

Abstract
Estrogens and androgens influence the growth and maintenance of bones and muscles and are responsible for their sexual dimorphism. A decline in their circulating levels leads to loss of mass and functional integrity in both tissues. In the article, we highlight the similarities of the molecular and cellular mechanisms of action of sex steroids in the two tissues; the commonality of a critical role of mechanical forces on tissue mass and function; emerging evidence for an interplay between mechanical forces and hormonal and growth factor signals in both bones and muscles; as well as the current state of evidence for or against a cross-talk between muscles and bone. In addition, we review evidence for the parallels in the development of osteoporosis and sarcopenia with advancing age and the potential common mechanisms responsible for the age-dependent involution of these two tissues. Lastly, we discuss the striking difference in the availability of several drug therapies for the prevention and treatment of osteoporosis, as compared to none for sarcopenia.
Keywords: estrogens, androgens, bone cells, muscle cells, osteoporosis, sarcopenia
1. Introduction
Bones and muscles are two of the largest tissues in mammals, comprising together 60 and 47 percent of lean body mass in men and women, respectively; 15 percent bone and 45 percent muscle in men, and 12 percent bone and 35 percent muscle in women. Starting as early as the third decade of life, both women and men alike experience a slow but progressive loss of mass and declining function in both tissues. In a large proportion of individuals, this eventually leads to osteoporosis and sarcopenia – the two most common contributors to the loss of independence and poor quality of life in older people. Indeed, musculoskeletal diseases are the leading cause of disability in the United States; and together account for more than 50 percent of all chronic conditions in people over 50 years of age in developed countries. The economic burden of these conditions for the US alone has been estimated (for the years 2004-2006) to be $950 billion dollars annually, or 7.4% of the national gross domestic product (http://www.boneandjointburden.org/; http://en.wikipedia.org/wiki/Musculoskeletal_disorder). In 2000, the cost of sarcopenia alone in the United States was 18.5 billion or 1.5% of total health expenditures [1].
Estrogens and androgens exert potent influences on the post-natal growth of bones and muscles and are responsible for their sexual dimorphism. In addition, estrogens and androgens are important for the homeostasis of either tissue during adulthood. A decline in the circulating levels of sex steroids leads to loss of mass and functional integrity in either tissue. The purpose of this article is to review our current state of understanding of the effects of sex steroids on bones and muscles, both from a basic science and a clinical perspective, and the implications of these effects in physiology and pathophysiology.
In particular, we highlight the similarities in the molecular and cellular mechanisms of action of sex steroids in the two tissues; the commonality of the important role of mechanical forces on tissue mass and function in bone and muscle; the emerging evidence for an interplay between mechanical forces and hormonal and growth factor signals; as well as the evidence for and against a cross-talk between muscles and bone. In addition, we review evidence for the parallels in the development of osteoporosis and sarcopenia with advancing age and the potential common mechanisms responsible for the age-dependent involution of these two tissues, including sex steroid deficiency, mitochondria dysfunction, and disuse.
2. Effects of estrogens and androgens on bone
Estrogens and androgens influence the shaping of the skeleton during growth and are largely responsible for its sexual dimorphism. In addition, estrogens and androgens contribute to the maintenance of bone homeostasis and strength during adult life. Deficiency of estrogens in females or both estrogens and androgens in males adversely affect skeletal development during growth and homeostasis during adulthood and contribute to the development of osteoporosis in either sex.
For many years, estrogen deficiency was considered the seminal factor for the involution of the skeleton later in life in both sexes. However, the estrogen-centric view of skeletal homeostasis has nowadays been revised by the recognition that the syndrome of fractures termed osteoporosis is the clinical manifestation of multiple slowly progressing and cumulative pathologies [2]. As in all other aging tissues, age-related mechanisms intrinsic to bone, including oxidative stress, declining autophagy, cell senescence, inflammation, ER stress, compromised unfolded protein response are apparently protagonists in the development of osteoporosis [3, 4]. Age-related changes in the ovaries, testicles, and adrenals as well as the decline of growth factor levels with age are contributory. The sharp decline of estrogen levels at menopause accelerates the age-dependent involution of the female skeleton and contributes to the loss of bone mass, architectural integrity, and strength. A slower and smaller decline of androgen levels may also contribute to the development of osteoporosis in elderly men as does late-onset hypogonadism. However, androgens may be less important than estrogens for the maintenance of bone health in elderly men.
3. Effects of estrogens and androgens on muscles
Skeletal muscles are the engines of locomotion and as such play also a major role in energy metabolism. Maintenance of an appropriate level of muscle mass (for a given body size) and function is critical for health and the quality of life, and a decrease of muscle mass contributes to morbidity and mortality in many disease states [5, 6]. Moreover, in a manner akin to bone fracture healing and the anabolic response of bone to mechanical strains, muscles regenerate following injury and become hypertrophic upon mechanical loading. As in the case of bone, systemic hormones, including sex steroids, growth factors, pro- and anti-inflammatory cytokines, as well as load bearing influence the mass of the muscle tissue and its function [7, 8]. In general, sex steroids promote the growth and maintenance of muscle mass and strength and exert beneficial effects on the metabolic function of skeletal muscles and their repair after injury [9, 10]. Thus, physiologic or pathologic changes in the levels of circulating sex steroids have important implications for muscle health in both males and females [11]. Nonetheless, there are substantial differences between the effects of androgens versus estrogens on skeletal muscles, the effects of either class of sex steroids in males versus females, and the sensitivity of one muscle type versus another (or even different cell types within the same muscle) to androgens and estrogens.
A decline in the circulating levels of sex steroids is associated with a decrease in lean striated muscle mass, muscle size, strength, and impaired muscle glucose homeostasis corresponding with an increase in fat mass [11]. Specifically, post-menopausal women or hypogonadal men, including patients with prostate cancer receiving androgen deprivation therapy, experience a decrease in lean mass, muscle size, and strength and a higher incidence of insulin resistance. As in the case of bone, loss of muscle strength in men and women with age exceeds the decline in muscle mass [12]. Notably, in men with suppressed endogenous testosterone and estradiol, androgen deficiency accounts for the decrease in lean mass, muscle size, and strength. On the other hand, a deficiency of estrogens (80% of which is derived from the aromatization of testosterone in males) is responsible for the increase in body fat; and loss of both estrogens and testosterone contribute to the decline in sexual function [13]. Nonetheless, in difference to the evidence for an important role of estrogens in skeletal homeostasis in both females and males, the evidence in support of a role of estrogens on muscle mass growth and maintenance (and of estrogen deficiency in sarcopenia later in life) in either males or females remains weak. Androgens may be the predominant sex steroid in the regulation of muscle homeostasis in both males and females. In any event, whereas estrogen replacement therapy for post-menopausal women has decreased dramatically during the last decade because of the appreciation of serious side effects, testosterone therapy is on the rise and prescription sales for testosterone have increased by 500% in the United States between 1993 and 2000 [14].
Low testosterone levels cause a decrease in muscle mass and strength in men [15-17]; and testosterone replacement therapy is very effective in increasing muscle mass and strength in both young and old men with androgen deficiency or castrated rodents [18-22]. Testosterone acts synergistically with mechanical loading to increase skeletal muscle mass [20]. Furthermore, testosterone as well as other androgenic steroids promote muscle growth in normal men [23-25] and also in females [26]. At their extreme, the very potent effects of androgens and related anabolic steroids on muscle mass are in plain view in the case of professional body builders. Albeit, low bioavailable testosterone may not relate to frailty in elderly men over the age of 65 [27]. Estrogens have positive effects on muscle contractile function and protection against post-exercise muscle damage and inflammation in the ovariectomized rat model, [28-30]. However, the evidence for similar positive effects in post-menopausal women receiving estrogen replacement therapy is far less convincing [31-34]. This may reflect differences in the sensitivity of muscle mass to estrogens in rodents as compared to humans, differences in systemic versus local (myofiber-specific) effects of estrogens, as well as the complexity of the interplay between effects of estrogens on energy metabolism and skeletal muscle mass.
The differences in the effects of androgens versus estrogens on skeletal muscles notwithstanding, there are major differences in the responsiveness to estrogens and androgens between muscles associated with copulation and reproduction versus weight bearing locomotive muscles [21]. Such differences are clear in the case of androgens and the androgen sensitive reproductive muscles, such as the bulbocavernosus or levator ani (LA), versus weight bearing hindlimb muscles [21, 35]. Further, weight bearing hindlimb muscles can exhibit their own differential responsiveness to both testosterone and estrogens [35, 36]. Estrogens exert potent influences on the other two major types of muscle: the cardiac muscle (known as myocardium) and smooth muscles. These effects may play an important role in the pathogenesis of cardiovascular diseases, including coronary heart disease in post-menopausal women. However, they are beyond the scope of this article and will not be discussed further. An excellent discussion of this topic can be found in the review articles by Crandall and Barrett-Connor [37, 38].
4. Cellular and molecular mechanisms of sex steroid action in bone
The actions of estrogens and androgens on bone result from binding of the ligands to classical nuclear hormone receptors – the estrogen receptor (ER) α and β and the androgen receptor (AR), respectively [39]. Binding of the ligand to the respective receptor stimulates transcription of target genes resulting from direct interactions of the ligand-activated receptor protein with DNA or with other transcription factors. Estrogen or androgen binding to a subpopulation of the ER and perhaps AR, which is localized at the plasma membrane, activates signal transduction pathways in the cytoplasm by triggering the production of cyclic nucleotides, calcium fluxes and cytoplasmic kinases. Kinase activation, in turn, causes the phosphorylation of transcription factors which mediate gene regulatory effects of sex steroids. In the case of estrogens, more genes are regulated through this non-genotropic mechanism than are regulated via the cis- or trans-interaction of the ERα with DNA [40]. A schematic illustration of the different modes of estrogen signaling via the ERα are depicted in Figure 1.
Figure 1.
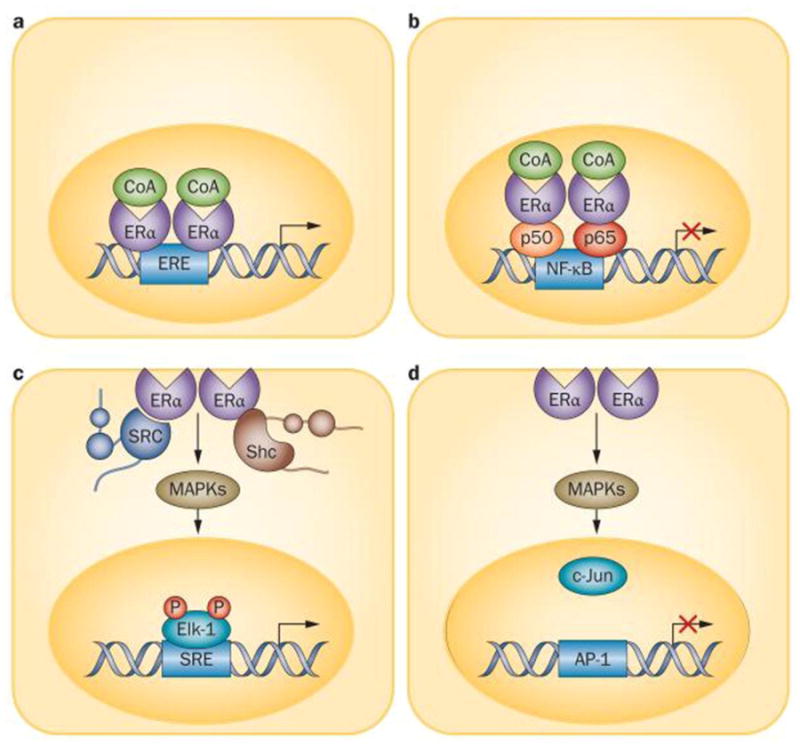
Molecular mechanisms of action of ERα. a) Classic genomic signalling, in which ligand-activated ERα dimers attach to EREs on DNA and activate or repress transcription. b) ERE-independent genomic signalling, in which ligandactivated ERα binds to other transcription factors (such as the p50 and p65 subunits of NF-κB), which prevent them from binding to their response elements. c,d) Nongenotropic mode of action, in which ligand-activated ERα (in the plasma membrane) activates cytoplasmic kinases which, in turn, induce the phosphorylation of substrate proteins and transcription factors (such as Elk-1 and AP-1) that (c) positively or (d) negatively regulate transcription. Abbreviations: AP-1, transcription factor AP-1; CoA, coenzyme A; Elk-1, ETS domain-containing protein Elk-1; ERα, estrogen receptor α; ERE, estrogen response element; Shc, Shc-transforming protein; SRE, serum response element. Reproduced from: Stavros C. Manolagas, Charles A. O'Brien, and Maria Almeida. Nature Reviews Endocrinology. 2013, 9: 699–712.
Recent studies with selective deletions of the ER or AR in specific cell types in mice have revealed that the effects of sex steroids on bone result from a complex interplay of actions on different cell types (Table 1) [39, 41]. Unexpectedly, the cell targets of the effects of sex steroids on the skeleton are different in the cancellous (also known as trabecular) and the cortical bone compartments. More surprisingly, the cellular targets of sex steroid action on bone are also different in females versus males. Briefly, estrogens acting through the ERα in chondrocytes of the growth plate influence their proliferation and thereby longitudinal bone growth. In particular, direct estrogen actions on proliferating chondrocytes are critical for the closure of the growth plate at the end of puberty in either sex. Thus, female or male animals or humans lacking ERα in chondrocytes have longer bones. In agreement with their well documented anti-remodeling effect, estrogens act directly on osteoclasts via the ERα to attenuate osteoclastogenesis and promote apoptosis [42, 43]. These effects, however, are responsible for the protective effect of estrogens on the cancellous, but not the cortical, bone. On the other hand, direct effects of estrogens on osteoblast progenitors mediated by ERα are responsible for the protective effects of estrogens against endocortical bone resorption [44]. The ERα of osteoprogenitors promotes bone formaton at the periosteal surface of the cortex and is required for optimal cortical bone accrual in mice; this effect, nonetheless, is independent of estrogens. Instead, it is due to the potentiation of Wnt/β-catenin signaling by the unliganded ERα. Be that as it may, the direct effects of estrogens on osteoclasts are only relevant to the female skeleton, because male mice lacking ERα in osteoclasts have normal cancellous bone mass [45].
Table 1. Cell targets of sex steroid action in bone and muscles.
| Bones | Muscles |
|---|---|
| 1. Chondrocytes | 1. Mesenchymal stem cells |
| 2. Osteoblast progenitors | 2. Satellite cells |
| 3. Osteoclast progenitors | 3. Myoblasts |
| 4. Mature osteoblasts | 4. Myocytes/myofibers |
| 5. Mature osteoclasts | 5. Fibroblasts |
| 6. Osteocytes | 6. Resident macrophages |
| 7. B-lymphocytes | 7. Cardiomyocytes |
| 8. T-lymphocytes |
Global deletion of the androgen receptor (AR) in male mice causes high bone turnover, increased bone resorption, and decreased cortical and cancellous bone mass. Targeted AR deletion in mature osteoblasts and osteocytes, however, decreases cancellous bone mass, but it has no effect on cortical bone [46-51]. Indeed, in the cancellous bone of male mice, the effects of androgens result from the restraining of osteoclast number via AR-signaling in mature osteoblasts and osteocytes – but not direct actions in osteoclasts or via aromatization to estrogens and ERα signaling [45]. Most surprisingly, in cortical bone, the effects of androgens do not require AR or ERα signaling in any cell type along the mesenchymal lineage or the myeloid lineage – from which osteoblasts and osteoclasts arise, respectively. Therefore, androgens must exert their effects indirectly by actions on some other cell types or one or more tissues other than bone, perhaps muscles. However, as it will be discussed below, this possibility remains unproven.
During the last ten years extensive evidence has implicated oxidative stress in the increased bone resorption associated with estrogen or androgen deficiency [2]. RANKL and MCS-F – the two cytokines that are essential for osteoclast generation – stimulate intracellular H2O2 accumulation, strongly suggesting that this event is a critical and purposeful adaptation for the differentiation and survival of osteoclasts [52]. Conversely, mice with osteoclast-targeted expression of human catalase – an enzyme that prevents H2O2 accumulation – in mitochondria, exhibit increased cortical and cancellous bone mass due to a decrease in osteoclast numbers. More strikingly, these mice are protected against the cortical, but not cancellous, bone loss caused by either ovariectomy or orchidectomy. Taken together with the evidence of the cell-specific deletions of the ERα and AR from cells of the mesenchymal and myeloid lineages these latest results suggest that estrogens (in both females and males) decrease bone resorption at the endocortical surface via an effect that is exerted on cells of the osteoblast lineage and causes the decrease in the production and/or release into the bone marrow of one or more heretofore unknown paracrine cytokines. This event, in turn, attenuates osteoclast formation, at least in part by decreasing H2O2 production in the mitochondria, and perhaps the recruitment of osteoclast precursors to the endocortical surface.
Selective activation of non-nuclear initiated actions of the ER dissociates the beneficial effects of estrogens on bone from their effects on reproductive organs [53-59]. This contention has been recently tested using an estrogen-dendrimer conjugate (EDC) that cannot enter the cell nucleus but it is very effective in stimulating non-nuclear signaling [60]. The compound prevented the loss of cortical bone mass caused by ovariectomy in mice as effectively as estradiol, but unlike estradiol, it did not prevent the loss of cancellous bone mass. These findings strongly suggest that the protection of cortical bone mass by estrogens results from a non-nuclear initiated mechanism of action of the ERα that is distinct from the classical nuclear-initiated actions of the estrogen-activated ERα on reproductive organs. In agreement with these conclusions, genetic manipulations of the ERα that prevent its nuclear-initiated actions abolish the protective effect of estrogens on cancellous bone, but have no effect on the cortical compartment [61, 62].
5. Cellular and molecular mechanisms of sex steroid action in muscles
Sex steroid receptor action in skeletal muscles
AR, ERα, and ERβ are expressed in several different cell types of skeletal muscles (Table 1). AR is expressed in mesenchymal stem cells, satellite cells, myocytes, and fibroblasts [63, 64]. The level of AR expression (and the sensitivity to androgens) is higher in levator ani and bulbocavernosis muscles – muscles associated with reproduction – than in weight bearing muscle [21]. The level of muscle AR expression is up-regulated by androgens; and intriguingly, it is also increased by resistance training in both rodents and humans [10, 21, 65-69]. Global deletion of the AR in mice causes a decrease of muscle mass in the males but not in the females; and it reproduces the effects of orchidectomy on the gene expression patterns of myoblasts [70]. Transgenic over-expression of the AR in rat muscle increases total lean body mass [71] and type IIb myofiber size but only in the EDL muscle. It also increases oxygen consumption and reduces fat body mass [72]. A better insight on the effects of androgens on bone has been obtained by the conditional deletion of the AR. Targeted AR deletion in satellite cells has no effect on skeletal muscles [73]. On the other hand, ubiquitous or selective deletion of the AR in post-mitotic myocytes in mice causes a decrease in body weight, lean body mass, intra-abdominal fat, and the weight of the androgen-sensitive musculus levator ani [35]. The weight of other peripheral skeletal muscles is either slightly reduced or not at all, and there are no changes in muscle strength or fatigue. Furthermore, the myocyte-specific AR deletion converts fast to slow fibers, but it has no effect on muscle strength or fatigue [35].
Both ERα and ERβ are expressed in skeletal muscle satellite cells, myofibers and endothelial cell of both females and males. Nonetheless, at first look global ERα or ERβ deletion in mice had no effect in skeletal muscle development or morphology in females [74, 75]. In subsequent studies, ERα, but not ERβ, deletion decreased the mass and contractile properties of some muscles, [36]. Similar effects are seen in mice with global loss of aromatase [36]. On the other hand, global ERβ deletion increases male skeletal muscle fatigue properties, but it has no effect in females [76]. Importantly, cortical or trabecular bone is not affected in these mice, indicating that direct actions of androgens in myocytes affects male body composition but not skeletal homeostasis.
Protein turnover regulation in skeletal muscles
Muscle growth or atrophy closely correlates with a positive and negative balance between protein accretion and degradation, respectively [77, 78]. Protein synthesis in muscles is activated by the IGF-1/Akt/mTORC1 pathway and involves glycogen syntheses kinase-3 beta (GSK3β) phosphorylation. Muscle protein synthesis, degradation, and cell survival are all regulated, in part, by Akt activation [79]. Akt signaling regulates muscle protein degradation through phosphorylation of the forkhead box O (FoxO) family of transcription factors, FoxO1 and 3 [8, 80]. FoxOs increase transcription of genes regulating ubiquitin proteasome protein degradation, muscle atrophy F-box (MAFbx; atrogin1) [81] and muscle ring Finger-1 (MuRF1) [82]. FoxO signaling in muscles can also induce DNA damage response 1 (REDD1, also referred to as Rtp801 and DDIT4) [83] and suppresses protein synthesis.
Both testosterone and mechanical loading increase muscle IGF-1 – a potent regulator of muscle mass – through the activation of Akt. Selective deletion of the AR in myocytes decreased muscle insulin-like growth factor (IGF-IEa) expression [84]. Testosterone deficiency in men is associated with a reduction in both circulating IGF-1 [15] and intramuscular IGF-1 expression [85]; and similar effects have been observed in castrated rodents [86]. Nonetheless, while the induction of muscle IGF-1 synthesis is thought to be a critical mechanism for testosterone-induced muscle growth, loss of androgens or androgen replacement in castrated animals is associated with either reduced or increased phosphorylation of Akt [87, 88]. Similarly, the response of skeletal muscle Akt to testosterone replacement in castrated animals is equivocal [89]. Part of the variability of the experimental findings may be due to differences in the dose, type, and duration of androgen deficiency, as well as the type of muscle examined. Estrogen deficiency decreases muscle Akt phosphorylation [90]. And, the recovery of skeletal muscle mass after disuse atrophy is impeded by estrogen deficiency. This phenomenon is associated with compromised skeletal muscle Akt phosphorylation, as well as decreased expression of the mTOR targets p70 s6k and S6 upon reloading [90]. Estrogen replacement therapy in post-menopausal women increases skeletal muscle IGF-1 related gene expression [91]. Ovariectomy suppresses skeletal muscle AMPK activation, and this effect is reversed by estrogen administration [92]. In addition to promoting fatty acid metabolism, AMPK activation can prevent mTORC1 induction by anabolic stimuli, including IGF-1, leucine, glucose and electrically-stimulated muscle contractions [93-95].
Energy metabolism in skeletal muscle
The capacity of skeletal muscles to utilize nutrient substrates is a critical determinant of muscle quality and viability and overall systemic health [96]. Determinants of skeletal muscle metabolic capacity include the type of fiber, metabolic enzymes expression level, and glycogen and lipid substrates abundance. Central to skeletal muscle metabolic homeostasis is mitochondria function, which has a critical role in apoptosis, autophagy, and protein turnover processes [97]. Since muscle mitochondria loss is a feature of many wasting conditions [98-100], increasing or maintaining muscle oxidative capacity is a potential therapeutic target for muscle wasting. Consistent with the anabolic effect of androgens on skeletal muscles, transgenic overexpression of the androgen receptor in the rat extensor digitorum longus (EDL) muscle increases myoglobin expression and mitochondrial enzyme activity [71]. Global deletion of the AR [70] (or ERβ [76]), on the other hand, increases muscle fatigue resistance in a muscle- and sex-specific manner. Myofiber specific loss of the AR has no effect on gastrocnemius glycogen stores, but it increases the percentage of type I myofibers in the soleus, while no changes are found in fiber type distribution in the EDL muscle [35]. Estrogen deficiency, on the other hand, adversely affects muscle oxidative metabolism or the regulation of protein turnover. Conversely, estrogen administration to ovariectomized mice induces the expression of these factors [101]. Furthermore, estrogen signaling through the ERα and ERβ regulates mitochondrial biogenesis and function [102]; and ovariectomy causes mitochondrial dysfunction, which is attenuated by estrogen replacement [103].
Mitochondrial function influences both anabolic and catabolic signal transduction pathways in muscles [97], including signaling involving AMP-activated protein kinase (AMPK) – a cellular energy sensor and activator of PGC1α – mTOR, and FOXOs [104, 105]. Peroxisome proliferator-activated receptors (PPARs) α, β, and δ, and their transcriptional co-activators PGC1α and PGC1β also play important roles in the metabolic homeostasis of muscles and muscle mass [106, 107]. In addition, PGC1α and PGC1β are involved in mitochondria biogenesis and function [97]. Testosterone induces muscle PGC1α and cytochrome c oxidase subunit IV (Cox4) expression in mice, while AR deletion suppresses them [108]. Intriguingly, both muscle AR and ER signaling is higher in some, but not all, resistance-trained humans [109]. Consistent with the role of estrogens on energy metabolism, ovariectomy increases the activation of PPARα and lean body mass in rats [110, 111] and suppresses the expression of skeletal muscle PPARδ, as well as PDK-4, UCP-2, and FOXO1 expression, which are also involved in oxidative metabolism [112].
The regulation of mitochondria dynamics, the balance of fusion and fission processes, is disrupted in many disease conditions and during the progression of muscle wasting [113]. Decreased muscle mitochondria fusion and increased fission has emerged as a central mechanism for skeletal muscle anabolic signaling suppression with the progression of wasting. Mitochondrial fusion protein 1 and 2 (Mfn1/Mfn2) and optic atrophy protein 1 (OPA1) serve as regulators of this process. In healthy individuals, mitochondrial protein content of PGC-1α, citrate synthase, and mitochondrial creatine kinase are directly correlated with mitochondrial fusion. Fusion is also associated with increased exercise capacity [114]. Additionally, mitochondria fission-sensitive regulation of AMPK can regulate FOXO3 independently of activation of Akt [99]. When FOXO activity is inhibited in the presence of altered mitochondria function, muscle atrophy is prevented. Further work is needed to establish a role of sex steroid regulation of mitochondria fission and fusion with muscle wasting disease and aging-induced sarcopenia.
The putative signaling pathways, paracrine mediators, and gene targets of sex steroid action in bone and muscle are summarized in Table 2.
Table 2. Putative signaling pathways, paracrine mediators, and gene targets of sex steroid action in bone and muscle.
| Bones | Muscles |
|---|---|
| 1. NF-kB | 1. NF-kB |
| 2. Kinases (ERK1/2; PI3K/Akt) | 2. Kinases (ERK1/2; PI3K/Akt) |
| 3. Cytokines (IL-1; IL-6; TNFα; FasL) | 3. Cytokines (IL-6; TNFα) |
| 4. Wnt/ß-catenin signaling | 4. Myogenic regulatory factors (MyoD; myogenin; cell cycle regulatory factors; CDK2; CDK4; P21; P57kip2) |
| 5. FoxOs | |
| 6. mTOR |
6. Interactions among sex steroids, mechanical forces, and growth factors
A. In bone
Bone responds to and adapts to changes in mechanical strains and ERα participates in the transduction of mechanical signals in bone. Indeed, mice with global ERα deletion have a defective response of bone to mechanical loading [115]; and ERα potentiates the activation of the Wnt/β-catenin pathway in response to mechanical stimulation in vitro [116]. In addition, ERα or ERβ may be required for the transduction of mechanical forces into pro-survival signals in osteocytes and osteoblasts, independently of estrogens [117]. Further, ERα expressed in osteoblast progenitors potentiates Wnt/β-catenin signaling and periosteal cell proliferation without the requirement of activation of the receptor protein by estrogens, i.e. in a ligand-independent manner [44]. In line with this evidence, activation of the LDL Receptor-Related Protein 5 (LRP5)/Wnt/β-catenin signaling pathway is required for the physiologic response of bone to mechanical loading and activation of this pathway enhances the sensitivity of osteoblastic cells to mechanical loading [118-120]. In contrast to the case with osteoblast progenitors, the actions of ERα on mature osteoblasts and osteocytes may not affect trabecular or cortical bone mass in females [121] or males [45]. Taken together, the evidence strongly suggests that ERα in osteoblast progenitors is responsible for transducing the effects of loading on cortical bone. Identical conclusions for the requirement of the ERα for the osteogenic response to mechanical loading in a ligand-independent manner have been derived from axial loading experiments of OVX mice [122, 123]. In contrast to females, the ERα is not required for the adaptive response to loading in male mice suggesting that in males other signals induced by loading override the anabolic actions of ERα on the periosteum [124].
The effects of estrogens on the development of reproductive organs, such as breast, are greatly influenced by growth factors, such as IGF [125]. An extensive cross-talk between growth factors and sex steroids seems also important for bone. Indeed, estradiol potentiates the effect of growth hormone in osteoblasts, via a non-nucleus-initiated mechanism of estrogen action [126]. Consistent with this in vitro finding, male mice experience greater periosteal and endocortical bone formation along with higher serum IGF levels during early puberty. Loss of estrogens in the female, on the other hand, increases bone expansion and the endocortical bone perimeter. Mice with very low IGF-1 levels (lacking the growth hormone receptor/binding protein gene) have limited bone expansion and no skeletal dimorphism, indicating that skeletal dimorphism during early puberty depends on the cross-talk between sex steroids and GH–IGF-1 action. Furthermore in middle-aged and elderly men, IGFBP-3 and IGF-I are positively correlated with BMD [127].
B. In muscles
As it is the case of bones, skeletal muscles are highly responsive to mechanical loading. Indeed, overloading or disuse dramatically increase and decrease muscle mass, respectively. The load response in muscle involves mechanical signaling and a corresponding remodeling response that comprises the regeneration of muscle tissue and the altered expression of protein forms related to shifts in the muscle phenotype. Myofiber interaction with the microenvironment related to components of the extracellular matrix is critical for both mechanical signaling and remodeling. The evidence for a role of IGF-1/AKT signaling in the anabolic effects of testosterone and mechanical loading notwithstanding, muscle protein synthesis and degradation can be regulated independently of Akt signaling. Indeed, there is evidence that IGF-1/AKT signaling is not involved in many loading conditions and disease states.
The mammalian target of rapamycin (mTOR) has emerged as a very important integration point for hormonal, nutritional, and mechanical signals involved in the regulation of muscle protein synthesis [104, 128]. Briefly, mTOR complex 1 (mTORC1) formation increases protein translation through phosphorylation of substrates 4EBP-1 and p70S6K. Activation of mTORC1 by mechanical stimuli involves activation of phospholipase D (PLD) [129, 130], extracellular regulated kinase 1/2 (ERK1/2) [131, 132], and p38 signaling [132-134].
Recently, a transcriptional target of Peroxisome proliferator-activated receptor gamma, coactivator 1-alpha 4 (PGC-1alpha4), has been implicated in load induced muscle hypertrophy [135]. PGC-1alpha4 is a protein isoform derived by alternative splicing of the PGC1alpha mRNA and regulates muscle hypertrophy through the transcriptional induction of G protein-coupled receptor 56 (GPR56) [136]. Further work is needed to establish the integration estrogen and androgen signaling with muscle mechanical signaling networks that activate muscle protein synthesis through mTOR dependent, and possibly mTOR independent mechanisms.
Mechanical overload induces muscle growth and involves an orchestrated cross talk between several cell types in muscle. All of these cell types are potential targets of androgen and estrogen action. Furthermore, a large body of evidence indicates that androgens and loading potentiates each other's effect on muscle growth. Load-induced hypertrophy involves edema, connective tissue proliferation, myogenic precursor cell proliferation, tissue repair, and inflammation [137-140]. These are all critical processes for the remodeling that results in overload-induced growth. Anabolic steroid administration simultaneously with mechanical overload increases the incidence of large diameter myofibers, and attenuates the increase in small fibers (< 500 μm2), which indicate muscle regeneration [141]. These findings suggest that anabolic steroids can target the muscle regeneration process, as well as fiber enlargement. Satellite cells, resident myogenic stem cells residing outside the sarcolemma of the myofiber, are the source of nuclei for growing and regenerating muscle fibers [142]. Muscle satellite cells express both the androgen receptor [64], and the estrogen receptors α and ß [143]. Estrogen and testosterone can stimulate satellite cell activity [29, 142, 143]. Myogenic regulatory factors (MRFs), including MyoD, myogenin, and cell cycle regulatory factors (CDK2, CDK4, and p21) regulate satellite cell activity and are induced in hypertrophying skeletal muscle [138, 144]. Androgens can induce CDK2, CDK4, and p21 expression, while repressing the CDK inhibitor p16 in many cell types. Myogenin expression is also induced by androgen receptor expression in cultured muscle cells, suggesting that androgens may regulate myoblast differentiation. The release of chemokines and growth factors can influence the satellite cell niche within the muscle and regulate the replication and differentiation of both satellite and fibroblast cells [145].
Microarray analysis of skeletal muscles from mice with global AR deletion demonstrates that genes involved in cell proliferation and differentiation, such as p57Kip2, are regulated, at least in part by AR signaling, and such signaling may serve to maintain satellite cells in a proliferative state [70]. Fibroblasts are responsible for the formation and maintenance of the extracellular matrix, which can feedback to alter myofiber intracellular signaling [146]. Additionally, resident and infiltrating immune cells have an important role in the remodeling response through the regulation of satellite cell and fibroblast cell activity. Androgens may also alter the inflammatory stage of muscle regeneration, as impairment of phagocytosis in implanted mouse skeletal muscle grafts and lower myosin heavy chain concentration in snake venom injected mouse soleus muscle has been reported in castrated mice [147, 148]. Snake venom induced-muscle injury responds favorably to testosterone administration in the predominantly slow-oxidative muscle, but not the fast-type glycolytic muscle. And, testosterone replacement in both young and old orchidectomized mice promotes regeneration after injury [149].
7. Sex steroids and the cross-talk between muscles and bones
It is widely believed that mechanical loads on bone arise, to a large extent, from muscle forces and that there exists a close functional relationship between the two tissues. In support of this, experimental evidence from animal models (and some, but not all, clinical observations in humans) suggest that the growth and maintenance of muscle and bone mass are interdependent [150]. Specifically, embryonic muscle paralysis or irregular myogenesis in chicken and mice causes poor bone mineralization and abnormal curvature [151]. Likewise, botulinum toxin-induced muscle paralysis in adult mice causes bone loss [152] and attenuates fracture healing [153]. Conversely, an increase in muscle mass caused by loss of function of myostatin – a member of the transforming growth factor-beta (TGF-beta) superfamily that is highly expressed in skeletal muscle –leads to an increase in bone formation and an increase in cross-sectional bone area in most anatomical regions, including the limbs, spine, and jaw in mice [154]; as well as in sites of large muscle attachment to bone [155]. Myostatin is also expressed in the early phases of fracture healing, and myostatin deficiency leads to increased fracture callus size and strength. Together, these lines of evidence suggest that myostatin may affect the proliferation and differentiation of osteoprogenitor cells, and that myostatin antagonists and inhibitors may be therapeutically useful in enhancing both muscle mass and bone. However, the effects of myostatin on the skeleton could be indirect, especially because modulation of this pathway after completion of growth has no effect on the adult skeleton. In any case, in agreement with the experimental observations in animal models, low bone density and increased incidence of bone fractures have been reported in patients with Duchenne muscular dystrophy [156].
As children grow they gain body weight and bone and muscle mass and strength. Body composition influences bone and the so-called muscle- bone unit. Acquisition of optimal bone mass at this stage is largely dependent on the increase in body size. Unfavorable body composition during sexual maturation results in sub-optimal bone mass and strength in both early adulthood and later life. Skeletal loads arising from muscle forces actuate bony levers such that limb muscle mass are a reasonable measure of skeletal load. It is, therefore, expected that optimizing muscle strength during this time will have a positive impact on both current and future risk of fracture. Consistent with this expectation, physical activity during childhood augments bone mass and density. Nonetheless, the positive effect of weight-bearing exercise on various estimates of bone strength is small and the specific contribution of muscle strength (or surrogates) to bone strength in pre-pubertal boys and girls has not been convincingly discerned. [157]. Moreover, muscular atrophy in children is not always accompanied by osteopenia [158]. A dissociation between muscle mass and bone mass is also implied by the dissimilar curves depicting the rate of gain and loss of bone mineral density as compared to the gain and loss of muscle mass as a function of age in humans [159]. Similarly, in aged mice receiving a myostatin inhibitor increased muscle mass and muscle fiber size is not accompanied by increased bone density or bone strength [160].
Exercise may promote bone mass acquisition through its direct loading effects on bone as well as through its effects on muscle mass and strength [161]. Additionally, exercise may be beneficial to bone through hormonal changes, including estrogen signaling through the ER, GH/IGF-1 (secondary to changes in serum glucose), and parathyroid hormone. The most opportune time for enhancing bone strength through physical activity is probably the pre- and peri-pubertal periods [157]. Nonetheless, the interaction of skeletal muscle and normal bone health may extend beyond exercise-induced muscle contraction [162-164]. Indeed, emerging evidence raises the possibility that the muscle and bone relationship is independent of mechanical loading. One mechanistic explanation could be that the positive effects of muscles on bone are the result of chemical factors released from muscle or myokines that affect bone metabolism [150]. In support of these ideas, muscle flaps can improve fracture repair from traumatic orthopaedic injury in animal models and humans [165-167]. Additionally, new bone formation can be directly stimulated by muscle implants alongside the periosteum [168]. Diminished bone healing is also seen when muscle surrounding these bone defects is damaged [169, 170]. In any event, the reduction in muscle mass with the loss of androgens does not seem to directly impact bone. Indeed, mice with muscle specific deletion of the AR display blunted growth, decreased intra-abdominal fat, but show no effect on trabecular and cortical bone parameters [26].
8. Parallels in the development of osteoporosis and sarcopenia
Osteoporosis
Soon after the attainment of peak bone mass– sometime during the third decade of life in humans–the balance between bone formation and bone resorption begins to progressively tilt in favor of the latter, in both women and men. This change begins long before and independently of any changes in sex steroid levels. Nonetheless, at menopause the loss of trabecular bone accelerates, causing perforation of entire trabeculae and loss of connectedness among trabeculae. The rate of bone loss in women slows within five to ten years after the menopause and is followed by a slower phase of bone loss that occurs also in men. This later phase affects primarily cortical bone, and a significant portion of it is due to increased endocortical resorption and intracortical porosity. The histologic hallmark of this later stage in either sex is a decrease in osteoblast number and/or function, consistent with the notion that aging per se causes a decrease in the supply of osteoblasts.
Similar to humans, sex steroid-sufficient female and male mice experience an age-dependent progressive decline in bone mass and strength, starting around four months of age; loss of bone strength precedes the loss of bone mass [171]. The progressive loss of bone mass and strength in mice are temporally associated with increased oxidative stress. Importantly, identical changes in oxidative stress are recapitulated in either sex of mice acutely upon loss of sex steroids. Furthermore, the adverse effects of the loss of sex steroids on murine bone can be prevented by the systemic administration of antioxidants or by genetically decreasing H2O2 production in osteoclast mitochondria [52, 171].
The risk of bone fractures in humans increases exponentially with advancing age, and to a much greater extent that can be accounted by the loss of bone mass alone. In fact for the same BMD, the risk of fracture increases six- fold between the age of 55 and 75 [172]. Age-related mechanisms intrinsic to bone, including oxidative stress, declining autophagy, cell (osteocyte) senescence are protagonists in involutional osteoporosis and age-related changes in other organs and tissues, such as ovaries and adrenals, are contributory [2-4]. Indeed, it is now well appreciated that apoptosis or dysfunction of osteocytes, defective mesenchymal cell differentiation toward the osteoblast phenotype, decreased growth factor production, declining physical activity, and perhaps declining muscle mass are all important pathogenetic mechanisms for the decline of bone mass and strength with age [39].
Apoptotic or dysfunctional osteocytes increase the release of RANKL – the rate limiting factor for osteoclast generation and survival – by healthy neighboring osteocytes [173]. Apoptotic osteocytes themselves and their cellular debris can also initiate “danger (or damage)-associated molecular events that trigger pro-inflammatory immune responses. In addition, osteocyte senescence or apoptosis causes a decrease in bone vascularity and hydration – mechanisms likely to reduce bone strength by altering the crystal structure of hydroxyapatite. Recent research findings from the mouse model also revealed that attenuation of Wnt/β-catenin signaling may be responsible for the decrease in bone formation and the increase in bone marrow adiposity associated with old age [2, 174]. FoxOs are members of a subclass of the forkhead family of transcription factors that provide important defense mechanisms against oxidative stress and growth factor deprivation by stimulating the transcription of antioxidant enzymes as well as genes involved in cell cycle, DNA repair, and lifespan [175, 176]. Importantly, in the process of stimulating these genes, FoxOs divert the limited pool of β-catenin in osteoblast progenitors from Wnt/TCF- to FoxO-mediated transcription and attenuated Wnt signaling [177]. Decreased Wnt signaling, in turn, causes a decrease in the production of osteoblasts and increases bone marrow adiposity [174]. In this way, FoxO activation worsens the effects of old age on bone and contributes to skeletal involution [178]. Lastly, a decrease in physical activity and muscle mass with old age, combined with a decrease in ERα expression in bone cells, may compromise the mechanosensitivity of bone and the ability of the skeleton adapts to meet mechanical needs.
Sarcopenia
Sarcopenia is a condition characterized by age-related loss of muscle mass and function, but its definition continues to evolve (Cederholm T, Morley JE. Curr Opin Clin Nutr Metab Care. 2015 Jan;18(1):1-4. doi: Sarcopenia: the new definitions.). Several distinct pathologies, including myofiber atrophy, motor unit loss, fibrosis, and intramuscular fat accumulation are thought to be responsible for the loss of muscle mass in sarcopenia [12, 179]. Collectively, these pathologic processes lead to deficits in muscle function. Such functional deficits are quantified by measuring walking speed or grip strength, and are used as criteria for the diagnosis of sarcopenia (Cederholm T, Morley JE. Curr Opin Clin Nutr Metab Care. 2015 Jan;18(1):1-4. doi: Sarcopenia: the new definitions.). The underlying biochemical culprits for the age-induced myofiber atrophy and disrupted muscle function are thought to be disrupted oxidative metabolism [180], resistance to anabolic stimuli [181], and a decline of myogenic stem cell (satellite cells) number and function [182]; each one of these biochemical changes have been proposed as potential therapeutic targets for the management of sarcopenia. At present, the role of other age-related changes in sarcopenia – such as the decrease in circulating sex steroid production or altered intracellular signaling pathways in muscles – is not well established. It is also remains unclear whether a single therapeutic intervention can counter the several pathologic mechanisms responsible for sarcopenia and thereby rescue muscle mass and function in old age. Nevertheless, various types of exercise and physical activity have been shown to reverse more than one muscle pathology associated with advancing age [183].
Inflammatory disease, autoimmune disorders, endocrine dysfunction, and aging cause muscle wasting and also can impede the skeletal muscle's robust capacity for regeneration [184]. While androgen administration attenuates muscle wasting with many chronic diseases [185], the systemic mediators of this wasting response remain elusive. Classical disease induced stimuli thought to induce muscle wasting involve circulating inflammatory cytokines, suppressed anabolic hormones, anemia, anorexia, and catabolic mediators related to systemic disruption of metabolic homeostasis [186]. Additionally, tumor derived factors can induce muscle wasting with cancer cachexia. For example, tumor-derived parathyroid-hormone-related protein (PTHrP) has recently been shown to cause muscle and adipose tissue loss in a mouse model of cancer cachexia in combination with an unknown factor [187]. Notably, aging related frailty does not usually involve the same systemic disruptions as cancer and other cachectic conditions.
Muscle wasting has been long thought as the result of accelerated muscle protein degradation though the ubiquitin proteasome system. A more recent network-based understanding of muscle protein turnover, however, indicates that abnormal muscle mitochondrial function, suppressed anabolic signaling, apoptosis, and autophagy also play important pathogenetic roles in cachexia [188, 189]. As mentioned earlier in this review, estrogens or androgens influence many of these processes in skeletal muscle. However, further work is needed to delineate exactly how the hormonal signals interact with the molecular pathways controlling these processes, and whether either of these hormones can be useful for the management of muscle wasting. Incomplete or extended muscle regeneration can lead to a suppressed growth response, decreased functional capacity, and even wasting. Testosterone supplementation improves early indices of muscle regeneration in castrated aged mice [149]. Inflammation and aging can also impede mechanical load-induced activation of muscle mTORC signaling, which is associated with muscle anabolic resistance [104, 190]. The chronic inflammation that is associated with muscle wasting induces ERK1/2 and p38 signaling [191-194]. Although PI3K/Akt signaling induces mTOR activity, Akt independent suppression of mTOR can occur during cancer cachexia [80]. Testosterone loss following castration in and of itself suppresses basal muscle mTOR. Once again, further work is needed to understand if decreased androgens levels contribute to this state, or whether androgen therapy can improve anabolic resistance in these conditions.
To summarize this section, advancing age leads to a loss of tissue mass and strength in both bone and skeletal muscles; and age-related mechanisms are protagonists in the development of both osteoporosis and sarcopenia. Specifically, mitochondria dysfunction, oxidative stress, decreased autophagy, inflammation, cellular senescence, stem cell exhaustion, as well as the age-associated decline in growth factor levels and physical activity are evidently shared mechanisms in both osteoporosis and sarcopenia. Like aging, sex steroid deficiency decreases mass and strength in either tissue and it may indeed accelerate the effects of aging by potentiating age-related pathogenetic mechanisms. Notably, glucocorticoid excess (due to endogenous production or pharmacologic administration) is a common secondary cause of both conditions [195].
9. Therapeutic modalities for osteoporosis and sarcopenia
As of the writing of this review article, there are seven classes of FDA approved therapeutic modalities for the prevention and treatment of osteoporosis; and three more are in late phase 3 clinical trials (Table 3). Within each class of modalities, there are several drugs. In sharp contrast, there is currently not a single approved therapy for the prevention or treatment of sarcopenia. Several pharmacologic interventions attempted so far have shown to have very limited efficacy. Part of the difficulty in developing anti-sarcopenia drugs could be the lack of diagnostic criteria and standardized primary outcomes [196]. As a result of the paucity of drug therapies, the only available options for the management of sarcopenia are exercise and resistance training and nutritional supplements. The effectiveness of such interventions, however, is limited and fraught with shortcomings, including the inability or unwillingness of elderly individuals to get involved in strenuous exercise training programs.
Table 3. Therapeutic modalities for osteoporosis and sarcopenia.
| Osteoporosis | Sarcopenia |
|---|---|
| A: FDA approved | A: FDA approved |
| 1. Estrogen replacement (conjugated estrogens +/- progesterone; ethinyl estradiol) | None B. Under investigation (Phase 1 or 2 Clinical Trials) |
| 2. Selective estrogen receptor modulators [SERMs] (Raloxifene) | 1. Testosterone |
| 3. Combination of conjugated estrogen and the SERM Bazedoxifene | 2. Megestrol acetate |
| 4. Salmon calcitonin (injectable; inhalable) | 3. Nandrolone |
| 5. Bisphosphonates (alendronate; ibandronate; risedronate; zolindronic acid) | 4. Dehydroepiandrosterone (DHEA). |
| 6. Anti-RANKL antibody (Denosumab) | 5. SARMs ? |
| 7. PTH 1-34 (teriparatide) | 6. Anabolic steroids ? |
| B. In Phase 3 Clinical Trials | 7. Growth hormone |
| 1. Kathepsin K inhibitor (Odanacatib) | 8. Angiotensin-converting enzyme inhibitors |
| 2. Sclerostin neutralizing antibodies (Romosozumab; | 9. PPAR-δ agonist GW1516 |
| AMG 785) | 10. Myostatin inhibitors |
| 3. PTHrP analog (abaloparatide) |
Importantly, in contrast to the dramatic decrease of estrogen replacement therapy for post-menopausal women during the last decade because of the appreciation of serious side effects, testosterone therapy for men with the so-called “low T” has increased in the United States by 500% between 1993 and 2000. This is a striking trend, but the extent to which it reflects “perceived” as opposed to “real” health benefits remains unclear, and of concern considering reports of serious side effects associated with such therapies [197].
10. Summary and concluding thoughts
Estrogens and androgens play important roles in the growth and maintenance of tissue mass and function in bones and muscles and are responsible for the sexual dimorphism of either tissue. The effects of sex steroids in both tissues result from direct actions on several different cell types via the classical sex steroid receptors (Table 1) and similar signaling pathways (Table 2); as well as indirectly via actions on other tissues. In both bones and muscles, the effects of sex steroids are additive and perhaps synergistic to the anabolic effects of mechanical loading and growth factors, such as IGF-1. Sex steroid deficiency leads to loss of tissue mass and functional integrity and contributes to the development of osteoporosis and sarcopenia – the two most common manifestations of aging in humans. A summary of the similarities of the effects of sex steroids on bone and muscles is provided in Figure 2.
Figure 2. Similarities of the effects of sex steroids in bones and muscles.

Estrogens have potent and well documented effects on skeletal growth and maintenance in females as well as males. By comparison to bone, the effects of estrogens on muscle mass growth and maintenance seem relatively weak; and consistent with a relatively weak effect, the contribution of estrogen deficiency to the development of sarcopenia later in life in females (or males) is uncertain. This is in contrast to the evidence that androgens have far more potent effects than estrogens on the muscle tissue in either sex, especially when combined with exercise. This apparent dichotomy raises the possibility that testosterone or dihydrotestosterone may be the more important sex steroid in the regulation of muscle homeostasis in both males and females. Against this possibility, however, is the evidence that global deletion of the androgen receptor in mice decreases muscle mass in the males but has no effect in the females. Unexpectedly, cortical or trabecular bone is not affected in male mice with global or myocyte-specific deletion of the androgen receptor, arguing against a role of androgens in the cross-talk between muscles and bones. More intriguingly, muscle mass in animals and humans can increase many-fold with androgenic steroids and exercise, but this cannot happen in bone. Why not? Could it be that homeostatic mechanisms, such as the so called “bone mechanostat” proposed several decades ago by Harold Frost, are far more restrictive against excessive tissue growth in bones than they are in muscles?
In any event, the contribution of a decline in sex steroid levels to the development of disease is far better documented in bone than in muscle, but it remains possible that this could well be due to the fact that osteoporosis is better defined than sarcopenia, and much easier to quantify by widely used and standardized techniques, such as dual energy x-ray absorptiometry (DEXA). At present, it is also unclear whether the dramatic difference in the availability of several effective drugs for the prevention and treatment of osteoporosis (including natural estrogens and SERMs) versus no drug therapy for sarcopenia is due to fundamental differences in the biologic causes of these two diseases; or simply to the easier diagnosis and quantification of the former versus the latter.
Mechanisms of cellular aging – such as mitochondria dysfunction and decreased autophagy– as well as the age-associated decline in growth factor levels and physical activity are inexorable culprits and likely the protagonists in the pathogenesis of osteoporosis and sarcopenia alike. Therefore, novel drugs that could target such common mechanisms of aging have the potential to be simultaneously beneficial for osteoporosis and sarcopenia.
Highlights.
Estrogens and androgens help to maintain mass and strength in bones and muscles.
Their effects result from direct as well as indirect actions on other tissues.
In both tissues, the effects of sex steroids are potentiated by mechanical loading.
Sex steroid deficiency contributes to osteoporosis and sarcopenia alike.
Cell aging and mitochondria dysfunction are shared seminal culprits in either disease.
Acknowledgments
The authors thank Leah Timmons for help with the preparation of the manuscript, and Robert L. Jilka, Charles A. Obrien, Robert S. Weinstein, and Maria Almeida for critically reviewing it prior to submission to the journal. The authors' research is supported by the NIH (P01 AG13918 and R01 AR56679 [to SCM]; and R01 CA121249A501 and P20 RR-017698 [to JAC]) and the Biomedical Laboratory Research and Development Service of the Veterans Administration Office of Research and Development (I01 BX001405 [to SCM]).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R. The healthcare costs of sarcopenia in the United States. J Am Geriatr Soc. 2004;52:80–5. doi: 10.1111/j.1532-5415.2004.52014.x. [DOI] [PubMed] [Google Scholar]
- 2.Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31:266–300. doi: 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manolagas SC, Parfitt AM. What old means to bone. Trends Endocrinol Metab. 2010;21:369–74. doi: 10.1016/j.tem.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Onal M, Piemontese M, Xiong J, Wang Y, Han L, Ye S, et al. Suppression of autophagy in osteocytes mimics skeletal aging. J Biol Chem. 2013;288:17432–40. doi: 10.1074/jbc.M112.444190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Argiles JM, Anker SD, Evans WJ, Morley JE, Fearon KC, Strasser F, et al. Consensus on cachexia definitions. J Am Med Dir Assoc. 2010;11:229–30. doi: 10.1016/j.jamda.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12:489–95. doi: 10.1016/S1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- 7.Carlson BM. Muscle regeneration and aging. Monogr Dev Biol. 1992;23:189–95. [PubMed] [Google Scholar]
- 8.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 9.McClung JM, Davis JM, Wilson MA, Goldsmith EC, Carson JA. Estrogen status and skeletal muscle recovery from disuse atrophy. J Appl Physiol (1985) 2006;100:2012–23. doi: 10.1152/japplphysiol.01583.2005. [DOI] [PubMed] [Google Scholar]
- 10.White JP, Baltgalvis KA, Sato S, Wilson LB, Carson JA. Effect of nandrolone decanoate administration on recovery from bupivacaine-induced muscle injury. J Appl Physiol (1985) 2009;107:1420–30. doi: 10.1152/japplphysiol.00668.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spangenburg EE, Geiger PC, Leinwand LA, Lowe DA. Regulation of physiological and metabolic function of muscle by female sex steroids. Med Sci Sports Exerc. 2012;44:1653–62. doi: 10.1249/MSS.0b013e31825871fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delmonico MJ, Harris TB, Visser M, Park SW, Conroy MB, Velasquez-Mieyer P, et al. Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am J Clin Nutr. 2009;90:1579–85. doi: 10.3945/ajcn.2009.28047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finkelstein JS, Yu EW, Burnett-Bowie SA. Gonadal steroids and body composition, strength, and sexual function in men. N Engl J Med. 2013;369:2457. doi: 10.1056/NEJMc1313169. [DOI] [PubMed] [Google Scholar]
- 14.Liverman CT, Blazer DG. Testosterone and Aging: Clinical Research Directions. Washington, DC: The National Academies Press; 2004. [PubMed] [Google Scholar]
- 15.Grinspoon S, Corcoran C, Lee K, Burrows B, Hubbard J, Katznelson L, et al. Loss of lean body and muscle mass correlates with androgen levels in hypogonadal men with acquired immunodeficiency syndrome and wasting. J Clin Endocrinol Metab. 1996;81:4051–8. doi: 10.1210/jcem.81.11.8923860. [DOI] [PubMed] [Google Scholar]
- 16.Garcia JM, Li H, Mann D, Epner D, Hayes TG, Marcelli M, et al. Hypogonadism in male patients with cancer. Cancer. 2006;106:2583–91. doi: 10.1002/cncr.21889. [DOI] [PubMed] [Google Scholar]
- 17.Burney BO, Hayes TG, Smiechowska J, Cardwell G, Papusha V, Bhargava P, et al. Low testosterone levels and increased inflammatory markers in patients with cancer and relationship with cachexia. J Clin Endocrinol Metab. 2012;97:E700–E709. doi: 10.1210/jc.2011-2387. [DOI] [PubMed] [Google Scholar]
- 18.Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95:2536–59. doi: 10.1210/jc.2009-2354. [DOI] [PubMed] [Google Scholar]
- 19.Bhasin S, Woodhouse L, Storer TW. Androgen effects on body composition. Growth Horm IGF Res. 2003;13(Suppl A):S63–S71. doi: 10.1016/s1096-6374(03)00058-3. [DOI] [PubMed] [Google Scholar]
- 20.Bhasin S, Storer TW, Berman N, Yarasheski KE, Clevenger B, Phillips J, et al. Testosterone replacement increases fat-free mass and muscle size in hypogonadal men. J Clin Endocrinol Metab. 1997;82:407–13. doi: 10.1210/jcem.82.2.3733. [DOI] [PubMed] [Google Scholar]
- 21.Antonio J, Wilson JD, George FW. Effects of castration and androgen treatment on androgen-receptor levels in rat skeletal muscles. J Appl Physiol (1985) 1999;87:2016–9. doi: 10.1152/jappl.1999.87.6.2016. [DOI] [PubMed] [Google Scholar]
- 22.Axell AM, MacLean HE, Plant DR, Harcourt LJ, Davis JA, Jimenez M, et al. Continuous testosterone administration prevents skeletal muscle atrophy and enhances resistance to fatigue in orchidectomized male mice. Am J Physiol Endocrinol Metab. 2006;291:E506–E516. doi: 10.1152/ajpendo.00058.2006. [DOI] [PubMed] [Google Scholar]
- 23.Bhasin S, Storer TW, Berman N, Callegari C, Clevenger B, Phillips J, et al. The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med. 1996;335:1–7. doi: 10.1056/NEJM199607043350101. [DOI] [PubMed] [Google Scholar]
- 24.Ferrando AA, Tipton KD, Doyle D, Phillips SM, Cortiella J, Wolfe RR. Testosterone injection stimulates net protein synthesis but not tissue amino acid transport. Am J Physiol. 1998;275:E864–E871. doi: 10.1152/ajpendo.1998.275.5.E864. [DOI] [PubMed] [Google Scholar]
- 25.Griggs RC, Kingston W, Jozefowicz RF, Herr BE, Forbes G, Halliday D. Effect of testosterone on muscle mass and muscle protein synthesis. J Appl Physiol (1985) 1989;66:498–503. doi: 10.1152/jappl.1989.66.1.498. [DOI] [PubMed] [Google Scholar]
- 26.Elbers JM, Asscheman H, Seidell JC, Gooren LJ. Effects of sex steroid hormones on regional fat depots as assessed by magnetic resonance imaging in transsexuals. Am J Physiol. 1999;276:E317–E325. doi: 10.1152/ajpendo.1999.276.2.E317. [DOI] [PubMed] [Google Scholar]
- 27.Cawthon PM, Ensrud KE, Laughlin GA, Cauley JA, Dam TT, Barrett-Connor E, et al. Sex hormones and frailty in older men: the osteoporotic fractures in men (MrOS) study. J Clin Endocrinol Metab. 2009;94:3806–15. doi: 10.1210/jc.2009-0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foryst-Ludwig A, Kintscher U. Metabolic impact of estrogen signalling through ERalpha and ERbeta. J Steroid Biochem Mol Biol. 2010;122:74–81. doi: 10.1016/j.jsbmb.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 29.Enns DL, Tiidus PM. Estrogen influences satellite cell activation and proliferation following downhill running in rats. J Appl Physiol (1985) 2008;104:347–53. doi: 10.1152/japplphysiol.00128.2007. [DOI] [PubMed] [Google Scholar]
- 30.Enns DL, Iqbal S, Tiidus PM. Oestrogen receptors mediate oestrogen-induced increases in post-exercise rat skeletal muscle satellite cells. Acta Physiol (Oxf) 2008;194:81–93. doi: 10.1111/j.1748-1716.2008.01861.x. [DOI] [PubMed] [Google Scholar]
- 31.Tiidus PM, Lowe DA, Brown M. Estrogen replacement and skeletal muscle: mechanisms and population health. J Appl Physiol (1985) 2013;115:569–78. doi: 10.1152/japplphysiol.00629.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Onambele-Pearson GL. HRT affects skeletal muscle contractile characteristics: a definitive answer? J Appl Physiol (1985) 2009;107:4–5. doi: 10.1152/japplphysiol.00448.2009. [DOI] [PubMed] [Google Scholar]
- 33.McClung JM, Davis JM, Carson JA. Ovarian hormone status and skeletal muscle inflammation during recovery from disuse in rats. Exp Physiol. 2007;92:219–32. doi: 10.1113/expphysiol.2006.035071. [DOI] [PubMed] [Google Scholar]
- 34.Enns DL, Tiidus PM. The influence of estrogen on skeletal muscle: sex matters. Sports Med. 2010;40:41–58. doi: 10.2165/11319760-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 35.Ophoff J, Van PK, Callewaert F, De GK, De BK, Vanden Bosch A, et al. Androgen signaling in myocytes contributes to the maintenance of muscle mass and fiber type regulation but not to muscle strength or fatigue. Endocrinology. 2009;150:3558–66. doi: 10.1210/en.2008-1509. [DOI] [PubMed] [Google Scholar]
- 36.Brown M, Ning J, Ferreira JA, Bogener JL, Lubahn DB. Estrogen receptor-alpha and -beta and aromatase knockout effects on lower limb muscle mass and contractile function in female mice. Am J Physiol Endocrinol Metab. 2009;296:E854–E861. doi: 10.1152/ajpendo.90696.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crandall CJ, Barrett-Connor E. Endogenous sex steroid levels and cardiovascular disease in relation to the menopause: a systematic review. Endocrinol Metab Clin North Am. 2013;42:227–53. doi: 10.1016/j.ecl.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 38.Mendelsohn ME. Estrogen actions in the cardiovascular system. Climacteric. 2009;12(Suppl 1):18–21. doi: 10.1080/13697130903020291. [DOI] [PubMed] [Google Scholar]
- 39.Manolagas SC, O'Brien CA, Almeida M. The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol. 2013;9:699–712. doi: 10.1038/nrendo.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rai D, Frolova A, Frasor J, Carpenter AE, Katzenellenbogen BS. Distinctive actions of membrane-targeted versus nuclear localized estrogen receptors in breast cancer cells. Mol Endocrinol. 2005;19:1606–17. doi: 10.1210/me.2004-0468. [DOI] [PubMed] [Google Scholar]
- 41.Vanderschueren D, Laurent MR, Claessens F, Gielen E, Lagerquist MK, Vandenput L, et al. Sex steroid actions in male bone. Endocr Rev. 2014:er20141024. doi: 10.1210/er.2014-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin-Millan M, Almeida M, Ambrogini E, Han L, Zhao H, Weinstein RS, et al. The estrogen receptor alpha in osteoclasts mediates the protective effects of estrogens on cancellous but not cortical bone. Mol Endocrinol. 2010;24:323–34. doi: 10.1210/me.2009-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130:811–23. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 44.Almeida M, Iyer S, Martin-Millan M, Bartell SM, Han L, Ambrogini E, et al. Estrogen receptor-alpha signaling in osteoblast progenitors stimulates cortical bone accrual. J Clin Invest. 2013;123:394–404. doi: 10.1172/JCI65910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ucer S, Iyer S, Bartell SM, Martin-Millan M, Han L, Kim HN, et al. The effects of androgens on murine cortical bone do not require AR or ERalpha signaling in osteoblasts and osteoclasts. J Bone Miner Res. 2015 doi: 10.1002/jbmr.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawano H, Sato T, Yamada T, Matsumoto T, Sekine K, Watanabe T, et al. Suppressive function of androgen receptor in bone resorption. Proc Natl Acad Sci U S A. 2003;100:9416–21. doi: 10.1073/pnas.1533500100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Callewaert F, Venken K, Ophoff J, De Gendt K, Torcasio A, van Lenthe GH, et al. Differential regulation of bone and body composition in male mice with combined inactivation of androgen and estrogen receptor-alpha. FASEB J. 2009;23:232–40. doi: 10.1096/fj.08-113456. [DOI] [PubMed] [Google Scholar]
- 48.Chiang C, Chiu M, Moore AJ, Anderson PH, Ghasem-Zadeh A, McManus JF, et al. Mineralization and bone resorption are regulated by the androgen receptor in male mice. J Bone Miner Res. 2009;24:621–31. doi: 10.1359/jbmr.081217. [DOI] [PubMed] [Google Scholar]
- 49.Notini AJ, McManus JF, Moore A, Bouxsein M, Jimenez M, Chiu WS, et al. Osteoblast deletion of exon 3 of the androgen receptor gene results in trabecular bone loss in adult male mice. J Bone Miner Res. 2007;22:347–56. doi: 10.1359/jbmr.061117. [DOI] [PubMed] [Google Scholar]
- 50.Sinnesael M, Claessens F, Laurent M, Dubois V, Boonen S, Deboel L, et al. Androgen receptor (AR) in osteocytes is important for the maintenance of male skeletal integrity: evidence from targeted AR disruption in mouse osteocytes. J Bone Miner Res. 2012;27:2535–43. doi: 10.1002/jbmr.1713. [DOI] [PubMed] [Google Scholar]
- 51.Maatta JA, Buki KG, Ivaska KK, Nieminen-Pihala V, Elo TD, Kahkonen T, et al. Inactivation of the androgen receptor in bone-forming cells leads to trabecular bone loss in adult female mice. Bonekey Rep. 2013;2:440. doi: 10.1038/bonekey.2013.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bartell SM, Kim HN, Ambrogini E, Han L, Iyer S, Serra US, et al. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation. Nat Commun. 2014;5:3773. doi: 10.1038/ncomms4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manolagas SC, Kousteni S, Chen JR, Schuller M, Plotkin L, Bellido T. Kinase-mediated transcription, activators of nongenotropic estrogen-like signaling (ANGELS), and osteoporosis: a different perspective on the HRT dilemma. Kidney Int Suppl. 2004;(91):S41–S49. doi: 10.1111/j.1523-1755.2004.09107.x. [DOI] [PubMed] [Google Scholar]
- 54.Kousteni S, Bellido T, Plotkin LI, O'Brien CA, Bodenner DL, Han K, et al. Nongenotropic, sexnonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–30. [PubMed] [Google Scholar]
- 55.Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O'Brien C, et al. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science. 2002;298:843–6. doi: 10.1126/science.1074935. [DOI] [PubMed] [Google Scholar]
- 56.Manolagas SC, Kousteni S. Perspective: Nonreproductive Sites of Action of Reproductive Hormones. Endocrinology. 2001;142:2200–4. doi: 10.1210/endo.142.6.8221. [DOI] [PubMed] [Google Scholar]
- 57.Page ST, Marck BT, Tolliver JM, Matsumoto AM. Tissue selectivity of the anabolic steroid, 19-nor-4-androstenediol-3 beta,17 beta-diol in male Sprague Dawley rats: Selective stimulation of muscle mass and bone mineral density relative to prostate mass. Endocrinology. 2008;149:1987–93. doi: 10.1210/en.2007-0956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wessler S, Otto C, Wilck N, Stangl V, Fritzemeier KH. Identification of estrogen receptor ligands leading to activation of non-genomic signaling pathways while exhibiting only weak transcriptional activity. J Steroid Biochem Mol Biol. 2006;98:25–35. doi: 10.1016/j.jsbmb.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 59.Otto C, Fuchs I, Altmann H, Klewer M, Schwarz G, Bohlmann R, et al. In vivo characterization of estrogen receptor modulators with reduced genomic versus nongenomic activity in vitro. J Steroid Biochem Mol Biol. 2008;111:95–100. doi: 10.1016/j.jsbmb.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 60.Bartell SM, Han L, Kim HN, Kim SH, Katzenellenbogen JA, Katzenellenbogen BS, et al. Non-Nuclear-Initiated Actions of the Estrogen Receptor Protect Cortical Bone Mass. Mol Endocrinol. 2013;27:649–56. doi: 10.1210/me.2012-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Modder UI, Sanyal A, Kearns AE, Sibonga JD, Nishihara E, Xu J, et al. Effects of loss of steroid receptor coactivator-1 on the skeletal response to estrogen in mice. Endocrinology. 2004;145:913–21. doi: 10.1210/en.2003-1089. [DOI] [PubMed] [Google Scholar]
- 62.Borjesson AE, Windahl SH, Lagerquist MK, Engdahl C, Frenkel B, Moverare-Skrtic S, et al. Roles of transactivating functions 1 and 2 of estrogen receptor-alpha in bone. Proc Natl Acad Sci U S A. 2011;108:6288–93. doi: 10.1073/pnas.1100454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dubois V, Laurent M, Boonen S, Vanderschueren D, Claessens F. Androgens and skeletal muscle: cellular and molecular action mechanisms underlying the anabolic actions. Cell Mol Life Sci. 2012;69:1651–67. doi: 10.1007/s00018-011-0883-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sinha-Hikim I, Taylor WE, Gonzalez-Cadavid NF, Zheng W, Bhasin S. Androgen receptor in human skeletal muscle and cultured muscle satellite cells: up-regulation by androgen treatment. J Clin Endocrinol Metab. 2004;89:5245–55. doi: 10.1210/jc.2004-0084. [DOI] [PubMed] [Google Scholar]
- 65.Carson JA, Lee WJ, McClung J, Hand GA. Steroid receptor concentration in aged rat hindlimb muscle: effect of anabolic steroid administration. J Appl Physiol (1985) 2002;93:242–50. doi: 10.1152/japplphysiol.01212.2001. [DOI] [PubMed] [Google Scholar]
- 66.Bamman MM, Shipp JR, Jiang J, Gower BA, Hunter GR, Goodman A, et al. Mechanical load increases muscle IGF-I and androgen receptor mRNA concentrations in humans. Am J Physiol Endocrinol Metab. 2001;280:E383–E390. doi: 10.1152/ajpendo.2001.280.3.E383. [DOI] [PubMed] [Google Scholar]
- 67.Lee WJ, McClung J, Hand GA, Carson JA. Overload-induced androgen receptor expression in the aged rat hindlimb receiving nandrolone decanoate. J Appl Physiol (1985) 2003;94:1153–61. doi: 10.1152/japplphysiol.00822.2002. [DOI] [PubMed] [Google Scholar]
- 68.Lee WJ, Thompson RW, McClung JM, Carson JA. Regulation of androgen receptor expression at the onset of functional overload in rat plantaris muscle. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1076–R1085. doi: 10.1152/ajpregu.00202.2003. [DOI] [PubMed] [Google Scholar]
- 69.Mitchell CJ, Churchward-Venne TA, Bellamy L, Parise G, Baker SK, Phillips SM. Muscular and systemic correlates of resistance training-induced muscle hypertrophy. PLoS One. 2013;8:e78636. doi: 10.1371/journal.pone.0078636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.MacLean HE, Chiu WS, Notini AJ, Axell AM, Davey RA, McManus JF, et al. Impaired skeletal muscle development and function in male, but not female, genomic androgen receptor knockout mice. FASEB J. 2008;22:2676–89. doi: 10.1096/fj.08-105726. [DOI] [PubMed] [Google Scholar]
- 71.Niel L, Shah AH, Lewis GA, Mo K, Chatterjee D, Fernando SM, et al. Sexual differentiation of the spinal nucleus of the bulbocavernosus is not mediated solely by androgen receptors in muscle fibers. Endocrinology. 2009;150:3207–13. doi: 10.1210/en.2008-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fernando SM, Rao P, Niel L, Chatterjee D, Stagljar M, Monks DA. Myocyte androgen receptors increase metabolic rate and improve body composition by reducing fat mass. Endocrinology. 2010;151:3125–32. doi: 10.1210/en.2010-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dubois V, Laurent MR, Sinnesael M, Cielen N, Helsen C, Clinckemalie L, et al. A satellite cell-specific knockout of the androgen receptor reveals myostatin as a direct androgen target in skeletal muscle. FASEB J. 2014;28:2979–94. doi: 10.1096/fj.14-249748. [DOI] [PubMed] [Google Scholar]
- 74.Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, et al. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci U S A. 1998;95:15677–82. doi: 10.1073/pnas.95.26.15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci U S A. 1993;90:11162–6. doi: 10.1073/pnas.90.23.11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Glenmark B, Nilsson M, Gao H, Gustafsson JA, Dahlman-Wright K, Westerblad H. Difference in skeletal muscle function in males vs. females: role of estrogen receptor-beta. Am J Physiol Endocrinol Metab. 2004;287:E1125–E1131. doi: 10.1152/ajpendo.00098.2004. [DOI] [PubMed] [Google Scholar]
- 77.Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol. 2005;37:1974–84. doi: 10.1016/j.biocel.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 78.Glass DJ. Signaling pathways perturbing muscle mass. Curr Opin Clin Nutr Metab Care. 2010;13:225–9. doi: 10.1097/mco.0b013e32833862df. [DOI] [PubMed] [Google Scholar]
- 79.Glass DJ. PI3 kinase regulation of skeletal muscle hypertrophy and atrophy. Curr Top Microbiol Immunol. 2010;346:267–78. doi: 10.1007/82_2010_78. [DOI] [PubMed] [Google Scholar]
- 80.White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, et al. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS One. 2011;6:e24650. doi: 10.1371/journal.pone.0024650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–8. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 83.Harvey KF, Mattila J, Sofer A, Bennett FC, Ramsey MR, Ellisen LW, et al. FOXO-regulated transcription restricts overgrowth of Tsc mutant organs. J Cell Biol. 2008;180:691–6. doi: 10.1083/jcb.200710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chambon C, Duteil D, Vignaud A, Ferry A, Messaddeq N, Malivindi R, et al. Myocytic androgen receptor controls the strength but not the mass of limb muscles. Proc Natl Acad Sci U S A. 2010;107:14327–32. doi: 10.1073/pnas.1009536107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mauras N, Hayes V, Welch S, Rini A, Helgeson K, Dokler M, et al. Testosterone deficiency in young men: marked alterations in whole body protein kinetics, strength, and adiposity. J Clin Endocrinol Metab. 1998;83:1886–92. doi: 10.1210/jcem.83.6.4892. [DOI] [PubMed] [Google Scholar]
- 86.Oner J, Oner H, Sahin Z, Demir R, Ustunel I. Melatonin is as effective as testosterone in the prevention of soleus muscle atrophy induced by castration in rats. Anat Rec (Hoboken) 2008;291:448–55. doi: 10.1002/ar.20659. [DOI] [PubMed] [Google Scholar]
- 87.Haren MT, Siddiqui AM, Armbrecht HJ, Kevorkian RT, Kim MJ, Haas MJ, et al. Testosterone modulates gene expression pathways regulating nutrient accumulation, glucose metabolism and protein turnover in mouse skeletal muscle. Int J Androl. 2011;34:55–68. doi: 10.1111/j.1365-2605.2010.01061.x. [DOI] [PubMed] [Google Scholar]
- 88.Ibebunjo C, Eash JK, Li C, Ma Q, Glass DJ. Voluntary running, skeletal muscle gene expression, and signaling inversely regulated by orchidectomy and testosterone replacement. Am J Physiol Endocrinol Metab. 2011;300:E327–E340. doi: 10.1152/ajpendo.00402.2010. [DOI] [PubMed] [Google Scholar]
- 89.Hourde C, Jagerschmidt C, Clement-Lacroix P, Vignaud A, Ammann P, Butler-Browne GS, et al. Androgen replacement therapy improves function in male rat muscles independently of hypertrophy and activation of the Akt/mTOR pathway. Acta Physiol (Oxf) 2009;195:471–82. doi: 10.1111/j.1748-1716.2008.01902.x. [DOI] [PubMed] [Google Scholar]
- 90.Sitnick M, Foley AM, Brown M, Spangenburg EE. Ovariectomy prevents the recovery of atrophied gastrocnemius skeletal muscle mass. J Appl Physiol (1985) 2006;100:286–93. doi: 10.1152/japplphysiol.00869.2005. [DOI] [PubMed] [Google Scholar]
- 91.Olivieri F, Ahtiainen M, Lazzarini R, Pollanen E, Capri M, Lorenzi M, et al. Hormone replacement therapy enhances IGF-1 signaling in skeletal muscle by diminishing miR-182 and miR-223 expressions: a study on postmenopausal monozygotic twin pairs. Aging Cell. 2014;13:850–61. doi: 10.1111/acel.12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim JY, Jo KJ, Kim OS, Kim BJ, Kang DW, Lee KH, et al. Parenteral 17beta-estradiol decreases fasting blood glucose levels in non-obese mice with short-term ovariectomy. Life Sci. 2010;87:358–66. doi: 10.1016/j.lfs.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 93.Ning J, Clemmons DR. AMP-activated protein kinase inhibits IGF-I signaling and protein synthesis in vascular smooth muscle cells via stimulation of insulin receptor substrate 1 S794 and tuberous sclerosis 2 S1345 phosphorylation. Mol Endocrinol. 2010;24:1218–29. doi: 10.1210/me.2009-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saha AK, Xu XJ, Lawson E, Deoliveira R, Brandon AE, Kraegen EW, et al. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes. 2010;59:2426–34. doi: 10.2337/db09-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thomson DM, Fick CA, Gordon SE. AMPK activation attenuates S6K1, 4E-BP1, and eEF2 signaling responses to high-frequency electrically stimulated skeletal muscle contractions. J Appl Physiol (1985) 2008;104:625–32. doi: 10.1152/japplphysiol.00915.2007. [DOI] [PubMed] [Google Scholar]
- 96.Chomentowski P, Coen PM, Radikova Z, Goodpaster BH, Toledo FG. Skeletal muscle mitochondria in insulin resistance: differences in intermyofibrillar versus subsarcolemmal subpopulations and relationship to metabolic flexibility. J Clin Endocrinol Metab. 2011;96:494–503. doi: 10.1210/jc.2010-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Romanello V, Sandri M. Mitochondrial biogenesis and fragmentation as regulators of muscle protein degradation. Curr Hypertens Rep. 2010;12:433–9. doi: 10.1007/s11906-010-0157-8. [DOI] [PubMed] [Google Scholar]
- 98.White JP, Baltgalvis KA, Puppa MJ, Sato S, Baynes JW, Carson JA. Muscle oxidative capacity during IL-6-dependent cancer cachexia. Am J Physiol Regul Integr Comp Physiol. 2011;300:R201–R211. doi: 10.1152/ajpregu.00300.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010;29:1774–85. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li P, Waters RE, Redfern SI, Zhang M, Mao L, Annex BH, et al. Oxidative phenotype protects myofibers from pathological insults induced by chronic heart failure in mice. Am J Pathol. 2007;170:599–608. doi: 10.2353/ajpath.2007.060505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.D'Eon TM, Souza SC, Aronovitz M, Obin MS, Fried SK, Greenberg AS. Estrogen regulation of adiposity and fuel partitioning. Evidence of genomic and non-genomic regulation of lipogenic and oxidative pathways. J Biol Chem. 2005;280:35983–91. doi: 10.1074/jbc.M507339200. [DOI] [PubMed] [Google Scholar]
- 102.Klinge CM. Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem. 2008;105:1342–51. doi: 10.1002/jcb.21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cavalcanti-de-Albuquerque JP, Salvador IC, Martins EL, Jardim-Messeder D, Werneck-de-Castro JP, Galina A, et al. Role of estrogen on skeletal muscle mitochondrial function in ovariectomized rats: a time course study in different fiber types. J Appl Physiol (1985) 2014;116:779–89. doi: 10.1152/japplphysiol.00121.2013. [DOI] [PubMed] [Google Scholar]
- 104.Frost RA, Lang CH. mTor signaling in skeletal muscle during sepsis and inflammation: where does it all go wrong? Physiology (Bethesda) 2011;26:83–96. doi: 10.1152/physiol.00044.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sandri M. New findings of lysosomal proteolysis in skeletal muscle. Curr Opin Clin Nutr Metab Care. 2011;14:223–9. doi: 10.1097/MCO.0b013e3283457a75. [DOI] [PubMed] [Google Scholar]
- 106.Brault JJ, Jespersen JG, Goldberg AL. Peroxisome proliferator-activated receptor gamma coactivator 1alpha or 1beta overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J Biol Chem. 2010;285:19460–71. doi: 10.1074/jbc.M110.113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, et al. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci U S A. 2006;103:16260–5. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Usui T, Kajita K, Kajita T, Mori I, Hanamoto T, Ikeda T, et al. Elevated mitochondrial biogenesis in skeletal muscle is associated with testosterone-induced body weight loss in male mice. FEBS Lett. 2014;588:1935–41. doi: 10.1016/j.febslet.2014.03.051. [DOI] [PubMed] [Google Scholar]
- 109.Thalacker-Mercer A, Stec M, Cui X, Cross J, Windham S, Bamman M. Cluster analysis reveals differential transcript profiles associated with resistance training-induced human skeletal muscle hypertrophy. Physiol Genomics. 2013;45:499–507. doi: 10.1152/physiolgenomics.00167.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Stunes AK, Westbroek I, Gustafsson BI, Fossmark R, Waarsing JH, Eriksen EF, et al. The peroxisome proliferator-activated receptor (PPAR) alpha agonist fenofibrate maintains bone mass, while the PPAR gamma agonist pioglitazone exaggerates bone loss, in ovariectomized rats. BMC Endocr Disord. 2011;11:11. doi: 10.1186/1472-6823-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Syversen U, Stunes AK, Gustafsson BI, Obrant KJ, Nordsletten L, Berge R, et al. Different skeletal effects of the peroxisome proliferator activated receptor (PPAR)alpha agonist fenofibrate and the PPARgamma agonist pioglitazone. BMC Endocr Disord. 2009;9:10. doi: 10.1186/1472-6823-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rogers NH, Perfield JW, Strissel KJ, Obin MS, Greenberg AS. Loss of ovarian function in mice results in abrogated skeletal muscle PPARdelta and FoxO1-mediated gene expression. Biochem Biophys Res Commun. 2010;392:1–3. doi: 10.1016/j.bbrc.2009.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009;89:799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 114.Garnier A, Fortin D, Zoll J, N'Guessan B, Mettauer B, Lampert E, et al. Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. FASEB J. 2005;19:43–52. doi: 10.1096/fj.04-2173com. [DOI] [PubMed] [Google Scholar]
- 115.Lee KC, Jessop H, Suswillo R, Zaman G, Lanyon LE. The adaptive response of bone to mechanical loading in female transgenic mice is deficient in the absence of oestrogen receptor-alpha and -beta. J Endocrinol. 2004;182:193–201. doi: 10.1677/joe.0.1820193. [DOI] [PubMed] [Google Scholar]
- 116.Armstrong VJ, Muzylak M, Sunters A, Zaman G, Saxon LK, Price JS, et al. Wnt/beta-catenin signaling is a component of osteoblastic bone cell early responses to load-bearing and requires estrogen receptor alpha. J Biol Chem. 2007;282:20715–27. doi: 10.1074/jbc.M703224200. [DOI] [PubMed] [Google Scholar]
- 117.Aguirre JI, Plotkin LI, Gortazar AR, Millan MM, O'Brien CA, Manolagas SC, et al. A novel ligand-independent function of the estrogen receptor is essential for osteocyte and osteoblast mechanotransduction. J Biol Chem. 2007;282:25501–8. doi: 10.1074/jbc.M702231200. [DOI] [PubMed] [Google Scholar]
- 118.Robinson JA, Chatterjee-Kishore M, Yaworsky PJ, Cullen DM, Zhao W, Li C, et al. Wnt/beta-catenin signaling is a normal physiological response to mechanical loading in bone. J Biol Chem. 2006;281:31720–8. doi: 10.1074/jbc.M602308200. [DOI] [PubMed] [Google Scholar]
- 119.Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, Dwyer D, et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012;50:209–17. doi: 10.1016/j.bone.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, et al. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. 2006;281:23698–711. doi: 10.1074/jbc.M601000200. [DOI] [PubMed] [Google Scholar]
- 121.Windahl SH, Borjesson AE, Farman HH, Engdahl C, Moverare-Skrtic S, Sjogren K, et al. Estrogen receptor-alpha in osteocytes is important for trabecular bone formation in male mice. Proc Natl Acad Sci U S A. 2013;110:2294–9. doi: 10.1073/pnas.1220811110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sugiyama T, Galea GL, Lanyon LE, Price JS. Mechanical loading-related bone gain is enhanced by tamoxifen but unaffected by fulvestrant in female mice. Endocrinology. 2010;151:5582–90. doi: 10.1210/en.2010-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Windahl SH, Saxon L, Borjesson AE, Lagerquist MK, Frenkel B, Henning P, et al. Estrogen receptor-alpha is required for the osteogenic response to mechanical loading in a ligand-independent manner involving its activation function 1 but not 2. J Bone Miner Res. 2013;28:291–301. doi: 10.1002/jbmr.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Callewaert F, Bakker A, Schrooten J, Van MB, Verhoeven G, Boonen S, et al. Androgen receptor disruption increases the osteogenic response to mechanical loading in male mice. J Bone Miner Res. 2010;25:124–31. doi: 10.1359/jbmr.091001. [DOI] [PubMed] [Google Scholar]
- 125.Kleinberg DL, Barcellos-Hoff MH. The pivotal role of insulin-like growth factor I in normal mammary development. Endocrinol Metab Clin North Am. 2011;40:461–71, vii. doi: 10.1016/j.ecl.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 126.Bolamperti S, Mrak E, Moro G, Sirtori P, Fraschini G, Guidobono F, et al. 17beta-Estradiol Positively Modulates Growth Hormone Signalling through the Reduction of SOCS2 Negative Feedback in Human Osteoblasts. Bone. 2013 doi: 10.1016/j.bone.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 127.Nordstrom P, Eklund F, Bjornstig U, Nordstrom A, Lorentzon R, Sievanen H, et al. Do both areal BMD and injurious falls explain the higher incidence of fractures in women than in men? Calcif Tissue Int. 2011;89:203–10. doi: 10.1007/s00223-011-9507-z. [DOI] [PubMed] [Google Scholar]
- 128.Goodman CA, Frey JW, Mabrey DM, Jacobs BL, Lincoln HC, You JS, et al. The role of skeletal muscle mTOR in the regulation of mechanical load-induced growth. J Physiol. 2011;589:5485–501. doi: 10.1113/jphysiol.2011.218255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hornberger TA, Chu WK, Mak YW, Hsiung JW, Huang SA, Chien S. The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc Natl Acad Sci U S A. 2006;103:4741–6. doi: 10.1073/pnas.0600678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.You JS, Frey JW, Hornberger TA. Mechanical stimulation induces mTOR signaling via an ERK-independent mechanism: implications for a direct activation of mTOR by phosphatidic acid. PLoS One. 2012;7:e47258. doi: 10.1371/journal.pone.0047258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–93. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 132.Miyazaki M, McCarthy JJ, Fedele MJ, Esser KA. Early activation of mTORC1 signalling in response to mechanical overload is independent of phosphoinositide 3-kinase/Akt signalling. J Physiol. 2011;589:1831–46. doi: 10.1113/jphysiol.2011.205658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Oh H, Fujio Y, Kunisada K, Hirota H, Matsui H, Kishimoto T, et al. Activation of phosphatidylinositol 3-kinase through glycoprotein 130 induces protein kinase B and p70 S6 kinase phosphorylation in cardiac myocytes. J Biol Chem. 1998;273:9703–10. doi: 10.1074/jbc.273.16.9703. [DOI] [PubMed] [Google Scholar]
- 134.Wu XN, Wang XK, Wu SQ, Lu J, Zheng M, Wang YH, et al. Phosphorylation of Raptor by p38beta participates in arsenite-induced mammalian target of rapamycin complex 1 (mTORC1) activation. J Biol Chem. 2011;286:31501–11. doi: 10.1074/jbc.M111.233122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ruas JL, White JP, Rao RR, Kleiner S, Brannan KT, Harrison BC, et al. A PGC-1alpha isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell. 2012;151:1319–31. doi: 10.1016/j.cell.2012.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.White JP, Wrann CD, Rao RR, Nair SK, Jedrychowski MP, You JS, et al. G protein-coupled receptor 56 regulates mechanical overload-induced muscle hypertrophy. Proc Natl Acad Sci U S A. 2014;111:15756–61. doi: 10.1073/pnas.1417898111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Carson JA, Nettleton D, Reecy JM. Differential gene expression in the rat soleus muscle during early work overload-induced hypertrophy. FASEB J. 2002;16:207–9. doi: 10.1096/fj.01-0544fje. [DOI] [PubMed] [Google Scholar]
- 138.Dangott B, Schultz E, Mozdziak PE. Dietary creatine monohydrate supplementation increases satellite cell mitotic activity during compensatory hypertrophy. Int J Sports Med. 2000;21:13–6. doi: 10.1055/s-2000-8848. [DOI] [PubMed] [Google Scholar]
- 139.Kelso TB, Shear CR, Max SR. Enzymes of glutamine metabolism in inflammation associated with skeletal muscle hypertrophy. Am J Physiol. 1989;257:E885–E894. doi: 10.1152/ajpendo.1989.257.6.E885. [DOI] [PubMed] [Google Scholar]
- 140.Roy RR, Baldwin KM, Martin TP, Chimarusti SP, Edgerton VR. Biochemical and physiological changes in overloaded rat fast- and slow-twitch ankle extensors. J Appl Physiol (1985) 1985;59:639–46. doi: 10.1152/jappl.1985.59.2.639. [DOI] [PubMed] [Google Scholar]
- 141.McClung JM, Mehl KA, Thompson RW, Lowe LL, Carson JA. Nandrolone decanoate modulates cell cycle regulation in functionally overloaded rat soleus muscle. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1543–R1552. doi: 10.1152/ajpregu.00285.2004. [DOI] [PubMed] [Google Scholar]
- 142.Schultz E, McCormick KM. Skeletal muscle satellite cells. Rev Physiol Biochem Pharmacol. 1994;123:213–57. doi: 10.1007/BFb0030904. [DOI] [PubMed] [Google Scholar]
- 143.Velders M, Schleipen B, Fritzemeier KH, Zierau O, Diel P. Selective estrogen receptor-beta activation stimulates skeletal muscle growth and regeneration. FASEB J. 2012;26:1909–20. doi: 10.1096/fj.11-194779. [DOI] [PubMed] [Google Scholar]
- 144.Adams GR, Haddad F, Baldwin KM. Time course of changes in markers of myogenesis in overloaded rat skeletal muscles. J Appl Physiol (1985) 1999;87:1705–12. doi: 10.1152/jappl.1999.87.5.1705. [DOI] [PubMed] [Google Scholar]
- 145.Conboy IM, Rando TA. Heterochronic parabiosis for the study of the effects of aging on stem cells and their niches. Cell Cycle. 2012;11:2260–7. doi: 10.4161/cc.20437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fry CS, Lee JD, Jackson JR, Kirby TJ, Stasko SA, Liu H, et al. Regulation of the muscle fiber microenvironment by activated satellite cells during hypertrophy. FASEB J. 2014;28:1654–65. doi: 10.1096/fj.13-239426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ferry A, Noirez P, Page CL, Salah IB, Daegelen D, Rieu M. Effects of anabolic/androgenic steroids on regenerating skeletal muscles in the rat. Acta Physiol Scand. 1999;166:105–10. doi: 10.1046/j.1365-201x.1999.00549.x. [DOI] [PubMed] [Google Scholar]
- 148.Grounds MD. Phagocytosis of necrotic muscle in muscle isografts is influenced by the strain, age, and sex of host mice. J Pathol. 1987;153:71–82. doi: 10.1002/path.1711530110. [DOI] [PubMed] [Google Scholar]
- 149.Serra C, Tangherlini F, Rudy S, Lee D, Toraldo G, Sandor NL, et al. Testosterone improves the regeneration of old and young mouse skeletal muscle. J Gerontol A Biol Sci Med Sci. 2013;68:17–26. doi: 10.1093/gerona/gls083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hamrick MW. The skeletal muscle secretome: an emerging player in muscle-bone crosstalk. Bonekey Rep. 2012;1:60. doi: 10.1038/bonekey.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nowlan NC, Sharpe J, Roddy KA, Prendergast PJ, Murphy P. Mechanobiology of embryonic skeletal development: Insights from animal models. Birth Defects Res C Embryo Today. 2010;90:203–13. doi: 10.1002/bdrc.20184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Warner SE, Sanford DA, Becker BA, Bain SD, Srinivasan S, Gross TS. Botox induced muscle paralysis rapidly degrades bone. Bone. 2006;38:257–64. doi: 10.1016/j.bone.2005.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hao Y, Ma Y, Wang X, Jin F, Ge S. Short-term muscle atrophy caused by botulinum toxin-A local injection impairs fracture healing in the rat femur. J Orthop Res. 2012;30:574–80. doi: 10.1002/jor.21553. [DOI] [PubMed] [Google Scholar]
- 154.Elkasrawy MN, Hamrick MW. Myostatin (GDF-8) as a key factor linking muscle mass and bone structure. J Musculoskelet Neuronal Interact. 2010;10:56–63. [PMC free article] [PubMed] [Google Scholar]
- 155.Green DJ, Hamrick MW, Richmond BG. The effects of hypermuscularity on shoulder morphology in myostatin-deficient mice. J Anat. 2011;218:544–57. doi: 10.1111/j.1469-7580.2011.01351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bianchi ML, Mazzanti A, Galbiati E, Saraifoger S, Dubini A, Cornelio F, et al. Bone mineral density and bone metabolism in Duchenne muscular dystrophy. Osteoporos Int. 2003;14:761–7. doi: 10.1007/s00198-003-1443-y. [DOI] [PubMed] [Google Scholar]
- 157.Tan VP, Macdonald HM, Kim S, Nettlefold L, Gabel L, Ashe MC, et al. Influence of physical activity on bone strength in children and adolescents: a systematic review and narrative synthesis. J Bone Miner Res. 2014;29:2161–81. doi: 10.1002/jbmr.2254. [DOI] [PubMed] [Google Scholar]
- 158.Crabtree NJ, Kibirige MS, Fordham JN, Banks LM, Muntoni F, Chinn D, et al. The relationship between lean body mass and bone mineral content in paediatric health and disease. Bone. 2004;35:965–72. doi: 10.1016/j.bone.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 159.Sayer AA, Syddall H, Martin H, Patel H, Baylis D, Cooper C. The developmental origins of sarcopenia. J Nutr Health Aging. 2008;12:427–32. doi: 10.1007/BF02982703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Arounleut P, Bialek P, Liang LF, Upadhyay S, Fulzele S, Johnson M, et al. A myostatin inhibitor (propeptide-Fc) increases muscle mass and muscle fiber size in aged mice but does not increase bone density or bone strength. Exp Gerontol. 2013;48:898–904. doi: 10.1016/j.exger.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bonnet N, Ferrari SL. Exercise and the skeleton: How it works and what it really does. IBMS BoneKEy. 2010;7:235–48. [Google Scholar]
- 162.Stein H, Perren SM, Cordey J, Kenwright J, Mosheiff R, Francis MJ. The muscle bed--a crucial factor for fracture healing: a physiological concept. Orthopedics. 2002;25:1379–83. doi: 10.3928/0147-7447-20021201-16. [DOI] [PubMed] [Google Scholar]
- 163.Liu R, Schindeler A, Little DG. The potential role of muscle in bone repair. J Musculoskelet Neuronal Interact. 2010;10:71–6. [PubMed] [Google Scholar]
- 164.Liu R, Birke O, Morse A, Peacock L, Mikulec K, Little DG, et al. Myogenic progenitors contribute to open but not closed fracture repair. BMC Musculoskelet Disord. 2011;12:288. doi: 10.1186/1471-2474-12-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Harry LE, Sandison A, Paleolog EM, Hansen U, Pearse MF, Nanchahal J. Comparison of the healing of open tibial fractures covered with either muscle or fasciocutaneous tissue in a murine model. J Orthop Res. 2008;26:1238–44. doi: 10.1002/jor.20649. [DOI] [PubMed] [Google Scholar]
- 166.Gopal S, Majumder S, Batchelor AG, Knight SL, De BP, Smith RM. Fix and flap: the radical orthopaedic and plastic treatment of severe open fractures of the tibia. J Bone Joint Surg Br. 2000;82:959–66. doi: 10.1302/0301-620x.82b7.10482. [DOI] [PubMed] [Google Scholar]
- 167.Utvag SE, Iversen KB, Grundnes O, Reikeras O. Poor muscle coverage delays fracture healing in rats. Acta Orthop Scand. 2002;73:471–4. doi: 10.1080/00016470216315. [DOI] [PubMed] [Google Scholar]
- 168.Zacks SI, Sheff MF. Periosteal and metaplastic bone formation in mouse minced muscle regeneration. Lab Invest. 1982;46:405–12. [PubMed] [Google Scholar]
- 169.Landry PS, Marino AA, Sadasivan KK, Albright JA. Effect of soft-tissue trauma on the early periosteal response of bone to injury. J Trauma. 2000;48:479–83. doi: 10.1097/00005373-200003000-00018. [DOI] [PubMed] [Google Scholar]
- 170.Duda GN, Taylor WR, Winkler T, Matziolis G, Heller MO, Haas NP, et al. Biomechanical, microvascular, and cellular factors promote muscle and bone regeneration. Exerc Sport Sci Rev. 2008;36:64–70. doi: 10.1097/JES.0b013e318168eb88. [DOI] [PubMed] [Google Scholar]
- 171.Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007;282:27285–97. doi: 10.1074/jbc.M702810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hui SL, Slemenda CW, Johnston CC., Jr Age and bone mass as predictors of fracture in a prospective study. J Clin Invest. 1988;81:1804–9. doi: 10.1172/JCI113523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.O'Brien CA. Control of RANKL gene expression. Bone. 2010;46:911–9. doi: 10.1016/j.bone.2009.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Iyer S, Ambrogini E, Bartell SM, Han L, Roberson PK, Cabo R, et al. FoxOs attenuate bone formation by suppressing Wnt signaling. J Clin Invest. 2013;123:3404–19. doi: 10.1172/JCI68049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8:440–50. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 176.Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008;20:126–36. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Almeida M, Han L, Martin-Millan M, O'Brien CA, Manolagas SC. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J Biol Chem. 2007;282:27298–305. doi: 10.1074/jbc.M702811200. [DOI] [PubMed] [Google Scholar]
- 178.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gonzalez-Freire M, de CR, Studenski SA, Ferrucci L. The Neuromuscular Junction: Aging at the Crossroad between Nerves and Muscle. Front Aging Neurosci. 2014;6:208. doi: 10.3389/fnagi.2014.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Marzetti E, Calvani R, Cesari M, Buford TW, Lorenzi M, Behnke BJ, et al. Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int J Biochem Cell Biol. 2013;45:2288–301. doi: 10.1016/j.biocel.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Walker DK, Dickinson JM, Timmerman KL, Drummond MJ, Reidy PT, Fry CS, et al. Exercise, amino acids, and aging in the control of human muscle protein synthesis. Med Sci Sports Exerc. 2011;43:2249–58. doi: 10.1249/MSS.0b013e318223b037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Alway SE, Myers MJ, Mohamed JS. Regulation of satellite cell function in sarcopenia. Front Aging Neurosci. 2014;6:246. doi: 10.3389/fnagi.2014.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ali S, Garcia JM. Sarcopenia, cachexia and aging: diagnosis, mechanisms and therapeutic options - a mini-review. Gerontology. 2014;60:294–305. doi: 10.1159/000356760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, et al. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest. 2013;123:4821–35. doi: 10.1172/JCI68523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kandarian S. The molecular basis of skeletal muscle atrophy--parallels with osteoporotic signaling. J Musculoskelet Neuronal Interact. 2008;8:340–1. [PubMed] [Google Scholar]
- 186.Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, et al. Cachexia: a new definition. Clin Nutr. 2008;27:793–9. doi: 10.1016/j.clnu.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 187.Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, et al. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature. 2014;513:100–4. doi: 10.1038/nature13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Narsale AA, Carson JA. Role of interleukin-6 in cachexia: therapeutic implications. Curr Opin Support Palliat Care. 2014;8:321–7. doi: 10.1097/SPC.0000000000000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584:1411–6. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- 190.Haran PH, Rivas DA, Fielding RA. Role and potential mechanisms of anabolic resistance in sarcopenia. J Cachexia Sarcopenia Muscle. 2012;3:157–62. doi: 10.1007/s13539-012-0068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Li H, Mittal A, Paul PK, Kumar M, Srivastava DS, Tyagi SC, et al. Tumor necrosis factor-related weak inducer of apoptosis augments matrix metalloproteinase 9 (MMP-9) production in skeletal muscle through the activation of nuclear factor-kappaB-inducing kinase and p38 mitogen-activated protein kinase: a potential role of MMP-9 in myopathy. J Biol Chem. 2009;284:4439–50. doi: 10.1074/jbc.M805546200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Penna F, Costamagna D, Fanzani A, Bonelli G, Baccino FM, Costelli P. Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS One. 2010;5:e13604. doi: 10.1371/journal.pone.0013604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Puppa MJ, Gao S, Narsale AA, Carson JA. Skeletal muscle glycoprotein 130's role in Lewis lung carcinoma-induced cachexia. FASEB J. 2014;28:998–1009. doi: 10.1096/fj.13-240580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang G, Jin B, Li YP. C/EBPbeta mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J. 2011;30:4323–35. doi: 10.1038/emboj.2011.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013;34:518–30. doi: 10.1016/j.tips.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Cruz-Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, et al. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing. 2010;39:412–23. doi: 10.1093/ageing/afq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Basaria S, Coviello AD, Travison TG, Storer TW, Farwell WR, Jette AM, et al. Adverse events associated with testosterone administration. N Engl J Med. 2010;363:109–22. doi: 10.1056/NEJMoa1000485. [DOI] [PMC free article] [PubMed] [Google Scholar]