

Abstract
In mice and humans, loss of myosin VI (Myo6) function results in deafness, and certain Myo6 mutations also result in cardiomyopathies in humans. The current studies have utilized the Snell’s waltzer (sv) mouse (a functional null mutation for Myo6) to determine if this mouse also exhibits cardiac defects and thus used to determine the cellular and molecular basis for Myo6-associated heart disease. Myo6 is expressed in mouse heart where it is predominantly expressed in vascular endothelial cells (VECs) based on co-localization with the VEC cell marker CD31. Sv/sv heart mass is significantly greater than that of sv/+ littermates, a result of left ventricle hypertrophy. The left ventricle of the sv/sv exhibits extensive fibrosis, both interstitial and perivascular, based on histologic staining, and immunolocalization of several markers for fibrosis including fibronectin, collagen IV, and the fibroblast marker vimentin. Myo6 is also expressed in lung VECs but not in VECs of intestine, kidney or liver. Sv/sv lungs exhibit increased peri-aveolar fibrosis and enlarged air sacs. Electron microscopy of sv/sv cardiac and lung VECs revealed abnormal ultrastructure, including luminal protrusions and increased numbers of cytoplasmic vesicles. Previous studies have shown that loss of function of either Myo6 or its adaptor binding partner synectin/GIPC results in impaired arterial development due to defects in VEGF signaling. However, examination of synectin/GIPC −/− heart revealed no fibrosis or significantly altered VEC ultrastructure, suggesting that the cardiac and lung defects observed in the sv/sv mouse are not due to Myo6 function in arterial development.
Key terms: Myo6, fibrosis, vascular endothelial cell, caveoli, GIPC
INTRODUCTION
Myosin VI (Myo6) is unique among characterized members of the myosin family of actin-based molecular motors in that it moves toward the minus (pointed) end of the actin filament [Buss and Kendrick-Jones 2011; Wells et al. 1999]. Myo6 has been implicated in a broad range of cellular functions. These include clathrin-mediated endocytosis, regulated trafficking of membrane proteins, Golgi organization, autophagy, actin dynamics, cell migration including tumor cell invasivity and transcription (for reviews see [Buss and Kendrick-Jones 2011; Chibalina et al. 2009; Tumbarello et al. 2013]).
The Myo6 mutant mouse, Snell’s waltzer (sv), has been a useful model for dissecting the in vivo functions for this myosin. Mice homozygous for the sv (sv/sv) mutation are deaf and lack vestibular function due to degeneration of the inner ear neurosensory epithelium [Avraham et al. 1995]. Patients with mutations in Myo6 are also deaf [Ahmed et al. 2003; Melchionda et al. 2001; Mohiddin et al. 2004]. Myo6 is expressed at the base of the stereo cilia of the neurosensory hair cells, and also is present on vesicles in a zone of presumed vesicular membrane trafficking termed the pericuticular necklace [Hasson et al. 1997]. Ultrastructural analysis of hair cells in neonatal sv/sv mice, before degeneration has occurred, revealed that the plasma membrane is detached from the base of stereo ciliary actin bundles resulting in fused stereo cilia [Self et al. 1999]. This observation suggests that Myo6 mediated minus-end directed tension between the plasma membrane and underlying actin bundle is required to maintain stereo ciliary membrane organization. Subsequent phenotypic studies of the sv/sv mouse have revealed a variety of defects in a number of other cell types. In the brain Myo6 is a component of the post synaptic density [Osterweil et al. 2005]. In the hippocampus of the sv/sv mouse, synapse number is reduced and dendritic morphology is abnormal. In cultured sv/sv hippocampal neurons, clathrin-mediated endocytosis of glutamate receptors is disrupted [Osterweil et al. 2005], although endocytosis of transferrin is not affected indicating that Myo6 is not universally involved in all modes of clathrin-mediated endocytosis. Moreover, the involvement of Myo6 in endocytosis can be developmentally regulated for the same ligand. In the neonatal intestine, lactoferrin uptake is normal in the sv/sv mouse, but completely blocked in the adult [Hegan et al. 2012]. In the renal proximal tubule epithelium, endocytic retrieval of serum proteins from glomerular filtrate is impaired resulting in proteinuria [Gotoh et al. 2010]. In the intestinal epithelial cell, where Myo6 is associated with the base of the microvilli of the apical brush border (BB), there are numerous structural and compositional defects in the BB of the sv/sv mouse [Collaco et al. 2010; Hegan et al. 2012] including a lifting of the membrane at the base of BB microvilli. In addition, the regulated endocytic trafficking of several proteins between the BB membrane and apical endosome is disrupted [Ameen and Apodaca 2007; Hegan et al. 2012].
In addition to deafness, a kindred of patients has been identified with an autosomal dominant mutation in the motor domain of Myo6 who also present with familial hypertrophic cardiomyopathy [Mohiddin et al. 2004]. Cardiac abnormalities observed included left ventricular hypertrophy, and prolongation of the QT interval. In order to investigate possible cellular bases for Myo6-associated cardiomyopathy, we have conducted a phenotypic characterization of the sv/sv heart and lung. Sv/sv mice exhibit increased heart mass/body mass ratios and pronounced left ventricular hypertrophy that is associated with extensive fibrosis. An increased heart/body mass ratio has also been reported for a novel Myo6 mutant mouse line [Williams et al. 2013]. Most familial cardiomyopathies result from mutations in cardiac sarcomeric proteins [Marston 2011; Watkins et al. 2011]. Localization of Myo6 associated with the sarcoplasmic reticulum (SR) has been reported [Karolczak et al. 2013]. However, our results indicate that Myo6 is predominantly expressed in the vascular endothelial cells (VECs) of the heart and lung, but not in the vasculature of other organ systems examined thus far including kidney, intestine and liver. Ultrastructural analysis of cardiac VECs revealed the presence of large numbers of cytoplasmic vesicles, suggesting a role for Myo6 in either clathrin or caveolin-dependent traffic between the vascular lumen and cardiac tissue space.
MATERIALS AND METHODS
Animals
Mice of the strain B6 x STOCK Tyrc-ch Bmp5se +/+ Myo6sv/J (stock #000578) were purchased from Jackson Labs (Bar Harbor, ME) and back-crossed to the wild type (WT) mouse line (129X1/SvJ) to establish mice that were mutant for Myo6. Mutant mice displayed the circling, head tossing, and hyperactivity phenotype that is associated with the Myo6 mutation. Heterozygous littermate mice were used as control animals for all experiments. All data shown for sv/+ and sv/sv was generated using male mice. The synectin/GIPC −/− mice used in this study were generated in the Simons laboratory, Yale School of Medicine as described in [Lanahan et al. 2010]. All procedures involving mice were performed in accordance with an approved Yale – IACUC animal protocol.
Electron microscopy
Tissues were processed for transmission electron microscopy (TEM) as described in [Mooseker and Tilney 1975]. Heart and lung was dissected and rinsed with phosphate buffered saline (PBS), cut into small pieces, and fixed in 2% glutaraldehyde, 0.2% tannic acid in 0.1M sodium phosphate buffer, pH 7.0 for 10 minutes at room temperature (RT), and then shifted to 4°C for an additional 50 minutes. Following fixation the tissue was washed three times with 0.1M sodium phosphate buffer, pH 7.0, and then incubated on ice for 1 hour in 1% OsO4 in 0.1M sodium phosphate buffer, pH 6.0, The tissue was then washed three times with cold water and incubated overnight at 4°C in 1% uranyl acetate. The samples were dehydrated with a 25 – 100% ethanol series at RT, followed by two 10 minute incubations in propylene oxide. The tissue was embedded in EmBed 812 (Electron Microscopy Sciences (EMS), Hatfield, PA) following established protocol. Imaging was done on a JEOL 1230 transmission electron microscope (JEOL, Tokyo, Japan), and digital images were recorded with a Hamamatsu ORCA-HR digital camera (Hamamatsu, Hamamatsu City, Japan).
Tissue preparation for histology and immunofluorescence microscopy
Heart and lung was dissected, rinsed with PBS, cut into large pieces, and fixed for 24 hours at 4°C in 10% paraformaldehyde (PFA) in PBS. Tissues were embedded in paraffin, sectioned, and stained with both hematoxylin and eosin (H&E) and Masson’s trichrome using standard methods (Research Histology, Yale University School of Medicine). Histological sections were viewed with a Nikon Diaphot 300 inverted microscope (Nikon Instruments, Melville, NY) equipped with a Zeiss Axiocam (Zeiss, Thornwood, NY), and digital images were captured with AxioVision v.4.6 (Zeiss).
For immunofluorescence microscopy, heart and lung were dissected, rinsed with PBS, cut into large pieces and fixed with 4 % PFA (prepared from a freshly opened vial of 16% PFA from EMS) in PBS for 10 minutes at RT and then shifted to 4°C for an additional 50 minutes. The tissues were washed twice with PBS, followed by 30 minutes in each of 5%, 8%, 12%, and 16% sucrose in PBS at RT. The tissue was incubated overnight in 20% sucrose in PBS at 4°C, and the following day was incubated in 12% sucrose, 35% OCT (Sakura Finetek USA, Torrance, CA) in PBS for 1 hour at RT. The tissue was embedded in 12% sucrose + 35% OCT in PBS and stored at −80°C. Five (heart) or 10μm (lung) sections were cut on a Leica CM 3050S cryostat (Leica, Wetzlar, Germany) and mounted on Superfrost + slides (Esco, Portsmouth, NH). The following steps were done with either PBS or TRIS buffered saline, 0.05% Tween-20 as the buffer, depending on the primary antibody used. The sections were washed 3×5 minutes with room temperature PBS to remove OCT. The tissue was blocked for 30 minutes at RT in 10% normal goat serum (NGS) in PBS. Primary antibodies were diluted in PBS, 5% NGS and incubated at RT for 1 hour, or overnight at 4°C. The slides were then washed 3×10 minutes with PBS. Alexa fluor labeled secondary antibodies (Invitrogen, Carlsbad, CA) were diluted 1:200 in PBS, 5% NGS, 0.33μM rhodamine phalloidin (Invitrogen) and 1μg/ml Hoechst dye (Sigma, St. Louis, MO) and incubated for 1 hour at RT in the dark. Slides were then washed 3×10 minutes with PBS and coverslips were mounted with Prolong Gold anti-fade media (Invitrogen).
All immunofluorescence microscopy was done using a Zeiss LSM510 META laser scanning confocal microscope, and digital images were captured using ZEN 2009 software (Zeiss). Both sv/+ and sv/sv tissue sections were mounted on the same slide, allowing equal treatment during preparation and were imaged with identical microscope settings. Contrast enhancement, cropping, and all other image manipulation was done using either Adobe Photoshop or ImageJ (http://rsb.info.nih.gov/ij/; NIH).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) labeling
Five (heart) or 10 (lung) μm cryosections were cut from sv/+ and sv/sv heart and lung tissue and washed 3×5 minutes with PBS. Tissue sections from both genotypes were mounted on the same slide in order to ensure equal treatment during the assay. The tissue sections were then blocked / permeabilized in 10% NGS, 0.1% Triton X-100 in PBS. The TUNEL assay (Roche Diagnostics, Indianapolis, IN) was done according to manufacturer’s instructions; enzyme and label were prepared at a 1:10 ratio and the tissue was incubated at 37°C for 1 hour, and then washed 3×5 minutes with PBS. F-actin was stained with 0.33μM rhodamine phalloidin (Invitrogen) for 30 minutes at RT in the dark, followed by 3×5 minute washes with PBS. Coverslips were mounted with Prolong Gold anti-fade media (Invitrogen), and confocal images were taken and processed as described above. To quantify the results of the TUNEL assay, low magnification images (450 μm × 450 μm ) were taken of heart and lung sections from both sv/+ and sv/sv. All microscope settings were identical for imaging of each genotype. Nuclei that were positively labeled by the TUNEL kit were scored per image and statistics and histograms were generated with Microsoft Excel (Microsoft, Redmond, WA).
Gel sample preparation, immunoblotting, differential sedimentation analysis and antibodies
For gel sample preparation heart and lung was dissected from sv/+ and sv/sv and rinsed with PBS. Tissue was then minced with a razor blade and homogenized in cytoskeleton stabilization buffer (CSB; 1mM EGTA, 75mM KCL, 2mM MgCl2, 1mM DTT, 1mM pefabloc, and 10mM imidazole, pH 7.2) with a hand-held Omni homogenizer (Omni International, Kennesaw, GA). An aliquot of the whole cell homogenate was taken and diluted 1:1 with SDS-PAGE gel sample buffer (final SDS concentration: 4%) and boiled for 5 minutes.
Samples were electrophoresed on 5–20% gradient gels and transferred to Hybond nitrocellulose membrane (GE Healthcare, Piscataway, NJ). Horseradish peroxidase conjugated secondary antibodies (Pierce, Rockford, IL) were visualized by Enhanced Chemo Luminescence (GE Healthcare).
For differential sedimentation/cell fractionation analysis of Myo6 expression, a single wild type heart was homogenized in CSB as described above, and the homogenate was first sedimented for 10 minutes at low speed (3000 × g). The low speed supernate was spun at high speed (15000 × g) for 10 minutes, and the high speed supernate was then spun at ultra-speed (250000 × g) for 20 minutes. The resulting low-, high-, and ultra-speed pellets were re-suspended to the original homogenate volumes in CSB and the re-suspended pellets and ultra-speed supernate were analyzed for Myo6 content by immunoblot as described above.
Primary antibodies directed against the following proteins were used in this study: Myo6 (Hasson and Mooseker 1994), CD31 (BD Pharmingen, San Diego, CA), vimentin, fibronectin, and collagen IV (Abcam, Cambridge, MA).
RESULTS
Myo6 is expressed in the VECs of the heart and lung, but not of other organs including kidney, liver and intestine
Localization of Myo6 with F-actin (Fig. 1A–C) in sections of sv/+ cardiac tissue indicates that it is not significantly expressed in cardiomyocytes, although the low intensity, diffuse staining of muscle cells is consistently slightly above the nonspecific background staining with anti-Myo6 in sv/sv cardiac tissue (Fig. 1D). Rather Myo6 is primarily expressed in VECs based on co-localization with the VEC marker, CD31, in capillaries (Fig. 2A–C) and larger vessels (Fig. 2D–F). Myo6 is also expressed in endothelial cells of capillaries and some but not all larger vessels of the lung (Fig. 3). In contrast to heart and lung, Myo6 is not expressed in VECs of the intestine (Fig. 4A–C), kidney (Fig. 4D–F) or liver (Fig. 4G–I).
Fig. 1. Myo6 is expressed in the heart.

Localization of Myo6 (A, C, E, F) and F-actin (B, C, E, F) in sv/+ (A–C) and sv/sv (D–F) heart. Merged images (C, F) also show Hoechst nuclear staining in blue. Myo6 is predominantly expressed in presumed VECs (see Fig. 2) although there is diffuse muscle staining above background levels observed in sv/sv heart. Inset Fig. 1A: Myo6 immunoblot of heart tissues homogenates from sv/+ and sv/sv mice. Bar: 35 μm.
Fig. 2. Myo6 is highly expressed in cardiac VECs.

Localization of Myo6 (A, C, D, F) and the VEC marker CD31 (B, C, E, F) in sv/+ heart. Low (A–C) and higher magnification (D–F) images show colocalization of Myo6 and CD31 in both capillaries and larger vessels. Merged images also show Hoechst stained nuclei in blue. Bar, A–C: 35 μm; D–F: 17 μm.
Fig. 3. Myo6 is expressed in VECs of the lung.
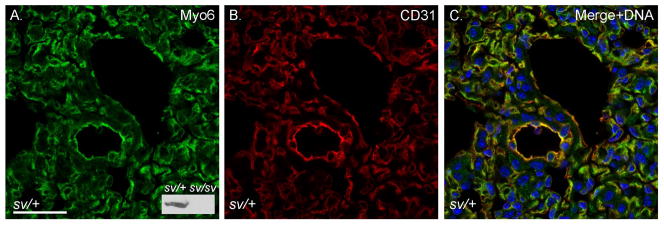
Localization of Myo6 (A, C) and CD31 (B, C) in sv/+ lung. Bar: 35 μm.
Fig. 4. Myo6 is not expressed in the VECs of intestine, kidney or liver.

Localization of CD31 (A, C, D, F, G, I) and Myo6 (B, C, E, F, H, I) in sv/+ intestine (A–C), kidney (D–F), and liver (G–I). Merged images show nuclear staining in blue. Bar: 35 μm.
SV/SV exhibit cardiac hypertrophy and left ventricular fibrosis
We have observed variable and statistically significant levels of increased heart mass/body mass (mg/gm) ratios (Fig 5A,B) in sv/sv mice, compared to age-matched sv/+ litter mates (4.8 vs 6.2);. In some animals, enlargement of the heart is quite apparent (Fig. 5A). The increased ratio in sv/sv mice is due in part to decreased average body weight ( sv/+ average mass of 37.0 gm vs. sv/sv average mass of 28.4 gm) although average heart mass of sv/sv is still greater than that of sv/+ mice ( 171 mg vs 178 mg). Nevertheless the increased body mass/heart mass in the sv/sv mice is not primarily due to the hyper-activity of the sv/sv mice, which engage in frequent spinning behavior due to vestibular dysfunction. Shaker-1 (sh-1/sh-1) mice, a mutant line for myosin VIIa, which is not expressed in VECs, engage in similar spinning behavior. Sh-1/sh-1 mice have both lower mean body mass (sh-1/+ mean 38.9 gm; sh-1/sh-1 mean 27.6 gm) and lower mean heart mass (sh-1/+ mean 179 mg; sh-1/sh-1 mean 160 mg) than sh-1/+ heterozygous controls. The sh-1/sh-1 do exhibit a slightly increased body mass/heart mass ratio (4.6 vs 5.4; p=0.05; Fig. 5C). Thus it is possible that the hyper activity could be a contributing factor for the increased ratio in sv/sv mice, particularly in those mice with smaller heart/body mass ratios.
Fig. 5. Sv/sv mice have enlarged hearts in comparison to sv/+ littermates.

(A) Images of dissected sv/+ and sv/sv hearts. B. The loss of myo6 causes a statistically significant increase in heart / body mass ratio. Sv/+ mean ratio = 4.8 ± 0.69, n = 12 mice vs. sv/sv mean ratio = 6.2 ± 0.80, n = 12 mice, *p < 0.00005. Mean body mass for sv/+ = 37.0g and mean heart weight = 171mg vs. sv/sv mean body weight = 28.4g and mean heart weight = 178mg. C. heart/body mass ratios in sh-1/+ and sh-1/sh-1 mice. Sh-1/+ mean ratio = 4.6 ± 0.50, n = 5 mice. sh-1/sh-1 mean ratio 5.4 ± 0.29, n = 5. P=0.05. Mean body mass for Sh-1/+ = 38.9g and mean heart mass = 179mg vs. sh-1/sh-1 mean body mass = 27.6 and mean heart mass = 160mg.
Histologic analysis (Fig. 6) revealed that the heart mass increase in the sv/sv mouse is primarily due to hypertrophy of the left ventricle. Masson trichrome staining of sv/+ (Fig. 6A) and sv/sv (Fig. 6B–D) heart tissue indicates that there are focal regions of left ventricular interstitial and perivascular fibrosis similar to that reported in caveolin-1 KO mice [Cohen et al. 2003]. Immunolocalization of CD31 and markers for fibrosis including collagen IV (Fig. 7A, D) and fibronectin (Fig. 7B, E) also indicate significant increases in extracellular matrix, particularly in perivascular regions. While these fibrotic lesions are devoid of myocytes, H&E staining reveals that these regions are populated with cells, most likely fibroblasts (Fig. 6H). Consistent with this possibility there are also elevated levels of vimentin staining in these regions indicative of increased numbers of fibroblasts (Fig. 7C, F). Although the histologic analysis suggests loss and disorganization of myocytes, TUNEL staining did not reveal significant differences in numbers of apoptotic nuclei in sv/sv heart (not shown).
Fig. 6. Left ventricular hypertrophy and fibrosis in the sv/sv heart.

Masson trichrome stained sections of sv/+ (A) and sv/sv (B–D) heart. Note the enlargement of the left ventricle (B) and trichrome (blue) stained interstitial and perivascular fibrotic regions in the sv/sv heart (C, D). (E, F)) H&E staining of non-fibrotic (E) and fibrotic regions (F) of sv/sv heart reveals the presence of non-cardiomyocte cells in fibrotic regions. Bar A, B: 1 mm; C, D: 100 μm; E, F: 25 μm.
Fig. 7. Elevated expression of markers for fibrosis in the sv/sv heart.

Localization of F-actin (A–F; red), collagen (A, D; green), fibronectin (B, E; green), and vimentin (C, F) in sv/+ (A–C) and sv/sv (D–F) heart. Hoechst-stained nuclei are shown in blue. Bar: 35 μm.
The above observations were performed on hearts from adult mice ages 6–12 months. Examination of heart tissue from sv/sv mice age 5 weeks revealed the presence of significant peri vascular fibrosis based on fibronectin staining indicating that the tissue damage resulting from loss of Myo6 function begins early and presumably progresses with age (Fig. 8A–C).
Fig. 8. Young sv/sv mice exhibit perivascular fibrosis.
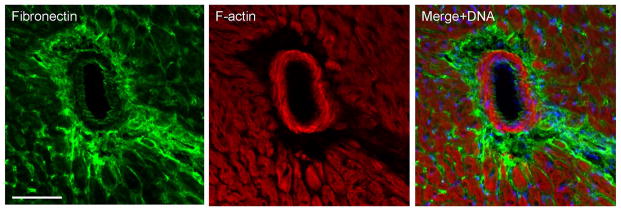
Localization of the fibrosis marker, fibronectin (A, C) and F-actin (B, C) in 5 week old sv/sv heart. Merged image (C) also shows nuclei in blue. Note the prominent perivascular fibrosis. Bar: 50 μm.
There is increased basement membrane deposition, air sac enlargement, fibrosis and elevated levels of apoptosis in the sv/sv lung
In the sv/sv lung, Masson-trichrome staining revealed slightly increased extra cellular matrix (ECM) staining both in terms of staining (blue) intensity and apparent thickness of ECM surrounding alveolar air sacs (Fig. 9A–D), an observation confirmed by TEM (see below). However, no large regions of fibrosis comparable to that observed in heart have been observed. The most apparent tissue alteration observed is the enlargement of aveolar air sac spaces (Fig 9). There is evidence for fibrosis in the lung of sv/sv mice, based on co-immunostaining with CD31 and fibronectin (Fig. 10). TUNEL staining revealed the presence of increased numbers of apoptotic aveolar epithelial nuclei (Fig. 11). Increased cell death could be the basis for the enlargement of air spaces observed.
Fig. 9. Increased fibrosis and enlarged air sacs in the sv/sv lung.

Low (A, B) and higher magnification (C, D) images of Masson trichrome stained sections of sv/+ (A, C) and sv/sv (B, D) lung. Note the increased peri-aveolar air sac trichrome ECM staining and enlarged air sacs in the sv/sv lung. Bar A, B: 50 μm; C, D: 20 μm.
Fig. 10. Elevated expression of fibronectin in the sv/sv lung.

Localization of CD31 (A, C, D, F) and the fibrosis marker fibronectin (B, C, E, F) in sv/+ (A–C) and sv/sv (D–F) lung. Merged images (C, F) show nuclei stained in blue. Bar: 35 μm.
Fig. 11. Increased apoptosis in sv/sv lung.

(A, B) TUNEL staining of apoptotic nuclei (green) in sv/+ (A) and sv/sv (B) lung. F-actin staining is shown in red. (C) Quantification of TUNEL positive nuclei/ 450 μm × 450 μm image (10 images/genotype). There is a significant increase (p=0.0004) in TUNEL positive nuclei in sv/sv lung.
Cardiac and pulmonary VECs exhibit altered ultrastructural organization including increased numbers of cytoplasmic vesicles
TEM examination of sv/sv heart tissue (Fig. 12B–F) revealed abnormal capillary VEC ultrastructure. There is a pronounced increase in the numbers of cytoplasmic vesicles (Fig. 12B–F). In contrast to the smooth luminal walls observed in capillaries of the sv/+ heart (Fig. 12A), the lumen of sv/sv capillaries often contain numerous thin projections (Fig. 12C–E) that are morphologically similar to those observed in male β3 integrin −/− mice. These mice exhibit defects in capillary maturation and in addition to luminal projections the VECs accumulate cytoplasmic vesicles [Weis et al. 2007]. Consistent with the LM cytological analyses above, there is also frequent increase in perivascular extracellular matrix around sv/sv capillaries (e.g. Fig. 12D).
Fig. 12. Cardiac VEC ultrastructure is perturbed in the sv/sv mouse.

TEM of capillaries in sv/+ (A) and sv/sv (B–F) heart. Note the luminal protrusions (Fig 12C–E) and increased numbers of cytoplasmic vesicles in the sv/sv (B–F). Bars: 500 nm
Ultrastructural analysis of the sv/sv lung (Fig. 13) revealed significant thickening and involution of the basement membrane between capillary VECs and the alveolar epithelium facing the air sac. Like sv/sv cardiac capillaries, pulmonary capillaries frequently have luminal projections (Fig. 13B, C) and increased numbers of cytoplasmic vesicles (Fig. 13B, D)
Fig. 13. Pulmonary VEC ultrastructure is perturbed in the sv/sv mouse.

TEM of capillaries in sv/+ (A) and sv/sv (B–D) lung. Note the thickening and involution of the basement membrane (B), luminal protrusions (C) and increased numbers of VEC cytoplasmic vesicles (B–D) in the sv/sv. Bars: 500 nm.
In non-Myo6 expressing VECs such as those in the kidney, the ultrastructure of sv/sv vasculature is indistinguishable from that of control animals (Supplementary Figs. 1, 2). For example, in capillaries of the kidney cortex, the endothelial lining consists of alternating regions of very thin and somewhat thicker segments. In both sv/+ and sv/sv, the thin regions are fenestrated. The thicker regions are non-fenestrated and in both genotypes surface expression of caveolae and cytoplasmic vesicles is comparable (Supplementary Fig. 2).
Sedimentation analysis indicates that cardiac VEC Myo6 is not associated with membrane/cytoskeleton sedimentable fractions
In cell fractionation studies on numerous cells and tissues we have analysed including, brain, intestinal epithelial cells, and kidney Myo6 co-sediments with actin enriched, sedimentable fractions [Gotoh et al. 2010; Hegan et al. 2012; Osterweil et al. 2005]. In contrast, Myo6 is present in the cytosolic, ultra-speed supernate fraction after differential sedimentation of heart tissue homogenates (using a buffer that stabilizes the actin filament cytoskeleton (Supplementary Fig. 3).
Myo6 is expressed in P1 cardiac but not renal VECs
As detailed in the Discussion, Myo6 is involved in VEGF-dependent arterial development in the kidney via endocytic regulation of VEGF Receptor 2 (VEGFR-2). Analysis of the sv/sv kidney revealed reduced arterial development and thus such impaired development might explain the molecular basis for the observed defects reported here (Lanahan et al. 2010]. Much of renal arterial growth and branching occurs during the first week after birth [Carmeliet and Collen 2000; Tomanek 2005]. However, no detectable VEC expression of Myo6 is observed in the vasculature of the P1 kidney (Supplementar;y Fig. 4B), while adult-like patterns of VEC expression are observed in P1 heart (Supplementary Fig. 4A). The expression of Myo6 in differentiating proximal tubules also serves as a positive control for Myo6 detection. This observation suggests that renal arterial development does not require post-natal Myo6 mediated trafficking of VEGFR-2 but must function at some earlier time in embryonic development.
The synectin/GIPC −/− mouse does not exhibit cardiac defects
Myo6 interacts with the adaptor protein synectin/GIPC to effect VEGFR-2 endocytosis and like the sv/sv mouse mice lacking synectin/GIPC exhibit impaired arterial development [Lanahan et al. 2010). To investigate whether the observed cardiomyopathy is due to Myo6 function in this process, we examined the cardiac expression of the fibrosis marker fibronectin (Fig. 14A, B) and VEC ultrastructure (Fig 14C, D) of the synectin/GIPC −/− mouse. No fibrosis was observed and cardiac VEC ultrastructure was normal, although as depicted in the examples shown in Fig. 14, many of the GIPC −/− VECs do have increased numbers of surface caveolae and cytoplasmic vesicles compared to the controls.
Fig. 14. Loss of synectin/GIPC function does not result in ventricular fibrosis or altered VEC ultrastructural organization.

(A, B) Localization of fibronectin (green) and F-actin (red) in WT (A) and synectin/GIPC −/− heart. (C, D). TEM of capillaries in WT (A) and synectin/GIPC −/− heart. Note the absence of fibrotic regions and normal VEC ultrastructure in the synectin/GIPC −/− although as shown here there are often increased numbers of surface caveolae and cytoplasmic vesicles. Bar A, B: 100 μm. Bar C, D: 500 nm
DISCUSSION
The results presented here demonstrate that cardiomyopathy associated with loss of Myo6 function is due to defects in cardiac VEC function since it is predominantly expressed in VECs and not myocytes. It is likely that the observed cardiac fibrotic tissue damage is due to impaired nutrient transport into the heart tissue from the vascular lumen and/or movement of metabolic wastes from the heart tissue into the vascular lumen. Consistent with this conclusion, the observed cardiac defects are strikingly similar to those observed in the caveolin 1 −/− mouse [Cohen et al. 2003]. There are three, not mutually exclusive, cellular mechanisms that could contribute to the observed tissue damage observed in both heart and lung. The first is that arterial vascular bed volume, resulting from impaired VEGFR-2 trafficking, leads to diminished vascular-tissue exchange of nutrients and toxins. The second is that there is direct inhibition of caveolin-mediated nutrient/toxin exchange in both the heart and lung. The build-up of metabolic wastes could lead to muscle cell death and subsequent interstitial “wound healing” by deposition of ECM by fibroblasts. The third is that previous studies have reported the association of Myo6 with the sarcoplasmic reticulum (SR) of the cardiac myocyte in both adult heart [Karolczak et al. 2013] and smooth endoplasmic reticulum in differentiating cardiac myocytes [Karolczak et al. 2015]. Although our localization studies indicate that Myo6 is predominantly expressed in VECs, this third possibility must be considered, as discussed below.
Two lines of evidence presented here argue against a predominant role of reduced arterial bed volume. First, Myo6 is not expressed in P1 renal VECs, thus it cannot be playing a major role at least at the time elaboration of the renal vasculature occurs during perinatal development. Second, and more compelling, is the observation that the synectin/GIPC −/− mouse does not exhibit sv/sv like cardiac defects. Presumably synectin/GIPC does not play an essential role in whatever loss of Myo6 function results in cardiac and pulmonary defects. This is not unreasonable given the multitude of functions uncovered for Myo6 that my not involve synectin/GIPC. Moreover, the vasculature-associated physiology of the sv/sv kidney, the VECs of which do not express Myo6, is essentially normal [Gotoh et al. 2010] despite the presumed reduction in renal vascular bed volume. On the other hand, VEGF and VEGFR-2 are also expressed in the adult, including in heart and lung [Maharaj and D’Amore 2007; Maharaj et al. 2006]. However, except for angiogenic functions of VEGF and its receptor functions in response to tissue damage and during vascularization of tumors, the roles of VEGF signaling in the adult are not well understood [Maharaj and D’Amore 2007]. Possible functions include maintaining vascularized tissue integrity/homeostasis (reviewed in [Luo et al. 2011]). For example, in the lung, inhibition of VEGFR-2 induces alveolar apoptosis and enlargement of airspaces [Kasahara et al. 2000; Maharaj and D’Amore 2007] which are consistent with the observations in sv/sv lung (Fig. 9). Inhibition of VEGF signaling in adult mice leads to variable, organ dependent regression of the vasculature, although this is not the case in the heart [Kamba et al. 2006].
As noted above the ventricular cardiomyopathy could also result from a direct loss of Myo6 function in caveolar-mediated nutrient/metabolite exchange. The observed accumulation of cytoplasmic vesicles in cardiac VECs could be due to defects in caveolar-mediated membrane trafficking. For example, the accumulation of cytoplasmic vesicles could be due to endocytic deficiencies in transport of caveolar derived vesicles [Tagawa et al. 2005], defective exocytic delivery of newly assembled caveolar membranes from the Golgi [Kiss 2012; Parton and Simons 2007] or defects in formation of transcellular vesiculo-vacuolar organelles (VVOs) [Dvorak and Feng 2001] given their similar diameter to surface caveolae. On the other hand, a role for Myo6 in caveolar function is not consistent with studies of caveolae in sv/sv fibroblasts. These cells exhibit normal caveolar profiles and VVOs which are “ectopically” recruited for endocytic uptake of the transferrin receptor, which in WT fibroblasts is internalized by clathrin-mediated endocytosis [Puri 2009]. Direct analysis of caveolar function, through studies using primary cultures of cardiac VECs will be required to distinguish between the two hypotheses enumerated here. For example, future studies could examine the localization of the albumin receptor, gp60 and endocytic uptake of albumin by sv/sv and WT (or sv/+) cardiac VECs [Vogel et al. 2001].
A third, VEC independent mechanism for at least the cardiac defects in sv/sv mice is that they are a consequence of loss of Myo6 function in the myocyte, where it has been reported to be associated with the SR. For example, defects in Ca2+ regulation by the SR could certainly lead to cardiac muscle damage. One as yet unresolved issue in this regard is that while there is Myo6 staining in myocytes that is above background levels observed in sv/sv heart, it is diffuse and not present in striated patterns observed by [Karolczak et al. 2013]. It is possible that the myocyte epitopes recognized by the antibody used in those studies are masked in our preparations. Alternatively, the differing observations could result from differences in fixation and/or staining protocols.
Supplementary Material
Acknowledgments
The authors thank Brian West, Department of Pathology, Yale School of Medicine for help with the histology evaluation and micrographs. This work was supported by NIH grant DK-25387 to MSM.
References
- Ahmed ZM, Morell RJ, Riazuddin S, Gropman A, Shaukat S, Ahmad MM, Mohiddin SA, Fananapazir L, Caruso RC, Husnain T, et al. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am J Hum Genet. 2003;72(5):1315–22. doi: 10.1086/375122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameen N, Apodaca G. Defective CFTR apical endocytosis and enterocyte brush border in myosin VI-deficient mice. Traffic. 2007;8(8):998–1006. doi: 10.1111/j.1600-0854.2007.00587.x. [DOI] [PubMed] [Google Scholar]
- Avraham KB, Hasson T, Steel KP, Kingsley DM, Russell LB, Mooseker MS, Copeland NG, Jenkins NA. The mouse Snell’s waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nat Genet. 1995;11(4):369–75. doi: 10.1038/ng1295-369. [DOI] [PubMed] [Google Scholar]
- Buss F, Kendrick-Jones J. Multifunctional myosin VI has a multitude of cargoes. Proc Natl Acad Sci U S A. 2011;108(15):5927–8. doi: 10.1073/pnas.1103086108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Collen D. Transgenic mouse models in angiogenesis and cardiovascular disease. J Pathol. 2000;190(3):387–405. doi: 10.1002/(SICI)1096-9896(200002)190:3<387::AID-PATH595>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Chibalina MV, Puri C, Kendrick-Jones J, Buss F. Potential roles of myosin VI in cell motility. Biochem Soc Trans. 2009;37(Pt 5):966–70. doi: 10.1042/BST0370966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AW, Park DS, Woodman SE, Williams TM, Chandra M, Shirani J, Pereira de Souza A, Kitsis RN, Russell RG, Weiss LM, et al. Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am J Physiol Cell Physiol. 2003;284(2):C457–74. doi: 10.1152/ajpcell.00380.2002. [DOI] [PubMed] [Google Scholar]
- Collaco A, Jakab R, Hegan P, Mooseker M, Ameen N. Alpha-AP-2 directs myosin VI-dependent endocytosis of cystic fibrosis transmembrane conductance regulator chloride channels in the intestine. J Biol Chem. 2010;285(22):17177–87. doi: 10.1074/jbc.M110.127613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak AM, Feng D. The vesiculo-vacuolar organelle (VVO). A new endothelial cell permeability organelle. J Histochem Cytochem. 2001;49(4):419–32. doi: 10.1177/002215540104900401. [DOI] [PubMed] [Google Scholar]
- Gotoh N, Yan Q, Du Z, Biemesderfer D, Kashgarian M, Mooseker MS, Wang T. Altered renal proximal tubular endocytosis and histology in mice lacking myosin-VI. Cytoskeleton (Hoboken) 2010;67(3):178–92. doi: 10.1002/cm.20435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson T, Gillespie PG, Garcia JA, MacDonald RB, Zhao Y, Yee AG, Mooseker MS, Corey DP. Unconventional myosins in inner-ear sensory epithelia. J Cell Biol. 1997;137(6):1287–307. doi: 10.1083/jcb.137.6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson T, Mooseker MS. Porcine myosin-VI: characterization of a new mammalian unconventional myosin. J Cell Biol. 1994;127(2):425–40. doi: 10.1083/jcb.127.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegan PS, Giral H, Levi M, Mooseker MS. Myosin VI is required for maintenance of brush border structure, composition, and membrane trafficking functions in the intestinal epithelial cell. Cytoskeleton (Hoboken) 2012;69(4):235–51. doi: 10.1002/cm.21018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR, Norberg SM, O’Brien SM, Davis RB, Gowen LC, et al. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol. 2006;290(2):H560–76. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- Karolczak J, Pavlyk I, Majewski L, Sobczak M, Niewiadomski P, Rzhepetskyy Y, Sikorska A, Nowak N, Pomorski P, Proszynski T, et al. Involvement of unconventional myosin VI in myoblast function and myotube formation. Histochem Cell Biol. 2015;144(1):21–38. doi: 10.1007/s00418-015-1322-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolczak J, Sobczak M, Majewski L, Yeghiazaryan M, Jakubiec-Puka A, Ehler E, Slawinska U, Wilczynski GM, Redowicz MJ. Myosin VI in skeletal muscle: its localization in the sarcoplasmic reticulum, neuromuscular junction and muscle nuclei. Histochem Cell Biol. 2013;139(6):873–85. doi: 10.1007/s00418-012-1070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106(11):1311–9. doi: 10.1172/JCI10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss AL. Caveolae and the regulation of endocytosis. Adv Exp Med Biol. 2012;729:14–28. doi: 10.1007/978-1-4614-1222-9_2. [DOI] [PubMed] [Google Scholar]
- Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, Carmeliet P, Simons M. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev Cell. 2010;18(5):713–24. doi: 10.1016/j.devcel.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Xiong Y, Han X, Lu Y. VEGF non-angiogenic functions in adult organ homeostasis: therapeutic implications. J Mol Med (Berl) 2011;89(7):635–45. doi: 10.1007/s00109-011-0739-1. [DOI] [PubMed] [Google Scholar]
- Maharaj AS, D’Amore PA. Roles for VEGF in the adult. Microvasc Res. 2007;74(2–3):100–13. doi: 10.1016/j.mvr.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharaj AS, Saint-Geniez M, Maldonado AE, D’Amore PA. Vascular endothelial growth factor localization in the adult. Am J Pathol. 2006;168(2):639–48. doi: 10.2353/ajpath.2006.050834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston SB. How do mutations in contractile proteins cause the primary familial cardiomyopathies? J Cardiovasc Transl Res. 2011;4(3):245–55. doi: 10.1007/s12265-011-9266-2. [DOI] [PubMed] [Google Scholar]
- Melchionda S, Ahituv N, Bisceglia L, Sobe T, Glaser F, Rabionet R, Arbones ML, Notarangelo A, Di Iorio E, Carella M, et al. MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am J Hum Genet. 2001;69(3):635–40. doi: 10.1086/323156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohiddin SA, Ahmed ZM, Griffith AJ, Tripodi D, Friedman TB, Fananapazir L, Morell RJ. Novel association of hypertrophic cardiomyopathy, sensorineural deafness, and a mutation in unconventional myosin VI (MYO6) J Med Genet. 2004;41(4):309–14. doi: 10.1136/jmg.2003.011973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooseker MS, Tilney LG. Organization of an actin filament-membrane complex. Filament polarity and membrane attachment in the microvilli of intestinal epithelial cells. J Cell Biol. 1975;67(3):725–43. doi: 10.1083/jcb.67.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil E, Wells DG, Mooseker MS. A role for myosin VI in postsynaptic structure and glutamate receptor endocytosis. J Cell Biol. 2005;168(2):329–38. doi: 10.1083/jcb.200410091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8(3):185–94. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- Puri C. Loss of Myosin VI No Insert Isoform (NoI) Induces a Defect in Clathrin-mediated Endocytosis and Leads to Caveolar Endocytosis of Transferrin Receptor. Journal of Biological Chemistry. 2009;284(50):34998–35014. doi: 10.1074/jbc.M109.012328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self T, Sobe T, Copeland NG, Jenkins NA, Avraham KB, Steel KP. Role of myosin VI in the differentiation of cochlear hair cells. Dev Biol. 1999;214(2):331–41. doi: 10.1006/dbio.1999.9424. [DOI] [PubMed] [Google Scholar]
- Tagawa A, Mezzacasa A, Hayer A, Longatti A, Pelkmans L, Helenius A. Assembly and trafficking of caveolar domains in the cell: caveolae as stable, cargo-triggered, vesicular transporters. J Cell Biol. 2005;170(5):769–79. doi: 10.1083/jcb.200506103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomanek RJ. Formation of the coronary vasculature during development. Angiogenesis. 2005;8(3):273–84. doi: 10.1007/s10456-005-9014-9. [DOI] [PubMed] [Google Scholar]
- Tumbarello DA, Kendrick-Jones J, Buss F. Myosin VI and its cargo adaptors - linking endocytosis and autophagy. J Cell Sci. 2013;126(Pt 12):2561–70. doi: 10.1242/jcs.095554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel SM, Minshall RD, Pilipovic M, Tiruppathi C, Malik AB. Albumin uptake and transcytosis in endothelial cells in vivo induced by albumin-binding protein. Am J Physiol Lung Cell Mol Physiol. 2001;281(6):L1512–22. doi: 10.1152/ajplung.2001.281.6.L1512. [DOI] [PubMed] [Google Scholar]
- Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011;364(17):1643–56. doi: 10.1056/NEJMra0902923. [DOI] [PubMed] [Google Scholar]
- Weis SM, Lindquist JN, Barnes LA, Lutu-Fuga KM, Cui J, Wood MR, Cheresh DA. Cooperation between VEGF and beta3 integrin during cardiac vascular development. Blood. 2007;109(5):1962–70. doi: 10.1182/blood-2005-10-038893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells AL, Lin AW, Chen LQ, Safer D, Cain SM, Hasson T, Carragher BO, Milligan RA, Sweeney HL. Myosin VI is an actin-based motor that moves backwards. Nature. 1999;401(6752):505–8. doi: 10.1038/46835. [DOI] [PubMed] [Google Scholar]
- Williams LH, Miller KA, Dahl HH, Manji SS. Characterization of a novel ENU-generated myosin VI mutant mouse strain with congenital deafness and vestibular dysfunction. Hear Res. 2013;299:53–62. doi: 10.1016/j.heares.2013.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.