29 nM and a Kd of 9.9 nM in a biolayer interferometry assay [34]. We observe similar IC50 values for both compounds in our assays: Fluorescence anisotropy (7: IC50 = 1.0 μM; 8: IC50 = 24 nM) and SPR (7: Kd = 1.7 μM; 8: Kd = 20 nM; see Table 1). The crystal structure (PDB ID: 4IQK) of 7 bound to the Kelch domain of Keap1 (Figure 2A) was reported previously by Marcotte et al. [33]. It highlighted key molecular interactions of 7 bound to Kelch, including a π-cation interaction of the naphthalene core with the guanidinium group of Arg 415, π-π stacking interactions of the flanking phenyl rings of 7 with Tyr 334 and Phe 577, and a hydrogen bond between Ser 508 and one of the sulfonamide oxygen atoms. In the present study, we have used a fluorescence anisotropy assay as a primary assay to explore the SAR around 7, supplemented with a confirmatory SPR assay. We have focused our studies around three regions of the molecule: A) the methoxyphenyl groups, B) the naphthalene core, and C) the sulfonamide linkers (see Figure 2B).
In our exploration of Region A, we found that removal of both terminal methoxy groups (i.e., 11a) resulted in an inactive compound. We replaced the methoxy groups with an electron-donating -CH3 or –N(CH3)2 group (i.e., 11h and 11i) or an electron-withdrawing substituent (e.g., 11c, 11e). The replacement of the p-OCH3 substituents with p-CH3 retained activity with an IC50 = 890 nM, while the activity increased by almost 5-fold in the presence of a strongly electron donating –N(CH3)2 (IC50 = 190 nM). An increase in potency by a stronger electron-donating group may be attributed to stronger cation-π interactions between the central naphthalene ring and Arg 415. We are investigating this possibility further and will report in due course. It appears as though large electron-withdrawing groups are not well tolerated at this position, as the IC50 values of 11c and 11e are beyond the highest concentration assayed (i.e., 25 μM), although the p-F substituent retained activity (i.e., 11j; IC50 = 660 nM), which could be used towards synthesis of metabolically stable analogs of 7.
The crystal structure of 7 bound to the Kelch domain of Keap1 showed that the para-methoxy groups were both solvent-exposed [33]. It appeared as though substitution at the meta-position of the phenyl rings might interact with the surface of the protein. Replacing both p-OCH3 substituents with m-OCH3 groups resulted in a similar potency analog (i.e., 11b: IC50 = 630 nM). When only one of the methoxy groups was moved to a meta position (i.e., 14d), there was a slight improvement in potency (IC50 = 410 nM). Combining both meta- and para-substitutions into one compound (i.e., 11g) did little to increase the affinity, and created a rather insoluble compound. Replacement of the m-methoxy groups with electron-withdrawing groups (-CF3 in 11f and –COOH in 11d) gave inactive compounds, in agreement with substitution at the p-position. We further desymmetrized the molecule by replacing one or both of the methoxy groups with a –CF3 or –H substituent. The only compound that retained activity within this series (i.e., 14a–c) was the one that replaced one methoxy with a hydrogen (14a), with an IC50 value of 2.5 μM. Although there was a loss of activity by 2.5-fold, it did reduce the molecular weight to improve ligand efficiency. Binding efficiency of a ligand, which is defined as binding energy per non-hydrogen atom of a molecule, provides a useful consideration for hit-to-lead optimization in a drug discovery program. Ligand efficiency can provide a meaningful comparison between multiple scaffolds and series of analogs [53].
To explore the naphthalene core region B of 7 (Figure 2B), we changed the substitution pattern on the naphthalene from 1,4 to 1,3 (19, 21 and 22) and 2,3 (20, 23). The loss of activity in each analog demonstrates the requirement of the 1,4-substitution pattern. The necessity of the fused system was apparent with the lack of activity seen when the naphthalene core was replaced with a phenyl ring (24). Additionally, we removed an entire benzenesulfonamide to give the 1-substituted analog (17, 18), which was inactive, demonstrating the need for the core and both of its flanking substituents. We are currently undertaking a more comprehensive replacement of the naphthalene scaffold with other heterocycles.
We also explored the SAR around the sulfonamide region C (Figure 2B), which connects the naphthalene core to the flanking aryl rings. The sulfonamide was replaced with an amide (i.e., 25), which was inactive. The S-N bond in a sulfonamide usually exhibits lower sp2 character than the corresponding C-N bond in a carboxamide [54], so that the topology of the two compounds is likely quite different.
In the native interaction of Nrf2 and Keap1, a type I β-turn comprising an ETGE motif binds to the Kelch domain of Keap1. In crystal structures of a Nrf2 peptide bound to the Kelch domain [55], Glu 79 and Glu 82 of the ETGE motif form ionic interactions with Arg 415 and 483 of Keap1 [55]. In an illuminating study, Jiang, et al. [34] showed that incorporating two acetic acid moieties into the structure of 7 (i.e., 8) improved the affinity by almost 100-fold. This effect was attributed to ionic interactions with Arg 415 and Arg 483 and H-bond interactions with Ser 508 residues. We observed a similar result in our assay upon synthesizing this compound (fluorescence anisotropy IC50 = 24 nM vs. literature value of 29 nM; SPR Kd = 20 nM vs. literature biolayer interferometry Kd = 9.9 nM) [34]. We also obtained further evidence for the importance of the polar nature of the interaction, as we saw that a set of di-alkyl substituted analogs (i.e., 12a–d and 12f) were inactive; however, it does not seem that the polar interactions need to be ionic, as the di-acetamide 12e was also quite active (IC50 = 63 nM). Our X-ray structure of 12e in complex with Kelch shows clearly that one of the di-acetamides makes two strong hydrogen bonds with the amide side chain of Asn 414 and the other makes a hydrogen bond with the backbone carbonyl of Ile 461. Similarly, attempts were made to prepare analogs of the 1,3- and 2,3-disubstituted naphthalenes containing acids or amides. The 1,3-substituted di-acid (i.e., 22) resulted in a gain of activity (IC50 = 5500 nM) compared to 19, although it is still almost 180-fold less potent than 8. We also attempted to prepare the disubstituted analog of 20, but repeated attempts at synthesizing this compound were unsuccessful and yielded either an unknown product upon basic saponification of the ethyl ester or degradation of the ethyl ester under acid-catalyzed hydrolysis. Our standard alkylation conditions, when applied to compound 20 using 2-bromoacetamide, gave a mono-acetamide analog 23 instead of the di-acetamide; the reasons for this are not clear.
We were curious whether both acetic acid groups of 8 were necessary for binding to the Kelch domain, so we prepared the singly substituted analogs 16a and 16b. We were gratified to find that the affinity of 16b (i.e., IC50 = 61 nM in the fluorescence anisotropy assay and Kd = 110 nM in SPR assay) is almost equivalent to that of the di-acid 8. The singly substituted acetic acid of 16b improves the ligand efficiency of the molecule and decreases the total charge density on the molecule, which may be important for more advanced assays (see immunoblot studies below). Intriguingly, the mono-ethyl ester 16a is much more active (i.e., IC50 = 85 nM and Kd = 400 nM) than the di-ester 12a (i.e., IC50 and Kd > 25 μM). This marked change in affinity may hint at an upper limit for the size of one of these substituents (see docking studies below). Based on the above results, we synthesized 1-substituted naphthalene 18, the acetic acid analog of 17, to see whether the acid functionality might be able to rescue the lack of a second aryl ring. Similarly, we prepared the di-acetamides of the 1,3- and 2,3-disubstituted naphthalenes (i.e., 21 and 23). All of these compounds were inactive, further confirming the importance of the 1,4-disubstitution pattern of the scaffold 7 for Keap1-Nrf2 inhibition.
Docking studies were performed to further investigate the influence of substitutions at the R2 and R3 positions in Region C for compounds 8, 12a, 12e, 16a–b and 7. In general, the docking results were found to be highly consistent with the measured IC50 values. The top-3 predicted compounds by docking were indeed those with the highest affinity (compound 8, 12e and 16b); furthermore, these compounds scored significantly better than those compounds which performed poorly (7) or not at all (12a), demonstrating a >3 unit difference in docking scores in the best case (8) (see Figure 3).
Nearly all of the compounds, with the exception of 12a, were predicted to bind in a mode similar to that of the parent compound 7 in the crystal structure 4IQK (Figure 2A). Analysis of the crystal structure suggests that binding of these compounds may rely heavily on π-π stacking and π-cation interactions. With the exception of the low-affinity inhibitor 12a, at least two of these three π-π/π-cation interactions were maintained in all compound poses. In 12a, the bulky ethyl carboxylate substituents at the R2 and R3 positions enforce an alternative binding conformation wherein the phenyl rings of Region A and the ethyl carboxylate groups of Region C is inverted in comparison to the crystallized ligand in 4IQK, resulting in loss of π-π stacking interactions between the phenyl rings of Region A and residues Phe 577 and Tyr 525 (Figure 3E). The steric bulk of this compound, along with the loss of these interactions may, in part, account for the poor IC50 value observed for 12a.
While π-π stacking appears to be one of the main types of interactions influencing the binding of 7, the best performing compounds also utilize extensive hydrogen bonding to the groups substituted in the R2 and R3 positions. Adding a carboxylic acid or amide substituent allows for hydrogen bonding to nearby Arg 483, Ser 508, Asn 414, and Ser 363. In the case of compounds 8 and 16b, there may also be charge-charge interactions of the carboxylates with Arg 483 and Arg 415, as has been previously modeled [34]. It is likely that these additional hydrogen bonds contribute to the increased IC50 values in comparison to the parent compound 7. These additional hydrogen bonds are made possible by substituents that protrude into the nearby water pockets resulting in the removal of one or both water molecules.
Modifications in Region C have the potential to protrude into two pockets near the bottom of the active site that are occupied by water molecules in the 7:Kelch domain crystal structure (Figure 2A). These water molecules are also present in the apo structure (1ZGK), but have been shifted by about 2.0 Å, and in a related ligand-bound structure (PDB ID: 4IN4), only water 1 is present. The different water patterns in the crystal structures suggest that the positions of these water molecules vary depending on the type of ligand bound and may contribute to ligand binding.
WATSite was used to evaluate the thermodynamic profile (ΔG) of binding site water molecules with the different compounds bound (Figure 3). In all cases, excluding 7, the WATsite analysis agreed with the ensemble docking for water selection (e.g. in 8 the docking routine selected a template where both water 1 and water 2 were removed, and the WATsite analysis also showed no waters in the nearby vicinity of water 1 or water 2). Interestingly, removal of these waters does not appear to have a high energetic penalty, and in the case of water 2 even seems to be slightly favorable. The low energetic cost of removing these waters coupled with the additional interactions that the compounds gain by occupying these pockets suggests that displacing at least one of these water molecules with substituents that protrude into these pockets and interact with nearby residues is more favorable than not doing so (e.g., 16b vs. 7).
We compared the ability of 7, 8, 11g, 12a, 16a and 16b to induce the expression of Nrf2 target genes HMOX1 and NQO1 in mouse lung type-II alveolar epithelial cells ex vivo (see Figure 5), and we compared these values to that of the well-studied electrophilic activator sulforaphane 1.
Although the differences between 7, 8, and 16a are not statistically significant, the data may imply that the di-acetic acid functionality of 8 is not essential for induction of HMOX1 and NQO1 and stabilization of Nrf2. Previously, Jiang et al. [34] showed activation of NQO1, GCLM, and HMOX1 in HCT116 cells treated with 8 for 16–32 hours. In the present study, we examined the induction of Nrf2 target gene expression 6 hours post-treatment with the activators, mainly in order to avoid indirect effects, such as metabolic activation of the small molecules to electrophilic species or interference at other pathways. We saw that the mono-carboxylic acid 16b was a rather poor inducer of HMOX1 and NQO1 after six hours; however, 16a, the ethyl ester of the mono-carboxylic acid, potently induced HMOX1 and stabilized Nrf2 levels after six hours in a manner similar to that seen with 7 and 8 (see Figure 5B). This is interesting because the acid 16b is more potent than the ester 16a in vitro. We believe 16a may facilitate membrane traversal, and, once in the cell, may be hydrolyzed by esterases to give the mono-carboxylic acid 16b. This may further imply that the carboxylic acids may be replaced to provide further leads to achieve induction of HMO1, NQO1 and Nrf2.
5. Conclusions
In conclusion, we have conducted an SAR study focusing on the structures of naphthalenes 7 and 8 in three regions (see Figure 2B). Region A shows optimum potency when substituted in the meta or para position with an electron-donating group. Region B is optimal with 1,4-substitution on the naphthalene core; further optimization of the core may be necessary and will be the focus of future studies. Region C provided the most interesting results, revealing new compounds that show promise as non-electrophilic activators of Nrf2: di-acetamides (i.e., 12e) and compounds containing a single acetic acid group on one of the N-substituents (i.e., 16b), a compound which has lower negative charge and better ligand efficiency. The importance of water displacement by these substituents in Region C may contribute to their high affinity, and this hypothesis is supported by our X-ray crystal structure of 12e bound to the Kelch domain (PDB ID: 4XMB). The mono-acid 16b stood out as a compound with high potency in vitro, with an IC50 of 61 nM in a fluorescence anisotropy assay, but the potency was lost ex vivo as we looked at the induction of Nrf2 target genes; however, the mono ethyl ester 16a, which showed moderate in vitro potency, was capable of inducing the expression of native Nrf2 target genes at levels similar to that of the well-known electrophilic activator sulforaphane 1. This data obtained with ester 16a may suggest that neutrally charged groups may be used to replace negatively charged carboxylates that have previously been shown to give high affinity compounds.
Supplementary Material
Highlights.
Neutral substituents can replace charged carboxylate groups
Crystal structure of amide 12e bound to Kelch domain
Unsymmetrically substituted analogs can have high affinity for the Kelch domain
Binding site waters can be displaced without incurring a large energetic penalty
Acknowledgments
The authors thank the University of Illinois at Chicago College of Pharmacy and the University of Illinois Cancer Center (to TWM), the National Institutes of Health (grants HL66109 and ES11863 to SPR) and the Walther Cancer Foundation (to ADM) for their support of this work. We thank Dr. Bernard Santarsiero for solving the crystal structure of compound 8. Protein crystallization and X-ray data collection were done through the Purdue Center for Cancer Research which is supported by P30 CA023168.
Abbreviations used
- BTB
Broad complex, Tramtrack and Bric-à-Brac
- LE
Ligand efficiency
- DMEM
Dubecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- HMOX1
heme oxygenase 1
- Keap1
Kelch-like ECH-associated protein 1
- NQO1
NAD(P)H quinone oxidoreductase 1
- MLE
mouse lung alveolar epithelial cell line
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- sMAF
small musculoaponeurotic fibrosarcoma oncogene homolog
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, De Galarreta CM, Cuadrado A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem. 2004;279:8919–8929. doi: 10.1074/jbc.M309660200. [DOI] [PubMed] [Google Scholar]
- 3.Hayes JD, Chanas SA, Henderson CJ, McMahon M, Sun C, Moffat GJ, Wolf CR, Yamamoto M. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem Soc Trans. 2000;28:33–41. doi: 10.1042/bst0280033. [DOI] [PubMed] [Google Scholar]
- 4.Namani A, Li Y, Wang XJ, Tang X. Modulation of NRF2 signaling pathway by nuclear receptors: implications for cancer. Biochim Biophys Acta. 2014;1843:1875–1885. doi: 10.1016/j.bbamcr.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Kansanen E, Kuosmanen SM, Leinonen H, Levonenn AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- 7.Jnoff E, Albrecht C, Barker JJ, Barker O, Beaumont E, Bromidge S, Brookfield F, Brooks M, Bubert C, Ceska T, Corden V, Dawson G, Duclos S, Fryatt T, Genicot C, Jigorel E, Kwong J, Maghames R, Mushi I, Pike R, Sands ZA, Smith MA, Stimson CC, Courade JP. Binding mode and structure-activity relationships around direct inhibitors of the Nrf2-Keap1 complex. ChemMedChem. 2014;9:699–705. doi: 10.1002/cmdc.201300525. [DOI] [PubMed] [Google Scholar]
- 8.Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem Res Toxicol. 2008;21:705–710. doi: 10.1021/tx700302s. [DOI] [PubMed] [Google Scholar]
- 9.Eggler AL, Small E, Hannink M, Mesecar AD. Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem J. 2009;422:171–180. doi: 10.1042/BJ20090471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006;387:1311–1320. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- 11.Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, Sasi S, Dalton TP, Anderson ME, Chan JY, Morrow JD, Freeman ML. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J Biol Chem. 2007;282:2529–2537. doi: 10.1074/jbc.M607622200. [DOI] [PubMed] [Google Scholar]
- 12.Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, Yamamoto M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol. 2007;27:7511–7521. doi: 10.1128/MCB.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem. 2006;281:24756–24768. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cleasby A, Yon J, Day PJ, Richardson C, Tickle IJ, Williams PA, Callahan JF, Carr R, Concha N, Kerns JK, Qi H, Sweitzer T, Ward P, Davies TG. Structure of the BTB Domain of Keap1 and Its Interaction with the Triterpenoid Antagonist CDDO. PLoS One. 2014;9:e98896. doi: 10.1371/journal.pone.0098896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J, Nussinov R. Flexible Cullins in Cullin-RING E3 Ligases Allosterically Regulate Ubiquitination. J Biol Chem. 2011;286:40934–40942. doi: 10.1074/jbc.M111.277236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baird L, Dinkova-Kostova AT. Diffusion dynamics of the Keap1-Cullin3 interaction in single live cells. Biochem Biophys Res Commun. 2013;433:58–65. doi: 10.1016/j.bbrc.2013.02.065. [DOI] [PubMed] [Google Scholar]
- 18.Baird L, Swift S, Lleres D, Dinkova-Kostova AT. Monitoring Keap1-Nrf2 interactions in single live cells. Biotechnol Adv. 2014;32:1133–1144. doi: 10.1016/j.biotechadv.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crunkhorn S. Deal watch: Abbott boosts investment in NRF2 activators for reducing oxidative stress. Nat Rev Drug Discov. 2012;11:96. doi: 10.1038/nrd3655. [DOI] [PubMed] [Google Scholar]
- 20.Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, Yang M, Raghupathi K, Novas M, Sweetser MT, Viglietta V, Dawson KT. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367:1087–1097. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]
- 21.Kikuchi N, Ishii Y, Morishima Y, Yageta Y, Haraguchi N, Itoh K, Yamamoto M, Hizawa N. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir Res. 2010;11:31. doi: 10.1186/1465-9921-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keum YS, Jeong WS, Kong AN. Chemoprevention by isothiocyanates and their underlying molecular signaling mechanisms. Mutat Res. 2004;555:191–202. doi: 10.1016/j.mrfmmm.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 23.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009;135:794–804. doi: 10.1378/chest.08-0492. [DOI] [PubMed] [Google Scholar]
- 24.Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci U S A. 2005;102:10070–10075. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo Y, Eggler AL, Liu D, Liu G, Mesecar AD, van Breemen RB. Sites of alkylation of human Keap1 by natural chemoprevention agents. J Am Soc Mass Spectrom. 2007;18:2226–2232. doi: 10.1016/j.jasms.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eggler AL, Luo Y, van Breemen RB, Mesecar AD. Identification of the highly reactive cysteine 151 in the chemopreventive agent-Sensor Keap1 Protein is method-dependent. Chem Res Toxicol. 2007;20:1878–1884. doi: 10.1021/tx700217c. [DOI] [PubMed] [Google Scholar]
- 28.Hu C, Eggler AL, Mesecar AD, van Breemen RB. Modification of Keap1 cysteine residues by sulforaphane. Chem Res Toxicol. 2011;24:515–521. doi: 10.1021/tx100389r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yore MM, Kettenbach AN, Sporn MB, Gerber SA, Liby KT. Proteomic analysis shows synthetic oleanane triterpenoid binds to mTOR. PLoS One. 2011;6:e22862. doi: 10.1371/journal.pone.0022862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M, Lambers Heerspink HJ, McMurray JJ, Meyer CJ, Parving H-H, Remuzzi G, Toto RD, Vaziri ND, Wanner C, Wittes J, Wrolstad D, Chertow GM. Bardoxolone Methyl in Type 2 Diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013;369:2492–2503. doi: 10.1056/NEJMoa1306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magesh S, Chen Y, Hu L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med Res Rev. 2012;32:687–726. doi: 10.1002/med.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson AJ, Kerns JK, Callahan JF, Moody CJ. Keap calm, and carry on covalently. J Med Chem. 2013;56:7463–7476. doi: 10.1021/jm400224q. [DOI] [PubMed] [Google Scholar]
- 33.Marcotte D, Zeng W, Hus JC, McKenzie A, Hession C, Jin P, Bergeron C, Lugovskoy A, Enyedy I, Cuervo H, Wang D, Atmanene C, Roecklin D, Vecchi M, Vivat V, Kraemer J, Winkler D, Hong V, Chao J, Lukashev M, Silvian L. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg Med Chem. 2013;21:4011–4019. doi: 10.1016/j.bmc.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 34.Jiang ZY, Lu MC, Xu LL, Yang TT, Xi MY, Xu XL, Guo XK, Zhang XJ, You QD, Sun HP. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J Med Chem. 2014;57:2736–2745. doi: 10.1021/jm5000529. [DOI] [PubMed] [Google Scholar]
- 35.Hu L, Magesh S, Chen L, Wang L, Lewis TA, Chen Y, Khodier C, Inoyama D, Beamer LJ, Emge TJ, Shen J, Kerrigan JE, Kong AN, Dandapani S, Palmer M, Schreiber SL, Munoz B. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg Med Chem Lett. 2013;23:3039–3043. doi: 10.1016/j.bmcl.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun H-P, Jiang Z-Y, Zhang M-Y, Lu M-C, Yang T-T, Pan Y, Huang H-Z, Zhang X-J, You Q-d. Novel protein-protein interaction inhibitor of Nrf2-Keap1 discovered by structure-based virtual screening. MedChemComm. 2014;5:93–98. [Google Scholar]
- 37.Richardson BG, Jain AD, Speltz TE, Moore TW. Non-electrophilic modulators of the canonical Keap1/Nrf2 pathway. Bioorg Med Chem Lett. 2015;25:2261–2268. doi: 10.1016/j.bmcl.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogura T, Tong KI, Mio K, Maruyama Y, Kurokawa H, Sato C, Yamamoto M. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc Natl Acad Sci U S A. 2010;107:2842–2847. doi: 10.1073/pnas.0914036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sato M, Aoki T, Inoue H, Tanaka T, Kunishima N. Keap1 protein binding compound, crystla of complex between the same and Keap1 protein and method for producing the same. JP2013028575. Jp Patent. 2013
- 40.Zhuang C, Narayanapillai S, Zhang W, Sham YY, Xing C. Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J Med Chem. 2014;57:1121–1126. doi: 10.1021/jm4017174. [DOI] [PubMed] [Google Scholar]
- 41.Wikenheiser KA, Clark JC, Linnoila RI, Stahlman MT, Whitsett JA. Simian virus 40 large T antigen directed by transcriptional elements of the human surfactant protein C gene produces pulmonary adenocarcinomas in transgenic mice. Cancer Res. 1992;52:5342–5352. [PubMed] [Google Scholar]
- 42.Wikenheiser KA, Vorbroker DK, Rice WR, Clark JC, Bachurski CJ, Oie HK, Whitsett JA. Production of immortalized distal respiratory epithelial cell lines from surfactant protein C/simian virus 40 large tumor antigen transgenic mice. Proc Natl Acad Sci U S A. 1993;90:11029–11033. doi: 10.1073/pnas.90.23.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inoyama D, Chen Y, Huang X, Beamer LJ, Kong AN, Hu L. Optimization of fluorescently labeled Nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of Keap1-Nrf2 interaction. J Biomol Screen. 2012;17:435–447. doi: 10.1177/1087057111430124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide:3 A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 45.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide:3 A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 46.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision Glide:3 Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 47.Hu B, Lill MA. WATsite: Hydration site prediction program with PyMOL interface. J Comput Chem. 2014;35:1255–1260. doi: 10.1002/jcc.23616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Briffett NE, Hibbert F. Alkaline hydrolysis of dibenzoylaminonaphthalenes in 70%(v/v) Me2SO-H2O and the effect of a neighbouring amide group. J Chem Soc, Perkin Trans. 1989;2:765–768. [Google Scholar]
- 49.Price CC, Voong S-T. 4-Nitro-1-Naphthylamine. Organic Syntheses Coll. 1955;3:664. [Google Scholar]
- 50.Xu F, Jia Y, Wen Q, Wang X, Zhang L, Zhang Y, Yang K, Xu W. Synthesis and biological evaluation of N-(4-hydroxy-3-mercaptonaphthalen-1-yl)amides as inhibitors of angiogenesis and tumor growth. Eur J Med Chem. 2013;64:377–388. doi: 10.1016/j.ejmech.2013.03.043. [DOI] [PubMed] [Google Scholar]
- 51.Proust N, Gallucci JC, Paquette LA. Effect of sulfonyl protecting groups on the neighboring group participation ability of sulfonamido nitrogen. J Org Chem. 2009;74:2897–2900. doi: 10.1021/jo900065q. [DOI] [PubMed] [Google Scholar]
- 52.Yang S, Denny WA. A new short synthesis of 3-substituted 5-amino-1-(chloromethyl)-1,2-dihydro-3H-benzo[e]indoles (amino-CBIs) J Org Chem. 2002;67:8958–8961. doi: 10.1021/jo0263115. [DOI] [PubMed] [Google Scholar]
- 53.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 54.Langenhan JM, Fisk JD, Gellman SH. Evaluation of hydrogen bonding complementarity between a secondary sulfonamide and an α-amino acid residue. Org Lett. 2001;3:2559–2562. doi: 10.1021/ol016237x. [DOI] [PubMed] [Google Scholar]
- 55.Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006;25:3605–3617. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.