

Abstract
Dopamine (DA) and serotonin (SRT) are monoamine neurotransmitters that play a key role in regulating the central and peripheral nervous system. Their impaired metabolism has been implicated in several neurological disorders, such as Parkinson’s disease and depression. Consequently, it is imperative to monitor changes in levels of these low-abundant neurotransmitters and their role in mediating disease. For the first time, a rapid, specific and sensitive isotope-dilution liquid chromatography-tandem mass spectrometry (LC-MS/MS) method was developed and validated for the quantification of DA and SRT in the nematode Caenorhabditis elegans (C. elegans). This model organism offers a unique approach for studying the effect of various drugs and environmental conditions on neurotransmitter levels, given by the conserved DA and SRT biology, including synaptic release, trafficking and formation. We introduce a novel sample preparation protocol incorporating the usage of sodium thiosulfate in perchloric acid as extraction medium that assures high recovery of the relatively unstable neurotransmitters monitored. Moreover, the use of both deuterated internal standards and the multiple reaction monitoring (MRM) technique allows for unequivocal quantification. Thereby, to the best of our knowledge, we achieve a detection sensitivity that clearly exceeds those of published DA and SRT quantification methods in various matrices. We are the first to show that exposure of C. elegans to the monoamine oxidase B (MAO-B) inhibitor selegiline or the catechol-O-methyltransferase (COMT) inhibitor tolcapone, in order to block DA and SRT degradation, resulted in accumulation of the respective neurotransmitter. Assessment of a behavioral output of the dopaminergic system (basal slowing response) corroborated the analytical LC-MS/MS data. Thus, utilization of the C. elegans model system in conjunction with our analytical method is well-suited to investigate drug-mediated modulation of the DA and SRT system in order to identify compounds with neuroprotective or regenerative properties.
Keywords: Caenorhabditis elegans, dopamine, serotonin, liquid chromatography-tandem mass spectrometry, isotope-dilution analysis
Graphical Abstract
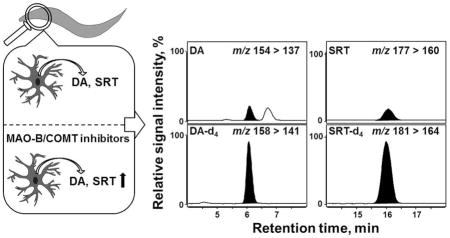
Introduction
Dopamine (DA) and serotonin (SRT) are monoamine neurotransmitters that play key roles in many aspects of mammalian nervous system function. The most studied functions of DA in the nervous system include cognition, motor function, brain-stimulation reward mechanisms, eating and drinking behaviors, sexual behavior, neuroendocrine regulation and selective attention [1, 2]. Alterations in DA neurotransmission result in major neurological and psychiatric disorders. The most recognized DA-related disorder is Parkinson’s disease, which originates from selective dopaminergic (DAergic) cell loss in the substantia nigra pars compacta [3]. Other disorders arising from abnormal DA function include addiction, schizophrenia, bipolar disorder, Huntington’s disease, attention deficit hyperactivity disorder and Tourette’s syndrome [2]. SRT is probably best known for its role in conveying a sense of contentedness and happiness and is implicated in practically every type of behavior, such as appetitive, emotional, motor, cognitive and autonomic functions [4, 5]. Alterations in the serotonergic (SRTergic) system play a part in many disorders, including depression, schizophrenia, migraines, anxiety and dementia [5–7].
Consequently, monitoring changes in the basal levels of DA and SRT are undoubtedly useful in modern neuroscience, with drug discovery research attempting to identify pathophysiological changes associated with these disorders. However, accurate detection of DA and SRT levels remains an analytical and technical challenge due to their low basal levels. Therefore, despite a variety of existing chromatographic methods coupled with fluorescence [8–11] or electrochemical detection [12, 13], mass spectrometry based techniques remain the method of choice. More specifically, liquid chromatography-tandem mass spectrometry (LC-MS/MS) methods represent a highly sensitive and selective tool for analysis and allow an unequivocal identification of the respective neurotransmitter by specific fragmentation patterns. In particular, use of the multiple reaction monitoring (MRM) mode, in combination with the usage of stable-isotopic labelled analogues of the respective analytes as internal standards, has become the gold standard for unambiguous quantification of not only neurotransmitters, but also various types of biomarkers in biological samples [14]. During the last 15 years, several publications have relied on the mass spectrometric analysis of neurotransmitter in brain tissues; LC-MS/MS methods have been developed to quantify DA in brain microdialysates [15–17], as well as in brain tissue following cleanup [13, 18]. These methods require a pretreatment step, with both recovery parameters and lower substance stability are partly unsatisfactory. However, Najmanova et al. published a rapid and precise LC-MS/MS method without cleanup for parallel identification of DA and SRT in brain tissue [19]. But, a LC-MS/MS method for quantification of DA and SRT in Caenorhabditis elegans (C. elegans), a common model organism in analytical neurochemistry, is still missing.
The simplicity and several key features of the small nematode C. elegans make it an appealing model organism for biomedical research. Advantages include its rapid life cycle, short life cycle and large brood size. Additionally, C. elegans is less complex than the mammalian system, while still showing high genetic homology. 60–80% of human disease genes have a corresponding C. elegans homolog and the detailed knowledge of the genomic architecture makes it easy to perform genetic manipulation [20]. A simple, but complete nervous system, in addition to the existence of DA-deficient mutants, both make C. elegans an attractive organism to study neuromodulation at the behavioral, cellular and molecular levels. In the hermaphrodite, the nervous system is comprised of 302 neurons and ~5,000 synapses [21] that uses an array of neurotransmitters, receptors and downstream signaling mechanisms that are similar to those in the mammalian brain [22]. C. elegans hermaphrodites have eight DAergic neurons; two pairs of cephalic (CEP) neurons, a pair of anterior deirid (ADE) neurons and a pair of postdeirid (PDE) neurons. They also contain all necessary genetic information encoding for DA biosynthesis, packaging and reuptake [23, 24] to control several behaviors, including habituation to mechanical stimuli [25], foraging [26], transitions between crawling and swimming behavior [27] and olfactory adaptation [28]. Mutants defective in DA signaling, such as cat-2 (tyrosine hydroxylase, TH) mutants or mutants lacking the D2-like receptor DOP-3, exhibit locomotor hyperactivity compared to wild-type (WT) animals on a bacterial lawn, indicating that DA plays a role in slowing to response to food stimuli [29]. In addition to DAergic neurons, the integrity of the SRTergic system can also be assessed by observing food-dependent modulation of feeding and locomotion [30, 31]. The conserved DA and SRT biology that shows high homology with mammalian systems makes C. elegans a unique system for studying the effect of various drugs and environmental conditions on the respective basal levels.
Within this study, a rapid, specific and sensitive isotope-dilution LC-MS/MS method for the simultaneous determination of DA and SRT in the genetically tractable, invertebrate C. elegans model system was developed and validated. A new extraction protocol assured high recoveries of both neurotransmitter characterized by certain instability. The usage of both deuterated internal standards and the selective and highly sensitive MRM technique allow for unequivocal identification of the analytes, with a detection sensitivity exceeding that of fluorescence and electrochemical quantification methods previously used for neurotransmitter monitoring in C. elegans. Moreover, we have applied our method to WT worms that were incubated with the monoamine oxidase B (MAO-B) inhibitor selegiline or the catechol-O-methyltransferase (COMT) inhibitor tolcapone.
Experimental Section
C. elegans strains and handling
C. elegans strains were handled and maintained at 20 °C as previously described [32]. Worms were grown on plates containing nematode growth medium (NGM) or 8P seeded with either Escherichia coli strain OP50 or NA22, respectively. The following strains were used in this study: WT N2 Bristol strain and CB1112 (cat-2(e1112)). All strains were provided by the Caenorhabditis Genetic Center (CGC; University of Minnesota).
Preparation of standard solutions
DA hydrochloride (Sigma-Aldrich, Taufkirchen, Germany) and DA-d4 hydrochloride (CDN Isotopes, Pointe-Claire, Canada) stock solutions were prepared in 0.2 M perchloric acid (PCA) (Sigma-Aldrich). SRT hydrochloride (Sigma-Aldrich) and SRT-d4 creatinine sulfate complex (CDN Isotopes) were dissolved in bidestilled water. The deuterated analogues of the respective neurotransmitter were taken as internal standards.
Treatment and extraction of DA and SRT from worm homogenates
Selegiline hydrochloride (Sigma-Aldrich) stock solutions were prepared in bidestilled water, whereas tolcapone (Sigma-Aldrich) stock solutions were prepared in dimethyl sulfoxide (DMSO). To prevent oxidation, fresh stock solutions were prepared shortly before each experiment. 40,000 synchronized L1 worms per tube were exposed to selegiline or tolcapone (50, 100, 250, 500 μM, respectively) in siliconized tubes for 3 h (selegiline treatment) or 4 h (tolcapone treatment) in M9 buffer. Worms were then pelleted by centrifugation at 4,000 rpm for 3 minutes and washed three times in M9 buffer. Afterwards, the worm pellet was re-suspended on ice in 100 μL extraction buffer (0.002 M sodium thiosulfate in 0.2 M PCA (both Sigma-Aldrich)) containing 25 nM DA-d4 and 25 nM SRT-d4, and were then temporarily frozen in liquid nitrogen. Finally, the extracts were homogenized by sonication and centrifugation, with an aliquot reserved for protein quantification using the bicinchoninic acid (BCA) assay-kit (Thermo Scientific, Schwerte, Germany). The remainder was subjected to LC-MS/MS analysis.
LC and ESI-MS/MS parameters
All analyses were conducted with an Agilent 1260 Infinity LC system coupled to an Agilent 6490 triple quadrupole-mass spectrometer (both from Waldbronn, Germany) interfaced with an electrospray ion source operating in the positive ion mode (ESI+). Analyte separation was carried out using an YMC-Triart PFP column (3 μm, 3 × 150 mm) guarded with a pre-column (3 μm, 3 × 10 mm) of the same material. Water and methanol (VWR, Darmstadt, Germany), both acidified with 10 mM formic acid, were used as eluents. Samples of 5 μL were injected into a mobile phase consisting of 100% water. Analytes were eluted from the column, which was tempered at 25 °C, with a 20-min linear gradient to 86:14 (v/v) water/methanol at a flow rate of 0.425 mL min−1. The total run time for one analysis was 24 min, including re-equilibration of the HPLC system. The following ion source parameters were determined after repeated injection of a DA standard solution using the Source Optimizer tool of the Agilent MassHunter Workstation Software (Version B.06.00): drying gas temperature = 80 °C, drying gas flow = 17 L min−1 of nitrogen, sheath gas temperature = 350 °C, sheath gas flow = 11 L min−1 of nitrogen, nebulizer pressure = 40 psi, capillary voltage = 2500 V, nozzle voltage = 0 V. The optimized ion funnel parameters were: high pressure RF voltage = 90 V and low pressure RF voltage = 60 V. Three mass transitions each were used for MRM analysis of DA and DA-d4, whereas two mass transitions each were applied for the analysis of SRT and SRT-d4. The optimized collision energies for each MRM transition, which were determined using the Optimizer tool of the MassHunter Software, are given in Table 1. DA and SRT co-eluted from the separation column with their deuterated analogues at 6.1 min and 16.2 min, respectively. Consequently, the LC-MS/MS run was subdivided in five time segments. In segments 1 (0–4 min), 3 (8–14 min) and 5 (18–24 min) the LC eluate was diverted to waste. In segments 2 (4–8 min) and 4 (14–18 min) the LC efflux was sprayed into the ion source. Segments 2 and 4 consisted of six (DA and DA-d4) and four (SRT and SRT-d4) MRM transitions, respectively. The dwell time for both segments was 100 ms.
Table 1.
MRM parameters for the detection of DA, SRT and their deuterated internal standard compounds.
| Analyte | MRM transition [m/z] a | Collision energy [V] |
|---|---|---|
| Dopamine | 154.1 > 137.0* | 8 |
| 154.1 > 91.0 | 24 | |
| 154.1 > 65.0 | 40 | |
|
| ||
| Dopamine-d4 | 158.1 > 141.0* | 8 |
| 158.1 > 95.0 | 24 | |
| 158.1 > 68.0 | 40 | |
|
| ||
| Serotonin | 177.1 > 160.0* | 4 |
| 177.1 > 115.1 | 32 | |
|
| ||
| Serotonin-d4 | 181.1 > 164.1* | 4 |
| 181.1 > 118.1 | 32 | |
Values determined using the Optimizer tool of the Agilent MassHunter Workstation Software. Asterisks indicate mass transitions used for quantification. Additional mass transitions were recorded for structure identification of the analytes.
Quantification of analytes and method validation
All quantitative results in worm treatment and method validation experiments were obtained using the isotope-dilution approach. The quantifier mass transitions of DA (m/z 154.1 > 137.0) and SRT (m/z 177.1 > 160.0) were used for quantification in relation to the quantifier mass transitions of their deuterated internal standards (m/z 158.1 > 141.0 for DA-d4 and m/z 181.1 > 164.1 for SRT-d4). The following method validation parameters were determined: linearity of detection, limit of detection (LOD), limit of quantification (LOQ), recovery, intra- and interday variation. All accordant experiments were conducted using matrix-matched samples. While the linear detection range, LOD and LOQ were determined using mutant cat-2 worms (deficient for DA biosynthesis [31]) of a larger pool (40,000 worms per sample), an equal number of individually handled WT N2 animals served as matrix for determination of recovery, intra- and interday variation. All worm extracts used for LC-MS/MS method validation were prepared as described in a previous section. cat-2 worms were spiked with varying amounts of DA and SRT (final: 0, 1, 5, 10, 50, 100 and 500 nM of both compounds) as well as constant amounts of DA-d4 and SRT-d4. Following LC-MS/MS analysis, peak areas of DA and SRT were normalized to those of their accordant deuterated analogues and plotted against the applied concentrations. Hereby, the actual amounts of DA and SRT in blank cat-2 matrix (only deuterated standards were added) were quantified. They amounted to 0.3 nM and 3.8 nM for DA and SRT, respectively. Spiked standard amounts were added to these values. Subsequently, calibration curves were subjected to linear regression analysis. The same set of samples was used to assess LOD and LOQ. Hereby, the signal-to-noise (S/N) ratios of the respective neurotransmitter peaks were plotted against the applied analyte concentrations. LOD and LOQ were defined as analyte amounts that produce signals with S/N ratios of 3 and 10, respectively. S/N ratios were calculated using the Agilent MassHunter Workstation Software Qualitative Analysis (Version B.06.00). The following calculation parameters were set: signal definition: height; noise definition: root-mean-square; length of noise region: 0.6 min for DA and 1.2 min for SRT (adapted to the respective peak widths). The recoveries were calculated by comparison of the DA-d4 and SRT-d4 peak areas in ten separate extracts of N2 worm homogenates with those in ten separately prepared stock solutions of the same concentration. Intraday variation was determined by quantification of DA and SRT in ten separate extracts of N2 worm homogenates that were prepared and analyzed on the same day. Quantified amounts of DA and SRT in five separate N2 worm samples that were pelleted, homogenized and extracted on different occasions were used to assess the interday variation. DA and SRT concentrations were normalized to the protein content of the individual worm extracts when intra- and interday variations were determined.
Basal slowing response
Assessment of DA-mediated behavior was performed using the basal slowing response assay, as previously described [31]. Briefly, worms were placed on bacteria-seeded plates following selegiline or tolcapone treatment as described above. Forty-eight hours post exposure, worms were washed off the plates with S basal buffer and ~ 10 worms were transferred to either unseeded or seeded 60 mm NGM plates. The seeded plates were prepared by spreading bacteria in a ring (inner diameter of ~ 1 cm and an outer diameter of ~ 3.5 cm) in the center of the plate. After a five-minute acclimation period, the locomotion rate was assessed as the number of body-bends per 20 s, and data are presented as the change in body bends (body bends on food – body bends off food); analysis was carried out after coding of plates. Worms deficient in cat-2 were used as positive control since they are TH-deficient [33].
Statistics
Dose-response curves and all histograms were generated using GraphPad Prism (GraphPad Software Inc.). Two-way ANOVAs were performed on the DA and SRT data and the basal slowing data, followed by Bonferroni’s multiple comparison post-hoc tests.
Results
Development of a LC-MS/MS method for MRM quantification of DA and SRT
The chromatographic conditions were optimized with respect to the optimal separation, as well as to the maximum sensitivity of the MS detection. The best separation was achieved on an YMC-Triart PFP (pentafluorophenyl) column (3 μm, 3 × 150 mm) with a flow rate of 0.425 mL min−1 using a gradient program of 10 mM formic acid and acidified methanol. ESI(+)-MS/MS in MRM mode was used to quantify DA and SRT in the C. elegans homogenates. First, CID (collision-induced dissociation) studies were carried out to investigate the fragmentation of the protonated molecular ions into their respective product ions and establish the collision energies required, so as to provide optimum intensity for each MRM transition. Using the collision energies provided in Table 1, all analytes were fragmented into characteristic product ions with optimal intensity. Second, instrumental settings of the ion source and ion funnel were optimized after repeated injection and MRM detection of a DA standard using the Source Optimizer tool of the acquisition software. In the end, three mass transitions each were monitored for DA and DA-d4, whereas two mass transitions each were applied for the analysis of SRT and SRT-d4 (Figure 1). The most abundant mass transition (quantifier) was chosen for quantification, with additional mass transitions (qualifier) used for unequivocal identification (Figure 1). The MRM transition m/z 154.1 > 137.0 was used for quantification of DA, m/z 158.1 > 141.0 for DA-d4, m/z 177.1 > 160.0 for SRT and m/z 181.1 > 164.1 for SRT-d4 (Table 1). The relative intensities of the three signals recorded for DA (100:77:43) were comparable to those of the internal standard. The same was true for the two signals recorded for SRT (100:17). The respective chemical structures and the underlying fragmentation reactions (loss of the primary amine function) yielding the most abundant product ions of the analytes are illustrated in Scheme 1. In order to quantify the analytes investigated in different worm lysates, deuterated internal standards of DA and SRT were used for internal calibration and unambiguous identification (Figure 2).
Figure 1.

MRM chromatograms of DA and SRT in a C. elegans homogenate. Three mass transitions were recorded for DA (left panel) whereas two mass transitions were optimized for the detection of SRT (right panel). Signal intensities of the qualifier transitions were scaled in relation to the respective quantifier transition (topmost chromatograms) that was set to 100%. The optimal collision energies for each depicted MRM transition are given in Table 1.
Scheme 1.
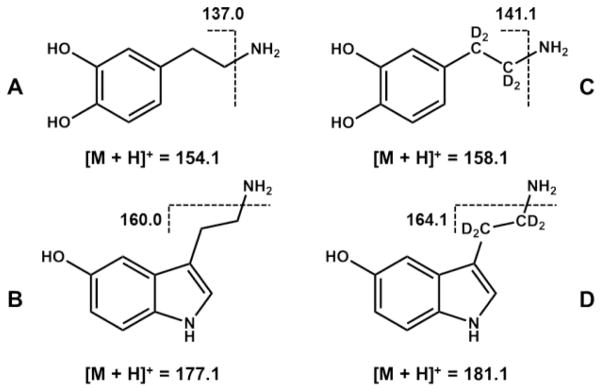
Chemical structures of (A) DA and (B) SRT, their respective deuterated internal standards, (C) DA-d4 and (D) SRT-d4, as well as underlying fragmentation reactions yielding the most abundant product ions used for MRM quantification.
Figure 2.

MRM chromatograms of DA, SRT and their respective deuterated internal standards in homogenates of cat-2 mutant worms (left panel), untreated WT N2 worms (middle panel) and WT N2 worms exposed to 100 μM of the MAO-B inhibitor selegiline (right panel). Only the quantifier mass transitions (see Table 1) of DA, SRT and the accordant internal standards are presented. For each neurotransmitter and type of worm homogenate analyzed, the signal intensity was scaled in relation to the appropriate internal standard (set to 100%).
Extraction of DA and SRT from C. elegans homogenates and analyte stability
After optimizing the LC-MS/MS conditions, the method was applied to C. elegans homogenates. Due to the instability and rapid oxidation of DA and SRT, especially in neutral or alkaline medium, acidified extraction methods were used. As an initial step, the stability of the deuterated analogues of the respective neurotransmitter in different extraction buffers used in the literature for tissue or C. elegans homogenates was evaluated [11, 34, 35]. Using 0.1 M PCA, 0.1 M PCA/ 0.1 μM ascorbic acid or 10 mM formic acid as extractants, the recoveries of DA-d4 and SRT-d4 were only 52, 49, 83% and 2, 1, 34%, respectively. However, the introduction of an extraction buffer containing 0.002 M sodium thiosulfate in 0.2 M PCA prevented significant degradation of DA-d4 (87% recovery) and SRT-d4 (74% recovery) in worm homogenates (Table 2). Further experiments revealed that extraction efficiency could be strongly improved by sonication of the prepared extract on ice. Stability studies revealed that a long-term storage (> 14 days) of worm pellets at −80 °C did not result in analyte degradation (data not shown). However, storing the extracted supernatant prior to LC-MS/MS analysis demonstrated a significant analyte reduction. Therefore, storage of the extracted worm homogenates was omitted in all further experiments, and analysis was carried out on the same day the samples were extracted.
Table 2.
Method validation parameters determined for the simultaneous quantification of DA and SRT in C. elegans homogenates.
| Parametera | Dopamine | Serotonin |
|---|---|---|
| Recovery [%]b | 87.4 ± 1.4 | 74.3 ± 0.8 |
| Intraday variation [%]c | 5.8 ± 1.4 | 6.3 ± 1.2 |
| Interday variation [%]c | 12.6 ± 3.5 | 11.5 ± 4.2 |
| Limit of detection [nM]d | 0.11 | 0.04 |
| Limit of detection on column [fmol]d | 0.56 | 0.20 |
| Limit of quantification [nM]e | 0.37 | 0.14 |
| Limit of quantification on column [fmol]e | 1.87 | 0.68 |
| Linearity in range LOD – 2.5 pmol [r2]f | 0.99984 | 0.99983 |
All method validation parameters were determined using matrix-matched samples (extract of 40,000 cat-2 or N2 worms per sample).
Data are means ± SE of 10 separate analyses.
|1 − (xi/x̄)| * 100 % with xi as result of each single measurement and x̄ as the mean result of all analyses. Data are means ± SE of 10 analyses (intraday variation) or 5 analyses (interday variation).
Defined as analyte amount that produces a peak with a S/N ratio of 3.
Defined as analyte amount that produces a peak with a S/N ratio of 10.
Worm extracts were spiked with fixed amounts of both deuterated internal standards as well as seven varying amounts of both analytes. The peak areas of the analytes, corrected for the responsiveness of the internal standards, were plotted against the concentrations of the analytes followed by linear regression analysis.
Method validation
The constructed calibration curves of the DA and SRT standard consisted of seven levels of spiked worm homogenates in the concentration range 0 – 500 nM. Linearity could be observed in the entire concentration range tested for each neurotransmitter, using the peak area ratios of the analyte to that of the respective deuterated standard. The slope of the regression line was 0.03573 ± 0.0027 for DA and 0.04736 ± 0.00285 for SRT, with a correlation coefficient (r2) in all experiments greater than 0.999. No interfering peaks from endogenous compounds were observed at the retention times of DA and SRT when WT or cat-2 mutant worms were investigated (Figure 2). This indicates that the method is specific. The LOD, defined as the amount of analyte that causes a peak with an S/N ratio of 3, was found to be 0.11 nM DA and 0.04 nM SRT. The LOQ, defined as the amount of analyte that causes a peak with an S/N ratio of 10, consequently amounted to 0.37 nM DA and 0.14 nM SRT (Table 2). The quantification of DA and SRT in homogenates of 40,000 WT worms, prepared as described in the Methods section, revealed average concentrations of 6.5 nM DA and 4.0 nM SRT (n = 35). These values are about 20-fold and 30-fold above the LOQ of DA and SRT, respectively. Thus, we should be able to unambiguously quantify DA and SRT in C. elegans even after reduction of the initially applied worm counts to about 2,000 species per sample. To our best knowledge, there is no method with a comparable sensitivity available yet. The recovery, expressing the remaining signal intensity after analyte loss and/or degradation during the sample preparation as well as ion suppression in the ESI source by co-extracted matrix components, amounted to values in a range of 74 – 87%, indicating a satisfactory recovery of the method. The relative standard deviation of quantitative results within a single assay and between independent experiments conducted on different occasions is expressed by the intra- and interday variation. As shown in Table 2, the variation coefficients of both intra- and interday results were less than 15%, demonstrating that the present method is both repeatable and reproducible.
Drug-mediated modulation of DA and SRT levels in C. elegans
The validated LC-MS/MS method was applied to evaluate the effects of the MAO-B inhibitor selegiline and the COMT inhibitor tolcapone on the DA and SRT levels in WT worms. MAO-B and COMT are crucial enzymes in the degradation of biogenic amines such as DA and SRT, not only in C. elegans but also in vertebrates such as humans. Consequently, an inhibition of these enzymes would result in an accumulation of respective substrates, resulting in increased detectable amounts. The DA values for non-treated worms amount to 1.1 ± 0.03 ng DA per mg protein (Figure 3). Selegiline treatment at 50, 100 and 250 μM resulted in a significant increase of DA levels, while the DA levels following a 500 μM treatment was indistinguishable from non-treated WT worms. DA levels increased dose-dependently after administration of tolcapone, reaching significance at ≥ 100 μM (Figure 3). The basal SRT level of WT worms is 0.89 ± 0.02 ng SRT per mg protein and did not change after selegiline incubation ≤ 250 μM (Figure 4). However, 500 μM selegiline resulted in a significant decrease of SRT levels in WT worms. In the case of tolcapone treatment, concentrations ≥ 250 μM were sufficient to significantly increase SRT levels compared to basal levels (Figure 4). Figure 2 shows respective MRM chromatograms of DA, SRT and the accordant deuterated internal standards in untreated WT worms (middle column) and selegiline-treated (100 μM) WT worms (right column). The signal intensity of DA in selegiline-treated worms is increased compared to non-treated WT worms. However, the SRT peaks show comparable intensities irrespective of selegiline treatment. These observations corroborate the quantitative results presented in Figures 3 and 4. The reliability of our method is further emphasized by the MRM chromatogram for the quantifier mass transition of DA in homogenates of unexposed cat-2 worms (Figure 2, left column). These mutants have an impaired DA biosynthesis and, likewise, the corresponding chromatogram lacks a clear DA signal. In fact, homogenates of cat-2 worms are not completely free of DA [33]. However, determined concentrations in our studies were between the LOD and LOQ.
Figure 3.

Impact of selegiline and tolcapone on the DA level in WT worms. Worms were exposed to selegiline (3 h) or tolcapone (4 h) at L1 stage, extracts were prepared and the DA level was quantified by isotope-dilution LC-MS/MS. Shown are mean values + SEM of at least three experiments each. Statistical analysis by two-way ANOVA, followed by Bonferroni’s multiple comparison post-hoc test. ***p<0.001, **p<0.01, *p<0.05 versus respective non-treated worms.
Figure 4.

Impact of selegiline and tolcapone on the SRT level in WT worms. Worms were exposed to selegiline (3 h) or tolcapone (4 h) at L1 stage, extracts were prepared and the SRT level was quantified by isotope-dilution LC-MS/MS. Shown are mean values + SEM of at least three experiments each. Statistical analysis by two-way ANOVA, followed by Bonferroni’s multiple comparison post-hoc test. **p<0.01, *p<0.05 versus respective non-treated worms.
Basal slowing behavioral analysis
Next we tested the translational value of the findings of the LC-MS/MS data and investigated whether the measured effect of selegiline and tolcapone on the DA and SRT levels persisted to alter behavioral outcomes of SRT and DA signaling. The basal slowing response is a DA-dependent behavior that affects the mechanosensation needed for proper food sensing in C. elegans, as worms slow their movement when encountering a bacterial lawn. Besides DA, SRT signaling also affects the slowing response of C. elegans. Changes (Δ) in number of body bends between plates with and without bacteria reflect the integrity of DAergic neurons. Higher values indicate functional, while lower values indicate dysfunctional DAergic neurons. cat-2 deletion mutants are defective in this response from the loss of DA synthesis. Consequently, they do not slow down on food and show a lower Δ value that is significantly different from the untreated and treated WT worms (Figure 5). The functional behavioral readout (Figure 5), which was measured 48 h following exposure, corroborates the LC-MS/MS data. An increased number of body bends in WT worms following selegiline or tolcapone treatment correlates with increased DA levels (Figure 3).
Figure 5.

Basal slowing response of WT worms following selegiline or tolcapone incubation. Behavioral data are expressed as the change in body bends per 20 seconds between treated and untreated WT animals placed on plates without food vs. plates with food. cat-2 mutants were used as a positive control. Shown are mean values + SEM of at least three experiments each. Statistical analysis by two-way ANOVA, followed by Bonferroni’s multiple comparison post-hoc test. ***p<0.001, **p<0.01, *p<0.05 versus respective non-treated worms.
Discussion
The purpose of this study was to establish a specific and sensitive isotope-dilution LC-MS/MS method for the quantification of DA and SRT in C. elegans. The developed methodology using the entire model organism incorporates significant advantages which employs adequate worm numbers; minimal sample preparation (e.g. protein precipitation, sonication and centrifugation); minimal sample loading capability; and a valid highly specific and sensitive LC-MS/MS method. In particular, the method allows an unequivocal identification of DA and SRT, as well as a detection sensitivity clearly exceeding that of chromatographic methods coupled with fluorescence or electrochemical detection commonly used for neurotransmitter monitoring in brain tissue and C. elegans samples. Although fluorescence and electrochemical detection has often been used in the past, both techniques occupy some limitations. Fluorescence detection requires laborious sample preparation due to the necessity of fluorescence derivatization protocols with highly precise reaction conditions [9–11]. Restrictions of electrochemical detection include its necessity of ion-pairing chemistry to enhance amine retention; the limited ability to accommodate changes in mobile phase composition; and the possible drift of the electrode response to time that requires more frequent standard calibrations [13, 36]. Furthermore, within both detection methods, the analytes can only be identified by their retention time, and quantification is limited to external calibration or internal standards like isoproterenol [37, 38]. However, in the LC-MS/MS method presented herein, the combination of the MRM mode with stable-isotopic labelled analogues of the respective analytes as internal standards allows an unequivocal identification and quantification of the respective neurotransmitter. Moreover, the presented method is characterized by its detection sensitivity and the minimal sample loading capability clearly exceeding that of other methods [39, 40] including the recently published DA detection using water-soluble photoluminiscent silicon nanoparticles incorporating an LOD of 0.3 nM [41]. Moriarty et al. provide a LC-ESI-MS/MS method for the analysis of SRT and related compounds in urine and achieved LODs of 8.8–18.2 nM and LOQs of 29.4 – 55.7 nM [42]. Only a few publications measuring neurotransmitters in brain microdialysates exist, including Suominen et al. presenting LODs for DA and SRT analyzed by LC-ESI-MS/MS of 0.20 nM (3 fmol injected onto the column) and an LOQ of 0.5 nM for DA and 1 nM for SRT [16, 17, 43]. Due to the high sensitivity of the developed method (LOQ DA: 0.37 nM; LOQ SRT: 0.14 nM; summarized in Table 2), the number of requisite worms (L1 stage) could be reduced to about 2,000 per sample compared to other HPLC methods requiring hundreds of thousands worms (200,000 worms) per sample [34, 44, 45]. While several methods extracting neurotransmitters from brain tissue are often laborious due to pretreatment or derivatization steps [13, 18, 35, 46], we developed a fast and simple extraction protocol allowing high-throughput analyses. In trying to adopt published tissue [11] and C. elegans extraction [34, 47] protocols, the stability of the isotopic labelled internal standards could only be insured using a new extraction medium containing 0.002 M sodium thiosulfate in 0.2 M PCA. The certain instability of the monitored neurotransmitter underlines the importance and advantage of the utilizing stable-isotopic labelled internal standards, as they allow for trustworthy correction of analyte loss when added directly to the extraction solvent.
The validated analytical method is particularly suited to investigate drug-mediated modulation of the DAergic and SRTergic system in C. elegans. Thereby, C. elegans incorporates advantages including its fast life cycle and the fully mapped nervous system with a conserved DA and SRT biology including synaptic release, trafficking and formation [23, 24]. Genes conserved in C. elegans which are involved in the DA synthesis and signaling have either been cloned, characterized or predicted from sequence. These include the biosynthetic as well as the catabolic enzymes, including aldehyde dehydrogenase (ALD-D), COMT, DOPA decarboxylase (DDC), GTP cyclohydrolase (GTPCH), MAO, and TH. Compounds in the DA pathway of C. elegans which have been already detected by HPLC include DA and its catabolic compounds 3-methoxytyramine, homovanillic acid and 3,4-dihydroxyphenylacetic acid [22, 48]. Whereas DA can be metabolized both by deamination via MAO and by methylation via COMT, SRT is degraded primarily by MAO [49]. Monitoring the levels of monoamines has traditionally been implicated for evaluation of the therapeutic efficacy of MAO and COMT inhibitors. The main function of both enzymes, MAO and COMT, is to lower the concentration of monoamines like DA and SRT in the central nervous system (CNS). Consequently, MAO and COMT inhibitors work to block the breakdown of monoamines in the brain, thus making them more available [11]. For example, applying phenelzine, a non-selective and irreversible MAO-A/B inhibitor, caused a gradual increase in extracellular SRT and DA levels in rat microdialysates, as quantified by HPLC coupled with fluorescence detection [11]. Overall, MAO and COMT inhibitors are frequently used for the treatment of neurological disorders like depression, Parkinson’s disease and Alzheimer’s disease [50–53]. The C. elegans genome encodes several proteins with homologies to MAO, with AMX-2 being the most similar to mammalian MAO-A and MAO-B and 5 COMT-like proteins which are all uncharacterized up to now [54]. In the literature, there are currently limited data available regarding the application and effects of MAO and COMT inhibitors on DA and SRT levels in C. elegans. To our knowledge, the present study reports, for the first time, the effect of an MAO-B and COMT inhibitor on DA and SRT levels in C. elegans. The quantification of DA and SRT in WT worms exposed to selegiline and tolcapone confirmed the suspected inhibition, resulting in an accumulation of the respective neurotransmitter. Selegiline (L-deprenyl), a selective, irreversible inhibitor of MAO-B is widely used in the treatment of Parkinson’s disease [55]. However, tolcapone is a potent, selective and reversible inhibitor of COMT in the brain; treatment with tolcapone has been shown to widen the therapeutic window for levodopa (L-DOPA) by reducing the doses needed for symptom control [56, 57].
The C. elegans model system is often used for behavioral studies, since neurotransmitters like DA and SRT modulate the nature of locomotory patterns [31]. The behavioral readout of WT worms following selegiline or tolcapone treatment (basal slowing) in this study corroborated the analytical LC-MS/MS data. With increasing DA and SRT levels, an increasing frequency of the worm’s moving events could be observed. In contrast, impaired DA signaling, like in cat-2 mutants, causes a defect in locomotor coordination that is signified by a reduced number of body bends [31, 33]. Since human patients with Parkinson’s disease display a constellation of movement disorders, C. elegans is an often used Parkinson’s disease model system. Although the anatomical DAergic circuitry in humans and worms is quite different, the C. elegans model system may help identify molecular and cellular mechanisms that are disrupted in humans with Parkinson’s disease [29]. Braungart et al. observed an ameliorating effect on mobility in worms treated with MPP+ (1-methyl-4-phenylpyridium) following selegiline incubation. In this study, selegiline showed different maximal beneficial concentrations, which is consistent with our data [58]. While maximum basal levels of DA and SRT are achieved following incubating with 50 μM, the highest concentration of administered selegiline results in DA content that is indistinguishable from untreated worms. Although data are limited regarding MAO and COMT inhibitors in C. elegans, exogenously applied DA has been previously shown to exacerbate manganese-induced toxicity. DA and Mn(II) are hypothesized to have a synergistic toxic effect in the worm since pre-treatment with 10 mM DA renders WT worms hypersensitive to subsequent MnCl2 exposure, bringing down the LD50 (lethal concentration, 50%) from 47 mM to 25 mM [37].
Conclusion
Altogether, the developed and validated specific and highly sensitive isotope-dilution LC-MS/MS method for the quantification of DA and SRT is particularly suited to investigate drug-mediated modulation of the DAergic and SRTergic system in C. elegans which was verified by exposing WT worms to an MAO-B and COMT inhibitor. Thereby, the advantages of this entire in vivo model organism, including its quick life cycle and a fast and simple extraction protocol, would allow for high-throughput analyses in order to identify neuroprotective or regenerative compounds. This may also be of central importance for the identification of disrupted molecular and cellular mechanisms in neurological disorders, since DA and SRT metabolism may be impaired. Additionally, animals can be examined for changes in phenotypes, especially those that are related to specific neurotransmitters. For example, altered serotonin levels would be expected to alter pharyngeal pumping, and altered dopamine levels would be expected to affect basal slowing response in the nematode.
Up to our knowledge this study exceeded the actually published LC-MS/MS methods quantifying DA and SRT in sensitivity and for the first time increased DA and SRT levels could be quantified exposing C. elegans towards MAO-B and COMT inhibitors. It should be noted that the presented methodology is not necessarily limited to studies in the field of C. elegans research. Preliminary tests show that our developed analytical approach may be easily transferred to other biological matrices, such as murine brain tissue, indicating its possible applicability to other model systems in molecular and analytical neuroscience.
Highlights.
Highly sensitive LC-MS/MS method for DA and SRT quantification in C. elegans
Applied sample preparation protocol assures high recovery rates of DA and SRT
Exposure of C. elegans to MAO-B or COMT inhibitors result in an accumulation of DA
C. elegans well-suited to study drug-mediated modulation of the DA and SRT system
Behavioral readout of the treated worms corroborated the analytical LC-MS/MS data
Acknowledgments
We would like to acknowledge the Caenorhabditis Genetic Center (CGC), which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440), for providing the strains used in this manuscript. MA was supported in part by grants from the NIH, R01ES07331 and R01ES10563.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wise RA. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- 2.Beaulieu JM, Gainetdinov RR. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 3.Lees AJ, Hardy J, Revesz T. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 4.Cloez-Tayarani I, Changeux JP. J Leukoc Biol. 2007;81:599–606. doi: 10.1189/jlb.0906544. [DOI] [PubMed] [Google Scholar]
- 5.Kema IP, de Vries EG, Muskiet FA. J Chromatogr B Biomed Sci Appl. 2000;747:33–48. doi: 10.1016/s0378-4347(00)00341-8. [DOI] [PubMed] [Google Scholar]
- 6.Alisky JM. Med Hypotheses. 2006;67:556–560. doi: 10.1016/j.mehy.2006.02.043. [DOI] [PubMed] [Google Scholar]
- 7.Mitani H, Shirayama Y, Yamada T, Kawahara R. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:531–534. doi: 10.1016/j.pnpbp.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 8.Fuxe K, Dahlstrom AB, Jonsson G, Marcellino D, Guescini M, Dam M, Manger P, Agnati L. Prog Neurobiol. 2010;90:82–100. doi: 10.1016/j.pneurobio.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 9.Umeda S, Stagliano GW, Borenstein MR, Raffa RB. J Pharmacol Toxicol Methods. 2005;51:73–76. doi: 10.1016/j.vascn.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Zhao XE, Suo YR. Talanta. 2008;76:690–697. doi: 10.1016/j.talanta.2008.04.032. [DOI] [PubMed] [Google Scholar]
- 11.Yoshitake T, Kehr J, Yoshitake S, Fujino K, Nohta H, Yamaguchi M. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;807:177–183. doi: 10.1016/j.jchromb.2004.03.069. [DOI] [PubMed] [Google Scholar]
- 12.Patel BA, Arundell M, Parker KH, Yeoman MS, O’Hare D. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;818:269–276. doi: 10.1016/j.jchromb.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 13.Tareke E, Bowyer JF, Doerge DR. Rapid Commun Mass Spectrom. 2007;21:3898–3904. doi: 10.1002/rcm.3295. [DOI] [PubMed] [Google Scholar]
- 14.Ciccimaro E, Blair IA. Bioanalysis. 2010;2:311–341. doi: 10.4155/bio.09.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hows ME, Lacroix L, Heidbreder C, Organ AJ, Shah AJ. J Neurosci Methods. 2004;138:123–132. doi: 10.1016/j.jneumeth.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 16.Cannazza G, Carrozzo MM, Cazzato AS, Bretis IM, Troisi L, Parenti C, Braghiroli D, Guiducci S, Zoli M. J Pharm Biomed Anal. 2012;71:183–186. doi: 10.1016/j.jpba.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Ji C, Li W, Ren XD, El-Kattan AF, Kozak R, Fountain S, Lepsy C. Anal Chem. 2008;80:9195–9203. doi: 10.1021/ac801339z. [DOI] [PubMed] [Google Scholar]
- 18.Tornkvist A, Sjoberg PJ, Markides KE, Bergquist J. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;801:323–329. doi: 10.1016/j.jchromb.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 19.Najmanová V, Rambousek L, Syslová K, Bubeníková V, Šlamberová R, Valeš K, Kačer P. Chromatographia. 2011;73:143–149. [Google Scholar]
- 20.Chen P, Martinez-Finley EJ, Bornhorst J, Chakraborty S, Aschner M. Frontiers in aging neuroscience. 2013;5:18. doi: 10.3389/fnagi.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leung MC, Williams PL, Benedetto A, Au C, Helmcke KJ, Aschner M, Meyer JN. Toxicol Sci. 2008;106:5–28. doi: 10.1093/toxsci/kfn121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chase DL, Koelle MR. WormBook. 2007:1–15. doi: 10.1895/wormbook.1.132.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bany IA, Dong MQ, Koelle MR. J Neurosci. 2003;23:8060–8069. doi: 10.1523/JNEUROSCI.23-22-08060.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bandyopadhyay J, Song HO, Park BJ, Singaravelu G, Sun JL, Ahnn J, Cho JH. Biochem Biophys Res Commun. 2009;390:136–141. doi: 10.1016/j.bbrc.2009.09.082. [DOI] [PubMed] [Google Scholar]
- 25.Sanyal S, Wintle RF, Kindt KS, Nuttley WM, Arvan R, Fitzmaurice P, Bigras E, Merz DC, Hebert TE, van der Kooy D, Schafer WR, Culotti JG, Van Tol HH. EMBO J. 2004;23:473–482. doi: 10.1038/sj.emboj.7600057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hills T, Brockie PJ, Maricq AV. J Neurosci. 2004;24:1217–1225. doi: 10.1523/JNEUROSCI.1569-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vidal-Gadea A, Topper S, Young L, Crisp A, Kressin L, Elbel E, Maples T, Brauner M, Erbguth K, Axelrod A, Gottschalk A, Siegel D, Pierce-Shimomura JT. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17504–17509. doi: 10.1073/pnas.1108673108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bazopoulou D, Tavernarakis N. Genetica. 2009;137:39–46. doi: 10.1007/s10709-009-9361-3. [DOI] [PubMed] [Google Scholar]
- 29.Omura DT, Clark DA, Samuel AD, Horvitz HR. PLoS One. 2012;7:e38649. doi: 10.1371/journal.pone.0038649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srinivasan S, Sadegh L, Elle IC, Christensen AG, Faergeman NJ, Ashrafi K. Cell Metab. 2008;7:533–544. doi: 10.1016/j.cmet.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sawin ER, Ranganathan R, Horvitz HR. Neuron. 2000;26:619–631. doi: 10.1016/s0896-6273(00)81199-x. [DOI] [PubMed] [Google Scholar]
- 32.Brenner S. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez-Finley EJ, Chakraborty S, Slaughter JC, Aschner M. Neurochem Res. 2013;38:1543–1552. doi: 10.1007/s11064-013-1054-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu RH, Harn HJ, Liu SP, Chen CS, Chang WL, Chen YM, Huang JE, Li RJ, Tsai SY, Hung HS, Shyu WC, Lin SZ, Wang YC. PLoS One. 2014;9:e85305. doi: 10.1371/journal.pone.0085305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song P, Mabrouk OS, Hershey ND, Kennedy RT. Anal Chem. 2012;84:412–419. doi: 10.1021/ac202794q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitton KP, Trevithick JR. Methods Enzymol. 1994;233:523–539. doi: 10.1016/s0076-6879(94)33058-1. [DOI] [PubMed] [Google Scholar]
- 37.Benedetto A, Au C, Avila DS, Milatovic D, Aschner M. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Echizen H, Itoh R, Ishizaki T. Clin Chem. 1989;35:64–68. [PubMed] [Google Scholar]
- 39.Kaplan KA, Chiu VM, Lukus PA, Zhang X, Siems WF, Schenk JO, Hill HH., Jr Analytical and bioanalytical chemistry. 2013;405:1959–1968. doi: 10.1007/s00216-012-6638-7. [DOI] [PubMed] [Google Scholar]
- 40.Rangiah K, Palakodeti D. Rapid communications in mass spectrometry : RCM. 2013;27:2439–2452. doi: 10.1002/rcm.6706. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Chen X, Kai S, Wang HY, Yang J, Wu FG, Chen Z. Anal Chem. 2015 doi: 10.1021/ac504520g. [DOI] [PubMed] [Google Scholar]
- 42.Moriarty M, Lee A, O’Connell B, Kelleher A, Keeley H, Furey A. Anal Bioanal Chem. 2011;401:2481–2493. doi: 10.1007/s00216-011-5322-7. [DOI] [PubMed] [Google Scholar]
- 43.Suominen T, Uutela P, Ketola RA, Bergquist J, Hillered L, Finel M, Zhang H, Laakso A, Kostiainen R. PLoS One. 2013;8:e68007. doi: 10.1371/journal.pone.0068007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caito SW, Valentine WM, Aschner M. J Neurochem. 2013;127:837–851. doi: 10.1111/jnc.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reckziegel P, Chen P, Caito S, Gubert P, Soares FA, Fachinetto R, Aschner M. Arch Toxicol. 2015 doi: 10.1007/s00204-015-1451-7. [DOI] [PubMed] [Google Scholar]
- 46.Greco S, Danysz W, Zivkovic A, Gross R, Stark H. Analytica chimica acta. 2013;771:65–72. doi: 10.1016/j.aca.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 47.Yin JA, Liu XJ, Yuan J, Jiang J, Cai SQ. J Neurosci. 2014;34:3947–3958. doi: 10.1523/JNEUROSCI.4013-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wintle RF, Van Tol HH. Parkinsonism Relat Disord. 2001;7:177–183. doi: 10.1016/s1353-8020(00)00055-9. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Berndt TJ, Gross JM, Peterson MA, So MJ, Knox FG. Am J Physiol Regul Integr Comp Physiol. 2001;280:R248–254. doi: 10.1152/ajpregu.2001.280.1.R248. [DOI] [PubMed] [Google Scholar]
- 50.Follmer C. Expert Rev Neurother. 2014;14:703–716. doi: 10.1586/14737175.2014.920235. [DOI] [PubMed] [Google Scholar]
- 51.Tai CH, Wu RM. Acta Med Okayama. 2002;56:1–6. doi: 10.18926/AMO/31725. [DOI] [PubMed] [Google Scholar]
- 52.Cai Z. Mol Med Rep. 2014;9:1533–1541. doi: 10.3892/mmr.2014.2040. [DOI] [PubMed] [Google Scholar]
- 53.Riederer P, Laux G. Exp Neurobiol. 2011;20:1–17. doi: 10.5607/en.2011.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.H. O., WormBook, editor. The C. elegans Research Community. WormBook. 2013 Aug 13; doi: 10.1895/wormbook.1.161.1. http://www.wormbook.org. [DOI]
- 55.Naoi M, Maruyama W, Inaba-Hasegawa K. Expert Rev Neurother. 2013;13:671–684. doi: 10.1586/ern.13.60. [DOI] [PubMed] [Google Scholar]
- 56.Truong DD. Clin Interv Aging. 2009;4:109–113. doi: 10.2147/cia.s3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marsala SZ, Gioulis M, Ceravolo R, Tinazzi M. Clin Neuropharmacol. 2012;35:185–190. doi: 10.1097/WNF.0b013e31825c034a. [DOI] [PubMed] [Google Scholar]
- 58.Braungart E, Gerlach M, Riederer P, Baumeister R, Hoener MC. Neurodegener Dis. 2004;1:175–183. doi: 10.1159/000080983. [DOI] [PubMed] [Google Scholar]