

Abstract
Necrotic cells passively release HMGB1, which can stimulate TLR4 in an autocrine fashion to potentially initiate “sterile” inflammation that maintains different disease states. We have shown that prooxidants can induce NF-κB activation through TLR4 stimulation. We examined whether prooxidants enhance HMGB1-induced TLR4 signaling through NF-κB activation. We used LPS-EK as a specific agonist for TLR4, and PPC and SIN-1 as in situ sources for ROS. As model systems, we used HEK-Blue cells (stably transfected with mouse TLR4), RAW-Blue™ cells (derived from murine RAW 264.7 macrophages) and primary murine macrophages from TLR4-KO mice. Both HEK-Blue and RAW-Blue 264.7 cells express optimized secreted embryonic alkaline phosphatase (SEAP) reporter under the control of a promoter inducible by NF-κB. We treated cells with HMGB1 alone and/or in conjunction with prooxidants and/or inhibitors using SEAP release as a measure of TLR4 stimulation. HMGB1 alone and/or in conjunction with prooxidants increased TNFα and IL-6 released from TLR4-WT, but not from TLR4-KO macrophages. Pro-oxidants increased HMGB1 release, which we quantified by ELISA. We used both fluorescence microscopy imaging and flow cytometry to quantify the expression of intracellular ROS. TLR4-neutralizing antibody decreased prooxidant-induced HMGB1 release. Prooxidants promoted HMGB1-induced NF-κB activation as determined by increased release of SEAP and TNF-α, and accumulation of iROS. HMGB1 (Box A), anti-HMGB1 and anti-TLR4-neutralizing pAbs inhibited HMGB1-induced NF-κB activation, but HMGB1 (Box A) and anti-HMGB1 pAb had no effect on prooxidant-induced SEAP release. The present results confirm that prooxidants enhance proinflammatory effects of HMGB1 by activating NF-κB through TLR4 signaling.
Keywords: Toll-like receptor 4, Prooxidants, Recombinant high mobility group box 1 protein, NF-κB activation, sterile inflammation
INTRODUCTION
Reactive oxygen species (ROS) [with the term used here to also include reactive nitrogen species (RNS)] can interact with and cause damage to lipids, DNA, and other cellular macromolecules [1]. Oxidative nitrosative stress (ONS), induced by ROS, is implicated in the pathogenesis of many human disorders [2]. Oxidative stress can be a reflection of a disturbance in the balance between systemic production/catabolism of ROS and antioxidant defenses against these free radicals [3], which can arise from both normal cellular processes as well as environmental oxidants. Oxidative stress is an adaptation to changing cellular milieu, which can cause impaired reversible or irreversible cellular function or pathological gain of function [3].
Our understanding and appreciation of the link between oxidative stress and inflammatory pathways are still obscure. It was recently shown that Toll-like receptors (TLRs) could mediate the inflammatory responses associated with experimental ischemia and reperfusion injury, a model of oxidative stress [4]. ROS are implicated as key mediators in the mechanism(s) that may underlie the cross talk between TLRs and interactions between neutrophils (PMNs) and alveolar macrophages (AM) as well as endothelial cells (ECs) [5]. TLRs are a family of germ-line encoded receptors designated as pattern recognition receptors (PRRs), and are expressed on both the cell surface and in endosomes [6]. TLRs recognize and respond to distinct molecular patterns associated with microbial pathogens called pathogen-associated molecular patterns (PAMPs) [7]. However, some members of the TLR, notably TLR2, TLR4 and TLR9, respond to various endogenous damage-associated molecular patterns (DAMPs), including heat shock proteins and extracellular matrix components such as heparin sulfate, mammalian DNA and high mobility group box 1 (HMGB1). These endogenous DAMPs are released in conditions of cellular stress or injury with the potential to drive “sterile” inflammatory responses [8,9] to a self-perpetuating condition that can initiate and/or maintain disease states.
HMGB1, a ubiquitous non-histone and highly conserved DNA-binding nuclear protein, is an archetypal DAMP molecule. It is highly expressed in mammalian cells and is involved in the stabilization of nucleosomes, and facilitates gene transcription [10]. Recent models of “sterile” injury and chronic inflammation have shown that HMGB1 is specifically recognized by many cell surface receptors including TLR4 and TLR2 [11], the receptors for advanced glycation end product (RAGE) [12], and most recently by T-cell immunoglobulin and mucin domain 3 proteins [13]. HMGB1 can trigger signal transduction through a range of receptors, but TLR4 appears to be the primary receptor for HMGB1 in defining its cytokine-like and cytokine-inducing properties [14]. In addition to stimulating cells directly, HMGB1 can form immunostimulatory complexes with inflammatory mediators [15] and other endogenous and exogenous factors [16,17].
Most cells, including monocytes and macrophages, constitutively express HMGB1 mRNA and its protein under basal conditions. Inflammatory cells including activated monocytes and macrophages [18], mature dendritic cells [19] actively secrete HMGB1, whereas necrotic cells passively secrete it into their extracellular milieu [20]. In contrast, apoptotic cells retain HMGB1 in their nuclei, and thus do not activate inflammation [21]. HMGB1 enhances LPS-induced production of proinflammatory cytokines including interleukin 6 (IL-6) and TNF-α in vitro [22]. In damaged tissue, extracellular HMGB1 acts as a necrotic signal, which alerts the surrounding cells and the immune system. HMGB1 acts in synergy with LPS in vivo in the mouse experimental arthritis model by activating macrophages to secrete cytokines, which may enhance phosphorylation of mitogen activated protein kinase (MAPK) p38 that provides the basis for NF-κB activation [22].
Recently, we showed that pro-oxidants induce NF-κB activation through the TLR4 signaling pathway [23]. Based on these findings, we hypothesized that prooxidants could enhance HMGB1-induced NF-κB activation through TLR4 signaling, a component of the pathway of innate immunity against pathogenic bacteria. Thus, we examined in vitro a potential mechanism of the interactions of reduced recombinant mouse/human HMGB1 [disulfide HMGB1] [24] with prooxidants on NF-κB activation through TLR4- coupled stimulation. One of our primary aims was to define a potential contribution in vivo for interactions between extracellular HMGB1 (exHMGB1) and oxidants in the mechanism of initiation and maintenance of “sterile” inflammation that may play a role in many disease states.
We used two distinct agents namely potassium peroxychromate (PPC) and SIN -1, as primary sources of exogenous reactive oxygen and nitrogen species. We chose to use PPC and SIN-1 because, in the final analysis, both agents produce functionally and biologically similar reactive intermediates that can induce oxidative/nitrosative stress (ONS). However, PPC is more efficient in generating ROS compared with SIN-1 in the same time interval. Under physiological conditions, PPC can decay to release oxygen-centered free radicals such as superoxide anion, hydrogen peroxide, and hydroxyl radicals [25] as produced by activated phagocytes [26]. On the other hand, SIN-1 produces peroxynitrite anions [27], which decompose to generate hydroxyl radicals and nitrogen dioxide that can oxidize redox-sensitive protein and non-protein cysteines by orders of magnitude greater than hydrogen peroxide [28].
A major finding of this manuscript is that exogenous prooxidants can promote extracellular disulfide HMGB1-induced NF-κB activation through the TLR4 signaling pathway with the potential to initiate “sterile” inflammatory responses. Our data confirm that HMGB1/prooxidant-coupled TLR4 stimulation can induce NF-κB activation to increase intracellular ROS (iROS), which enhances ONS with a consequent release of inflammatory cytokines. Thus, therapeutic targeting of extracellular HMGB1 has the potential to provide health benefits that ameliorate ONS-mediated inflammatory disease states, especially in situations with release of excessive amounts of HMGB1 [29].
MATERIALS AND METHODS
Chemicals
HEK-Blue selection medium, selection antibiotic Zeocin, Quanti-Blue detection reagent (alkaline phosphatase detection medium), LPS from Escherichia coli (LPS-EK Ultrapure) and RAW-Blue™ cells are derived from the murine RAW 264.7 macrophages with chromosomal integration of a secreted embryonic alkaline phosphatase (SEAP) reporter construct inducible by NF-κB, were obtained from InvivoGen (San Diego, CA). 3-Morpholinylsydnoneimine chloride or Linsidomine chloride (SIN-1) was obtained from Cedarlane Inc (Burlington, NC). Low endotoxin, azide-free (LEAF) affinity purified rat IgG2a, κ-isotype anti-mouse TLR4 (CD284)/MD-2 complex pAb for TLR4 neutralization and kits for sandwich ELISA of human-specific TNF-α and IL-10 were purchased from BioLegend (San Diego, CA). CellROX® Deep Red and NucBlue® Live ReadyProbes™ reagents were obtained from Invitrogen. Parameter™ TBARS assay kit was purchased from R & D Systems Inc (Minneapolis, MN). Recombinant disulfide high mobility group box 1 (HMGB1) [or disulfide HMGB1] (LPS-free) [complete notation: HMGB1C23-C45C106h] [24], fully reduced HMGB1 (LPS-free), Box A portion of HMGB1 (human & mouse, LPS-free), HMGB1 ELISA kit, anti-HMGB1 Chicken IgY neutralizing pAb and chicken IgY (polyclonal, isotype control antibody) were obtained from IBL International Corp (Toronto, ON, Canada). We prepared and characterized PPC as previously published [23,26]. Both PPC and SIN-1 used in our assays were confirmed to be LPS-free.
Cell lines and culture conditions
We have used three different cell types as model cultures to characterize ROS/HMGB1/TLR4-coupled activation. We first used cells derived from the human embryonic kidney-293 (HEK-293) cell line. HEK-Blue Null1-v and HEK-Blue mTLR4 cells were purchased from InvivoGen (San Diego, CA). HEK-Blue Null1-v is the parental cell line of HEK-Blue mTLR4, which does not express mTLR4, whereas HEK-Blue mTLR4 cells are stably transfected to express mTLR4 gene at high levels with MD-2 and CD14 co-receptor genes involved in TLR4 recognition and presentation. In addition, both HEK-Blue cells stably express an optimized secreted alkaline phosphatase (SEAP) reporter gene under the control of a promoter inducible by NF-κB transcription factor. The level of SEAP protein released into the culture media was used as a measure of NF-κB activation through TLR4 stimulation with QUANTI-Blue™ detection reagent.
RAW-Blue™ cells were derived from the murine RAW 264.7 macrophages with chromosomal integration of a SEAP reporter construct inducible by NF-κB. RAW-Blue™ cells express many pattern-recognition receptors (PRRs), including all toll-like receptors (TLRs) except for TLR5, but with TLR4 and TLR8 expressed at the highest levels. However, TLR8 is endosomal and a different ligand activates it from TLR4. Thus, RAW-Blue™ cells are useful as TLR reporter cells for inducing NF-κB, with subsequent release of SEAP after treatment with appropriate TLR-specific ligands, including ROS for TLR4 [23]. Therefore, we used RAW-Blue™ cells with the same read-out metric as HEK-Blue mTLR4 cells to affirm that the synergistic/additive effects of oxidants with HMGB1 can also occur in cells that express multiple TLRs.
Both HEK and RAW-Blue cells were grown in a 37 °C, 100 % humidified incubator in Dulbecco’s Modified Eagle’s Medium (DMEM, 4.5 g of glucose/L) without pyruvate but supplemented with 2 mM L-glutamine, 10 % (v/v) fetal bovine serum (FBS), 50 units/ml penicillin, 50 μg/ml streptomycin and 100 μg/ml Normocin™. HEK-Blue-Null1-v, HEK-Blue mTLR4 and RAW-Blue cells were maintained in growth medium supplemented with Zeocin.
Isolation, characterization and treatment of peritoneal primary macrophages
We also used primary murine macrophages to further clarify and confirm the mechanism of the proposed signaling pathway in ROS/HMGB1-coupled NFκB activation through TLR4 stimulation. Primary murine peritoneal macrophage cultures were derived from mouse strain B6.B10ScN-Tlr4 lps-del/JthJ (Jackson Labs) with complete deletion of TLR4 gene (knock out)[TLR4-KO] [30] with a corresponding heterozygous wild-type (TLR4-WT) control strain C57BL/10ScSnJ. These mice were cared for and maintained as approved by UMKC-IACUC in accordance with NIH guidelines. We isolated and characterized primary peritoneal macrophages from TLR4-WT and TLR4-KO mice strains according to a standard published method [31].
Macrophages (5×105 cells/well) were seeded in 12-well plates containing DMEM/F-12 (500 μl/plate) with 10 % (v/v) fetal bovine serum (FBS), 10 mM L-glutamine, 100 IU/ml penicillin and 100μg/ml streptomycin. Cells were preincubated with LPS-free disulfide HMGB1 (1 μg/ml) for 30 min followed by treatment with either [PPC (5 μM) or SIN-1 (500 μM) in the continued presence of HMGB1 for 16 h. As a positive control for TLR4 activation, we also treated cells with LPS-EK (0.5μg/ml) alone for ~16 h. At the end of the incubation time, we collected cell supernatants for use to quantify murine TNF-α and IL-6 by ELISA.
Measurement of extracellular HMGB1 (exHMGB1) levels
HEK-Blue mTLR4 cells were stimulated overnight (~16 h) with varying concentrations of PPC and SIN-1. We determined the extent by which the inhibition of TLR4 could affect oxidant-induced HMGB1 release. We preincubated HEK-Blue mTLR4 cells for 2 h with TLR4 (CD284)/MD2 complex polyclonal antibody (pAb) followed by stimulation overnight with the highest concentration of each prooxidant used in the present study in the continued presence of the pAb. Cells were also stimulated overnight with LPS-EK as a positive control, but with no prior treatment with TLR4 pAb. Levels of HMGB1 in the conditioned media were measured using HMGB1 ELISA kit according to the manufacturer’s instructions.
Quanti-Blue® SEAP reporter assay
HEK-Blue-Null1-v and HEK-Blue mTLR4 cells (2 ×104) were plated in 96-well plates and grown to 70 % confluence. Cells were then pretreated with HMGB1 (BoxA) for 30 min, anti-HMGB1 neutralizing pAb or TLR4 pAb for 2 h, followed by stimulation with HMGB1, PPC or SIN-1 for 16 h in the continued presence of the inhibitors. Aliquots of the culture medium (20 μl) were removed and added to new 96-well plates containing 180 μl of pre-warmed Quanti-Blue™, a SEAP colorimetric detection medium, as per manufacturer’s instructions. Color was allowed to develop for 1 h, and absorbance was read at 650 nm in a Bio-Tek ® microplate reader (Burlington, VT).
We also determined the extent to which varying concentrations of HMGB1 would affect SEAP released into the media. In each case, cells were also stimulated overnight with LPS-EK as a positive control. Furthermore, we verified whether prooxidants would synergize with HMGB1 to enhance the SEAP released into the media. We pretreated cells with HMGB1 for 30 min followed by further treatment overnight with PPC or SIN-1 alone or in combination in the continued presence of HMGB1.
Measurement of intracellular ROS (iROS) accumulation by fluorescence microscopy
HEK-Blue mTLR4 cells were treated with culture medium (control), HMGB1, PPC and SIN-1 alone or in combination in Lab-Tek II 8-well slides for ~16h. We determined treatment-mediated accumulation of intracellular ROS (iROS) by staining with CellROX® Deep Red reagent according to the manufacturer’s instructions. Briefly, cells were incubated with CellRox® and NucBlue live cell stain (nuclear) for 30 min at 37 °C followed by washes with PBS and fixation with 4% paraformaldehyde for 15 min. Following subsequent washes with PBS, images were acquired using fluorescence microscope (Axiovert 200M; Zeiss) at excitation and emission wavelengths (λs) of ~644/655 nm for CellROX® Deep Red reagent and 405/410-550 nm for NucBlue®. Finally, acquired images were merged and composite histograms were obtained and analyzed [32] with ImageJ (V1.44, http://rsbweb.nih.gov/ij/) software.
Quantification of iROS by flow cytometry
To confirm the accuracy of the fluorescence microscopy data, we also used flow cytometer to determine the fluorescence intensity of iROS with the same dye. HEK-Blue mTLR4 cells were grown in 6-well dishes to 80 % confluency. Cells were treated with culture medium (control), HMGB1, PPC and SIN-1 alone or in combination for ~16h after which cells were incubated with CellROX® Deep Red reagent (1 μM) for 30 min at 37 °C followed by subsequent washes (2X) in PBS. Washed cells were scraped and resuspended in warm PBS for flow cytometer.
Acquisition and analysis of flow cytometric data were conducted on FACSCanto™ II flow cytometer (BD Biosciences, San Jose, CA). Electronic gating was used to exclude doublets and subcellular debris with a forward scatter threshold set at 500. The fluorescence intensity corresponding to iROS-labeled CellROX® Deep Red reagent was determined using Allophycocyanin (APC) filter at excitation/emission λs of ~ 645/660 nm. Unstained cells were used as negative controls for iROS. For each parameter investigated, at least 104 events (cells) were analyzed /sample. The fluorescence intensities as logarithmically amplified data were compared between different treatments.
Thiobarbituric Acid Reactive Substances (TBARS) assay
HEK-Blue mTLR4 cells were plated in 6-well plates at a density of 5×105 cells/well. Cells were allowed to reach 70% confluence, following which they were pre-incubated alone with HMBG1 for 1 h, or treated in combination with either, PPC and SIN-1. After over night (~16 h) stimulation in the continued presence of stimulants, cell were scraped and lysed in 150-μl lysis buffer (Mammalian cell-PE-LB™) supplemented with protease inhibitors. The lysates were frozen at -20° C for 4 h, then thawed once and centrifuged at 10,000-× g for 10 min.
In the absence of oxidative stress, free malonyldialdehyde (MDA), an oxidative degradation product of lipids, is typically low and not detectable. Thus, sample supernatants require acid treatment of proteins, and breakdown of peroxides by heat and acid to facilitate color development in the thiobarbituric acid reactive substances (TBARS) assay. MDA forms a 1:2 adduct with thiobarbituric acid (TBA). The optical density (OD) of MDA-TBA adduct formed in sample supernatants was quantified colorimetrically using a Bio-Tek® microplate reader set at 532 nm. The adducts were analyzed with a Parameter™ TBARS assay kit according to manufacturer’s instructions. The TBARS levels in unknown samples were determined from MDA equivalence standard carried out simultaneously in the assay with unknown samples. Total protein concentrations in each sample used for normalization were determined by BCA protein assay kit (Pierce).
Quantitation of TNF-α, IL-10 and IL-6 levels
HEK-Blue mTLR4 cells were preincubated with HMGB1 (Box A) fragment or TLR4 (CD284)/MD2 complex pAb for 2h followed by stimulation for 16 h with HMGB1, PPC or SIN-1 singly, and/or in combination [HMGB1+PPC], or HMGB1+SIN-1]. We also stimulated cells for 16 h with LPS-EK as a positive control, but without prior treatment with HMGB1 (BoxA) or TLR4 pAb. The levels of TNF-α and IL-10 released into the conditioned media were measured using human TNF-α and IL-10 ELISA kits as per manufacturer’s instructions.
In addition, following different treatments for primary peritoneal macrophages derived from TLR4-WT and TLR4-KO mice, we measured levels of TNF-α and IL-6 released into the conditioned media with TNF-α and IL-6 ELISA kits as per manufacturer’s instructions.
Statistical Analysis
We used Statistical Package for the Social Sciences (SPSS) to perform data analysis in all experiments. Data are presented as mean ± SEM from at least 3 – 7 independent experiments carried out in duplicate, where applicable, and analyzed by 1- or 2-way analysis of variance (ANOVA) followed by Tukey’s post hoc tests. Significance was assigned at p ≤ 0.05 %.
RESULTS
PPC and SIN-1increase HMGB1 release in HEK-Blue mTLR4 cells
Treatment with 1 μg/ml LPS-EK (a TLR4-specific agonist) overnight increased HMGB1 release by a mean of 16.8-fold over untreated control cells. Similarly, overnight treatment with 0.1, 1.0 and 5 μM of PPC increased HMGB1 release by a mean of 2.3- and 4.3-fold, respectively, over the untreated control cells incubated under the same conditions. Treatment with 0.1, 0.5 and 1 mM of SIN-1 increased HMGB1 release by a mean of 2.3- and 3.6-fold, respectively. Furthermore, preincubation of cells for 2 h with TLR4 neutralizing pAb or HMGB1 neutralizing pAb prior to PPC (5 μM) or SIN-1 (0.5 mM) treatment decreased HMGB1 release by 47.8 and 53.3 %, respectively. Neither PPC (0.1 μM) nor SIN-1 (0.1 mM) had a discernable effect on HMGB1 release (Fig. 1).
Fig. 1. Effect of pro-oxidants on disulfide HMGB1 isoform release in HEK-Blue mTLR4 cells.
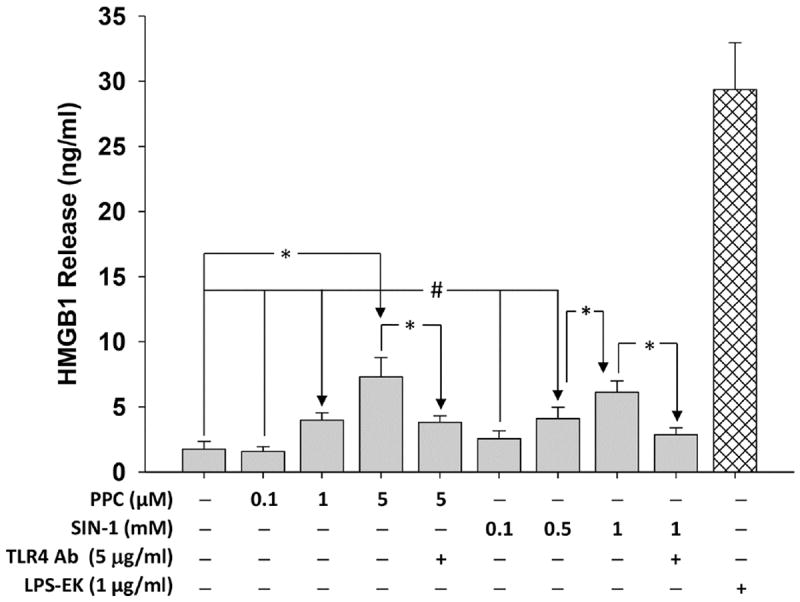
Cells were treated overnight (~16 h) with varying concentrations of PPC or SIN-1. HMGB1 released into the conditioned media was determined using HMGB1 ELISA kit according to the manufacturer’s instructions. The effect of LPS-EK on HMGB1 release was used as positive control. The data represent 3 independent experiments carried out in duplicate, #p ≤ 0.05 and *p ≤ 0.01.
HMGB1 increases TLR4 signaling in HEK-Blue mTLR4 cells
Overnight treatment with 1 μg/ml of LPSEK for 16 h induced SEAP release by a mean of 10-fold over control cells under the same treatment conditions. However, treatment with 0.001, 0.01, 0.1, or 1 μg/ml of HMGB1 overnight induced SEAP release by a mean of 1.2-, 1.8-, 2.5- and 3.5-fold over untreated control cells incubated under the same conditions (Fig. 2). The data indicates that SEAP release is HMGB1 concentration dependence.
Fig. 2. Effect of disulfide HMGB1 isoform on secreted embryonic alkaline phosphatase (SEAP) release in HEK-Blue mTLR4 cells.
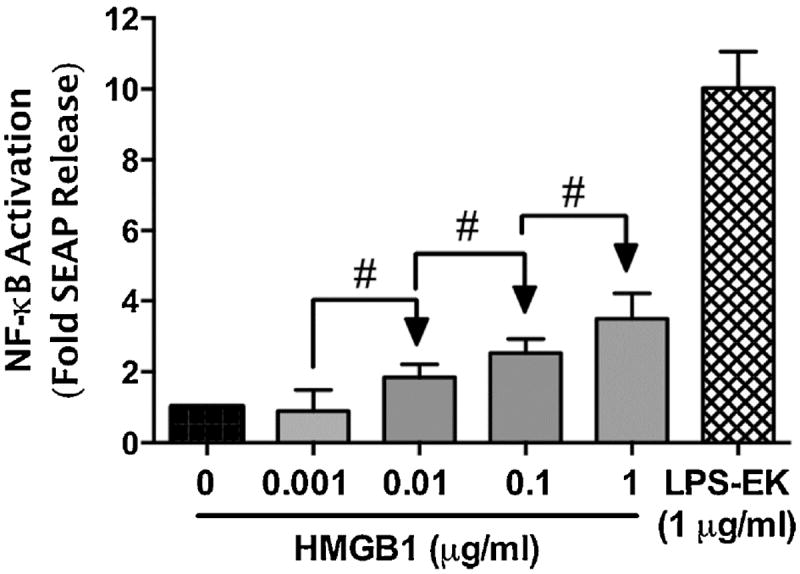
Cells were treated overnight with varying concentrations of HMGB1. SEAP released into the conditioned medium was determined using Quanti-Blue, and the absorbance was read at 650 nm. The effect of LPS-EK on SEAP release was used as positive control. The data represent 5 independent experiments carried out in duplicate. #p ≤ 0.05
HMGB1 enhances prooxidant-mediated TLR4 signaling in HEK-Blue mTLR4 cells
Treatment overnight with HMGB1 (1 μg/ml), PPC (1 μM) or SIN-1 (500 μM) increased SEAP release by a mean of 3.6-, 4.5- and 2.9-fold, respectively, over untreated control cells (Fig. 3). However, co-treatment of PPC or SIN-1 with HMGB1 (1 μg/ml) increased prooxidant-induced SEAP release by 66 and 47 %, respectively. The data confirm that HMGB1 acts additively with prooxidants to increase NFκB activation through TLR4 signaling pathway.
Fig. 3. Effect of co-treatment of disulfide HMGB1 and prooxidants on secreted embryonic alkaline phosphatase (SEAP) release in HEK-Blue mTLR4 cells.
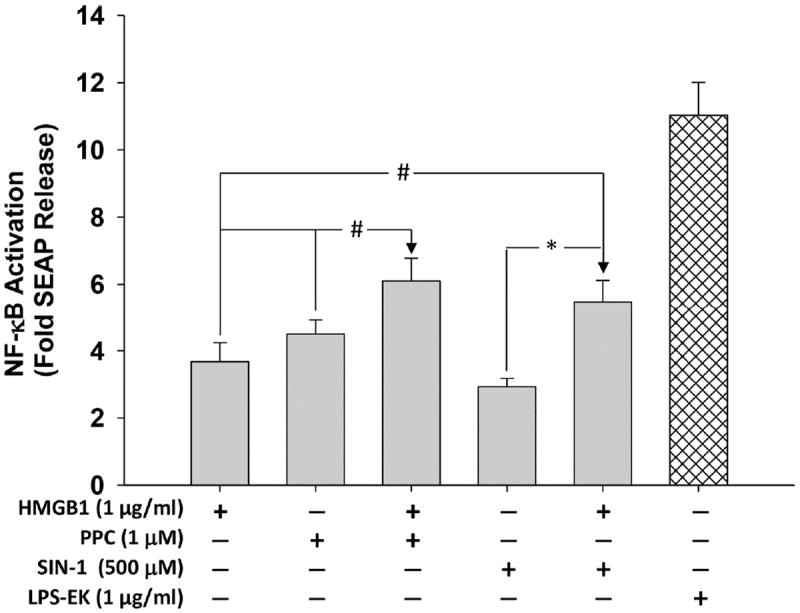
Cells were treated with HMGB1, PPC and SIN-1 alone and/or in combination for 16 h. SEAP released into the conditioned medium was determined using Quanti-Blue according to the manufacturer’s instructions, and the absorbance was read at 650 nm. The effect of LPS-EK on SEAP release was used as positive control. The data represent 5 independent experiments carried out in duplicate. #p ≤ 0.05 and *p ≤ 0.01
Neither HMGB1 nor any of the pro-oxidants (PPC and SIN-1) alone or in combination had any significant effect on SEAP release from HEK-Blue null1-v cells [data not shown]. The data not only demonstrated the inactivity of the reporter gene insert in the null 1-v cells, but also confirmed that SEAP release was due to activation of functional TLR4 expressed in HEK-Blue mTLR4 cells.
Specificity of HMGB1 and prooxidants in NF-κB activation through TLR4 stimulation
We used pharmacological approaches to further clarify that TLR4 stimulation by HMGB1 or prooxidants would play a role in SEAP release through NF-κB activation (Fig. 4). HMGB1, PPC or SIN-1 treatment alone increased SEAP release by about 3.5-, 4- and 2.7-fold, respectively. TLR4 neutralizing antibody decreased HMGB1-, PPC- and SIN-1-induced SEAP release by 42.8, 66.5 and 60.5 %, respectively, which further indicates that HMGB1 acts through TLR4 like the prooxidants. However, HMGB1 neutralizing pAb decreased HMGB1-mediated SEAP release with no effect on either PPC- or SIN-1-induced release. The data suggest that even though HMGB1 and prooxidants act through TLR4, but the mechanism of interactions may be quite different. Chicken isotype-specific IgY had no effect (data not shown), which confirmed the specificity of HMGB1 pAb in the treatment. Thus, only preincubation with TLR4 neutralizing pAb, but not with HMGB1 neutralizing pAb or with HMGB1 (BoxA), inhibited PPCand SIN-1-induced SEAP release, which confirm HMGB1 pAb and HMGB1 (BoxA) as a specific HMGB1 blockers on TLR4 (Fig. 4).
Fig. 4. Effect of TLR4 neutralizing polyclonal antibody (TLR4 pAb), HMGB1 pAb and HMGB1 (Box A) on SEAP release in HEK-Blue mTLR4.
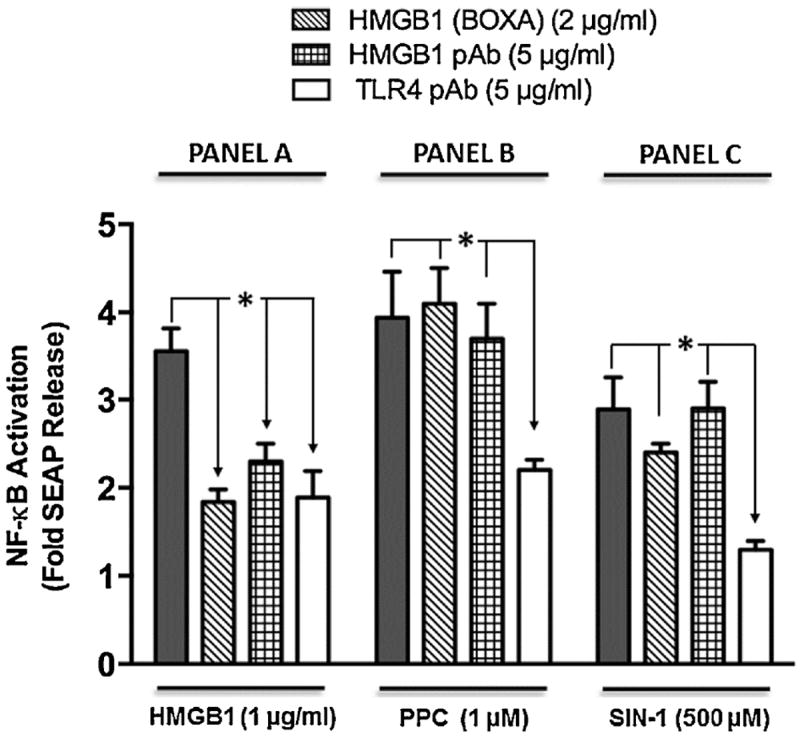
Cells were pre-incubated with TLR4 neutralizing antibody, HMGB pAb or HMGB1 (BoxA) for 2 h followed by treatment overnight with HMGB1 (Panel A), PPC (Panel B) or SIN-1 (Panel C) in the continued presence of TLR4 pAb, HMGB1 pAb or HMGB1 (BoxA). SEAP released into the conditioned medium was determined using Quanti-Blue™ with absorbance read at 650 nm. The data represent 3 - 7 independent experiments that were carried out in duplicate. +p ≤ 0.001
HMGB1 promotes prooxidant-induced intracellular ROS (iROS) accumulation
Treatment of HEK-Blue mTLR4 cells with HMGB1, PPC or SIN-1 increased iROS accumulation as fluorescence intensity by a mean of 4.7-, 6- and 4.2-fold, respectively, over untreated control cells. Co-treatment of these cells with PPC or SIN-1 in continued presence of HMGB1 increased fluorescence intensity by 93 and 70 %, respectively (Fig. 5). The data suggest that HMGB1 may act additively with prooxidants to increase intracellular oxidative stress.
Fig. 5. Immunofluorescence representation of the levels of intracellular ROS (iROS) accumulated following stimulation of HEK-Blue mTLR4 cells.
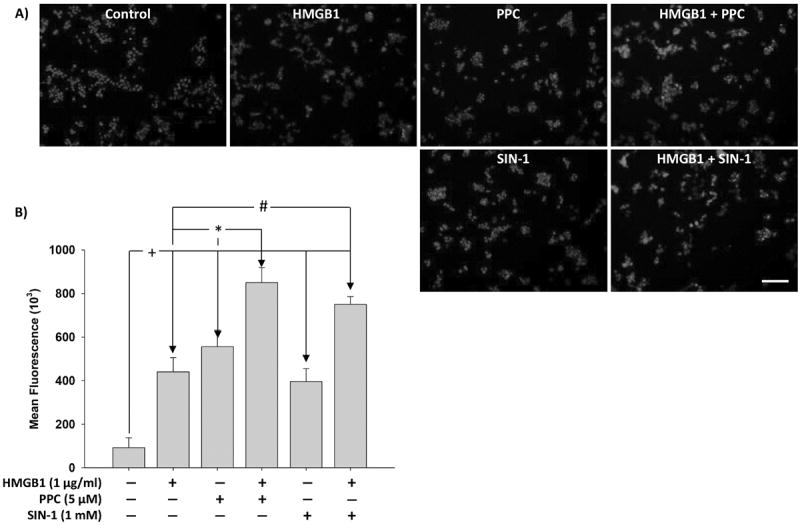
Cells were treated overnight with HMGB1, PPC or SIN-1 alone and/or in combination. Cells were then stained with CellRox® Deep Red Reagent and NucBlue® Live ReadyProbes® Reagent (to counter stain cellular nuclei) for 30 min at 37 °C followed by subsequent PBS washes and fixation with 4 % paraformaldehyde for 15 min. After subsequent rinses with PBS, images were acquired using fluorescence microscope and analyzed using the ImageJ® software. (Fig. 5A) are merged representative pictures with scale bar = 50 μm. (Fig. 5B) represents semiquantitative histograms generated using ImageJ® software. The data represent 3 independent experiments carried out in duplicate. #p ≤ 0.05, *p ≤ 0.01 and +p ≤ 0.001.
With the flow cytometer, we observed significant differences only in increased iROS in HMGB1 alone when compared with [HMGB1+ PPC] or [HMGB1+ SIN-1] treatments (Figs. 6A & 6B). The mean flow cytometric histograms (Fig. 6B) indicate that all treatments produced enhanced fluorescence intensity when compared with the untreated control group. Nonetheless, in all treatment groups, only PPC and [HMGB1 + PPC] treatments produced the most intense iROS fluorescent intensities.
Fig. 6. Flow cytometric analysis of intracellular ROS (iROS) accumulation in HEK-Blue mTLR4 cells following different treatment.
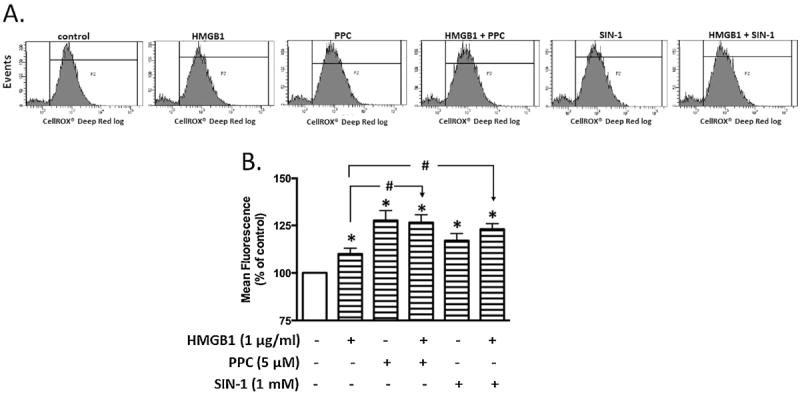
Cells were treated overnight with HMGB1, PPC or SIN-1 alone and/or in combination. Following the removal of the incubation media, cells were incubated with CellRox® Deep Red Reagent for 30 min. Cells were rinsed with PBS, then scraped and resuspended into warm PBS according to the manufacturer’s instructions for flow cytometric analysis. Representative tracings (Fig 6A) following treatments with media (control), disulfide HMGB1, PPC or SIN-1 alone or in combination i.e., [HMGB + PPC] or [HMGB1 + SIN-1]. Quantitative analyses of tracings from (Fig. 6A) show changes in fluorescence intensity after different treatments (Fig. 6B). The data in Fig. 6B represent multiple independent experiments conducted in duplicate (n = 5-6; #p ≤ 0.05; *p ≤ 0.05).
HMGB1 and prooxidants increase lipid peroxides quantified as TBARS
We also used TBARS assay to evaluate the combinatorial effects of ROS as lipid peroxides and/or protein carbonyl adducts. Thus TBARS assay cannot accurately dissect out the effects of treatment-induced iROS levels. However, treatment of HEK-Blue mTLR4 cells overnight with HMGB1, PPC and SIN-1 individually increased MDA produced by 2-, 4- and 3-fold, respectively, compared with untreated control cells under the same conditions (Fig. 7). The level of MDA produced by [HMGB1 + SIN-1] combination was unchanged compared to SIN-1 treatment alone. However, [HMGB1 + PPC] combination treatment increased MDA level by 5-fold compared to untreated control cells, which was only 1-fold higher compared to PPC-treatment alone.
Fig. 7. Levels of malonyldialdehyde (MDA) quantified as thiobarbituric acid reactive substances (TBARS) following stimulation of HEK-Blue mTLR4 cells.
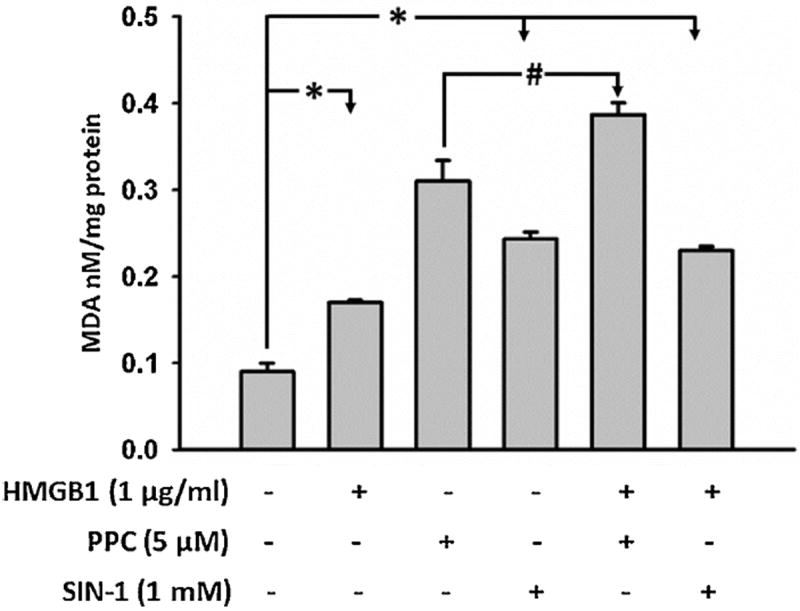
Cells were stimulated overnight with HMGB1, PPC or SIN-1 alone and/or in combination in continuous presence of the stimulators. At the end of incubation, the conditioned media was removed and cells rinsed with phosphate buffered saline (PBS). Attached cells were scraped, lysed, freeze thawed once, and centrifuged. The lysates were used to quantify MDA as TBARS according to the manufacturer’s instructions. The data represent 3 independent experiments carried out in duplicate (#p = 0.5, *p ≤ 0.01).
Effects of HMGB1 and prooxidants on TNF-α and IL-10 release in HEK-Blue-mTLR4 cells
To affirm the biological relevance of ROS/HMGB1 coupling, we measured the effect of TLR4 activation on TNF-α and IL-10 release in addition to SEAP release. Treatment with HMGB1 induced TNF-α and IL-10 release into the culture media by a mean of 6.3- and 4.9-fold, respectively, over untreated control cells (Fig. 8A & 8B). Treatment with PPC alone increased TNF-α and IL-10 release by a mean of 9.9- and 5.8- fold, respectively, whereas SIN-1 increased TNF-α and IL-10 release by a mean of 5- and 2.8-fold, respectively.
Fig. 8. Effect of disulfide HMGB1 alone and /or in combination with prooxidants on TNF-α and IL-10 release in HEK-Blue mTLR4 cells.
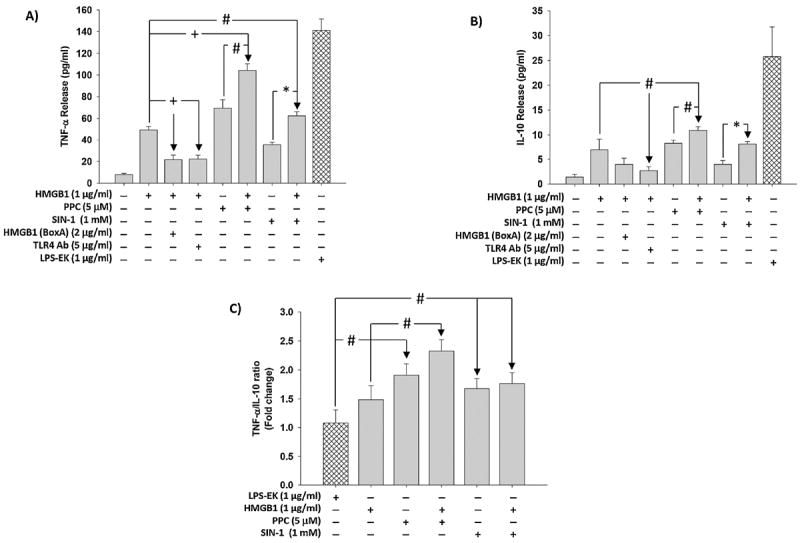
Cells were pre-incubated with HMGB1 (BoxA) or TLR4 pAb followed by stimulation overnight with HMGB1 in continued presence of the inhibitors. In the same set of experiments, cells were also incubated overnight with HMGB1, PPC or SIN-1 alone and/or in combination. Levels of TNF-α (Fig. 8A) and IL-10 (Fig. 8B) released into the culture medium were quantified using the ELISA kits of the relevant cytokine according to manufacturer’s instructions. The ratios of TNF-α to IL-10 (Fig. 8C) were calculated. The effect of LPS-EK on TNF-α and IL-10 release was used as positive control. The data represent 3 independent experiments. (#p ≤ 0.05, *p ≤ 0.01 and +p ≤ 0.001).
In addition, co-treatment of PPC with HMGB1 increased TNF-α and IL-10 release by 108.8 and 57 %, respectively, whereas co-treatment of SIN-1 with HMGB1 increased TNF-α and IL-10 release by 25 and 16.9 %, respectively. However, pretreatment with TLR4 neutralizing pAb and HMGB1 (BoxA) inhibited HMGB1-induced TNF-α release by 56.7 and 55.5 %, respectively, and decreased IL-10 release by 42.4 and 61 %, respectively (Fig. 8A & 8B).
When we expressed the levels of TNF-α and IL-10 released into the media after treatment as TNF-α/IL-10 ratios (Fig. 8C), a different picture emerged. We first set the TNF-α/IL-10 ratios following treatment with LPS-EK to 1.0 to which all TNF-α/IL-10 ratios after different treatments could be compared. The ratios are suggestive of the level of maintenance of proinflammatory compared with antiinflammatory processes post treatment. We compared the ratios between and within treatments with HMGB1, PPC, or SIN-1 singly and /or in combination. [HMGB1 +PPC] increased TNF-α/IL-10 ratio by 33 % compared with HMGB1 treatment alone, whereas [HMGB1 + SIN-1] had a minimal effect (< 5 %). However, the ratios were significantly increased in all treatments when compared with the ratios obtained after LPS-EK treatment alone. The values of these ratios are suggestive of a mechanistic role for HMGB1/ROS/TLR4 interactions in initiating and maintaining inflammation through the TLR4 activation pathway.
Combined HMGB1 and prooxidant treatment increases TLR4 signaling in RAW-Blue™ cells
Treatment of RAW-Blue™ cells with HMGB1, PPC or SIN-1 individually increased SEAP release equally (~ by 2-fold) over cells treated with the control media. Pretreatment of HMGB1followed by PPC or SIN-1, increased SEAP release by 48 and 37 %, respectively, compared to PPC or SIN-1 alone (Fig. 9). The data confirmed that ROS/HMGB1-coupled stimulation of TLR4 through NF-κB activation could selectively occur in cells with multiple TLR expression. Pretreatment with a pure TLR4 antagonist, LPS-RS, that does not have any effect on other TLRs including TLR2, followed by stimulation with HMGB1, PPC or SIN-1 reduced SEAP release by 6-fold. Furthermore, pretreatment with anti-TLR4 pAb (10 μg/ml) followed by PPC (5 μM) also specifically decreased SEAP release by 3-fold) (data not shown), which further supports specificity of the ROS/HMGB1/TLR4-coupled interaction.
Fig. 9. The effect of prooxidants or HMGB1 alone and /or in combination on SEAP release in RAW-Blue™ macrophage cells that express multiple TLRs (except TLR5).
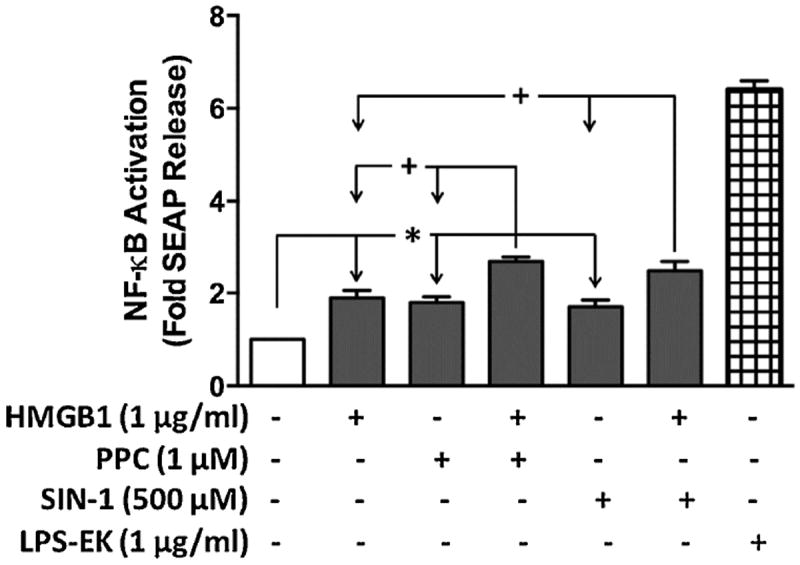
RAW-Blue™ cells are useful as TLR reporter cells for induction of NF-κB, with a subsequent release of SEAP after treatment with appropriate TLR-specific ligands, including ROS for TLR4 [23]. The QUANTI-Blue™ detection system was used to provide an easy and rapid means to quantify SEAP released into the culture media (based on the extent of TLR4 stimulation). *p ≤ 0.01 and +p ≤ 0.001 with n = 3 independent experiments conducted in duplicate.
Combined HMGB1 and prooxidant treatment increase TNF-α and IL-6 in primary murine macrophages derived from wild-type TLR4-WT, but not those from TLR4-KO mice
To gain further insight into a potential functional relevance of ROS/HMGB1-coupled activation of TLR4, we used primary macrophages from TLR4-KO and TLR4-WT mice in the presence and absence of prooxidants and/or HMGB1 combination. We analyzed the production of TNFα (Fig. 10A) and IL-6 (Fig. 10 B) as proinflammatory mediators, produced by the interactions between HMGB1 and prooxidants through TLR4 stimulation. Treatment with PPC, SIN-1 or HMGB1 individually significantly increased both TNFα (by 3 – 6X) and IL-6 (by 2.5 – 4X) in primary macrophages derived from TLR4-WT compared to those TLR4-KO. Combined [HMGB1+PPC] or [HMGB1+SIN-1] treatment produced significantly higher increases in both TNFα (by 5 –7X) and IL-6 (by 3 – 4X) release in TLR4-WT compared with the same treatments in TLR4-KO macrophages.
Fig. 10. Effect of HMGB1 alone and /or in combination with prooxidants on TNF-α and IL-6 release in primary murine macrophages derived from TLR4-WT and TLR4-KO mice.
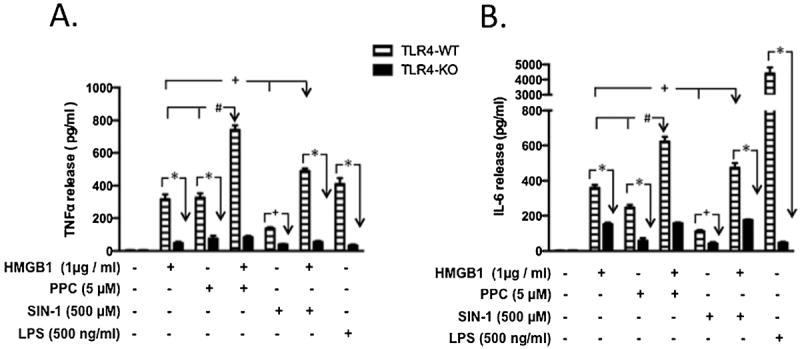
Primary macrophages derived from TLR4-WT and TLR4-KO mice were incubated overnight with disulfide HMGB1, PPC or SIN-1 alone and/or in combination as HMGB1+PPC] and HMGB1+SIN-1]. In all cases of [HMGB1+prooxidant] treatments, cells were preincubated with HMGB1 for 1h followed by overnight stimulation with either PPC or SIN-1 in the continued presence of HMGB1. The levels of TNF-α (Fig. 10A) and IL-6 (Fig. 10B) released into the culture media were quantified using the ELISA kits of the relevant murine cytokine according to manufacturer’s instructions. The effect of LPS-EK on TNF-α and IL-6 release was used as positive control. The data represent 3 - 6 independent experiments conducted in duplicate with *p < 0.0001; +p ≤ 0.01; #p ≤ 0.001, PPC or HMGB1 in TLR4-WT compared with [HMGB1 + PPC] combination; +p < 0.01, SIN-1 or HMGB1 in TLR4-WT compared with [HMGB1 + SIN-1] combination.
We also treated primary macrophages with LPS-EK, a TLR4-specific agonist, to confirm and delineate the fidelity of macrophages derived from both TLR4-WT and TLR4-KO mice. As is clearly evident (Figs. 10 A & 10 B), LPS-EK produced very increases in TNFα (by 7X) and IL-6 (by 20X) only in TLR4-WT macrophages compared with TLR4-KO-derived cells. Overall the data indicate that increased release of both TNFα and IL-6 was due to an intact and functional TLR4 expression. There were no significant differences whatsoever in the basal release of TNFα and IL-6 in macrophages derived from both TLR4-WT and TLR4-KO animals in the absence of HMGB1 and/or prooxidant treatments. Our data confirm that synergistic increase in TNFα and IL-6 release was due to prooxidant/HMGB1-coupled activation of TLR4.
DISCUSSION
In the present set of studies, we had hypothesized that HMGB1 would promote prooxidant-induced NF-κB activation through the TLR4 signaling pathway. To test this hypothesis, we used a culture model of HEK-Blue mTLR4 cells stably transfected with functional TLR4-CD14-MD2 complex and optimized to express SEAP reporter gene under the control of NF-κB transcription factor. We treated these cells with ROS/RNS-producing agents alone and/or in combination with disulfide HMGB1 isoform, followed by measurement of SEAP release as a marker for NF-κB activation. We also used primary murine macrophages derived from TLR4-WT and TLR4-KO mice. The novelty of our finding is the confirmation that extracellular disulfide HMGB1 can promote prooxidant-induced NF-κB activation through the TLR4 signaling pathway. Interestingly, we also confirmed (data not shown) that fully reduced HMGB1 isoform [compared with the disulfide HMGB1 isoform] was not effective either alone or in combination with prooxidants in enhancing the release of TNF-α and IL-6 from primary peritoneal TLR4-WT macrophages. This is in agreement with a recently published work in which the extracellular TLR4 adaptor, myeloid differentiation factor 2 (MD-2), was shown to bind specifically to the cytokineinducing disulfide isoform of HMGB1 to the exclusion of other isoforms [33]. Thus, our findings have broad functional implications for a mechanistic understanding of the role of HMGB1/ROS/TLR4-coupled stimulation in initiating, propagating and maintaining oxidative stress-induced disease states through NF-κB activation. Ample evidence suggests that HMGB1, when actively secreted by activated immune cells or passively released from dying cells, is a mixture of several isoforms with distinct posttranslational modifications [34].
The data we have presented here support an essential role for additive and/or synergistic interactions of prooxidants- and HMGB1-mediated TLR4 stimulation that activates NF-κB. First, prooxidants and LPS-EK (a specific agonist for TLR4) efficiently induced HMGB1 release, an endogenous TLR4 agonist. HMGB1 and prooxidants also induced SEAP and TNF-α release (both markers of NF-κB activation and proinflammatory processes), but neither TLR4 agonist (LPS-EK), nor HMGB1 and prooxidants (PPC and SIN-1) induced SEAP release in HEK-Blue null cells. Furthermore, TLR4 neutralizing pAb and HMGB1 (Box A) fragment inhibited both HMGB1-induced SEAP and TNF-α release. Neither HMGB1 nor prooxidants efficiently stimulated TNF-α and IL-6 in murine macrophages derived from TLR4-KO, but in combination they synergistically stimulated TNF-α and IL-6 release from primary murine macrophages derived from TLR4-WT mice compared with either HMGB1 or prooxidants alone. Finally, HMGB1 augmented prooxidant-induced NF-κB activation and iROS accumulation to contribute to enhanced oxidative/nitrosative.
We had previously shown in vivo and in vitro that PPC could activate NF-κB mainly through a TLR4-dependent pathway, which was attenuated by prior treatment with antioxidants [23,32]. Our initial experimental evidence for the involvement of prooxidants in promoting HMGB1-induced NF-κB activation is based on SEAP and TNF-α release, and inhibition of their release with TLR4 neutralizing pAb and HMGB1 (BoxA) fragment, a specific antagonist of HMGB1. In addition, murine macrophages that are devoid of TLR4 expression do not respond to HMGB1 or prooxidants stimulation, which confirms a level of specificity for the involvement of both agonists in this model for TLR4 stimulation.
Our present data clearly support interaction of structurally unrelated and diverse agents, namely ROS and disulfide HMGB1 with TLR4 for which LPS-EK is the native/specific agonist. As far as we are aware, LPS-EK/TLR4-coupled activation involves the native reversible “molecular” specificity that is the basis for complimentary LPS-EK binding to TLR4. Furthermore, MD-2 is shown to possess a hydrophobic pocket folded by two antiparallel β-sheets for binding LPS and confers molecular specificity for LPS interaction and TLR4 signaling [35]. Thus, it may be reasoned that MD-2 can similarly discriminate different HMGB1 isoforms to facilitate TLR4-dependent signaling. It appears that only the disulfide HMGB1 binds to MD-2, an interaction that is of critical importance for HMGB1-mediated cytokine/chemokine production and the development of subsequent tissue injury [34]. On the other hand, ROS/TLR4-coupled activation could be due to a reversible “atomic” specificity in which specific sulfur atoms on cysteine residues [36, 37] uniquely expressed in the intracellular domains of TLR4 [38] may play a crucial role.
We used a combination of fluorescent microscopy imaging, flow cytometer and TBARs [39] to provide insight on the contributory factors that may regulate ROS/HMGB1-coupled activation of TLR4 in our cell model systems. We used CellROX® Deep Red reagent for cell labeling in both fluorescence microscopy and flow cytometer. Even though the trend towards enhanced fluorescence were the same in both imaging microscopy and flow cytometer, but the fluorescence intensity readouts for imaging microscopy were more robust compared with those for flow cytometer for the same treatments. The apparent differences between iROS-readout derived from fluorescence microscopy and flow cytometer could be due to a loss of or a reduction in cell fluorescence inherent in sample preparation for the flow cytometric assay. Sample preparation for flow cytometer may introduce errors by eliminating fragile cells with potentially higher levels of iROS, which may result in lower fluorescence readout in the dispersed cells. This may be due to the fact that broken fluorescent cells lose their fluorescence, and thus cannot be counted. These cells are regarded as “debris” as they pass through the aperture, which reads the fluorescence of intact cells only.
Thus on one hand fluorescence microscopy would appear to encompass ROS in both extra- and intra-cellular compartments. On the other hand, flow cytometer measures only iROS by combining the advantages of microscopy and biochemical assay. However, a major advantage of flow cytometry remains its inherent use to determine iROS levels on a cell-by-cell basis in the absence of cell “debris” background. Functionally, prooxidants in combination with HMGB1 produced synergistic and significant enhancements in TNFα and IL-6 only in peritoneal macrophages derived from TLR4-WT compared with those from TLR4-KO mice. However, the robustness of the levels of TNFα and IL-6 released into the conditioned media after treatment with prooxidants and/or HMGB1 in the murine macrophages from TLR4-WT corresponds to the fluorescent microscopy data from HEK-Blue mTLR4 cells.
As a DAMP molecule, HMGB1 released extracellularly can function as a signal of tissue damage [9]. Indeed, during sepsis the serum levels of HMGB1 are markedly elevated [40]. In response to proinflammatory stimuli such as LPS, interferon gamma (INF-γ) and nitric oxide (NO), HMGB1 is actively released from a number of different cell types including macrophages, pituicytes, dendritic cells, NK cells and fibroblasts [40, 41]. In the present study, we report for the first time that exogenous prooxidants induced HMGB1 release in HEK-Blue mTLR4 cells. Mechanistically, this is of significance because it confirms that HMGB1 can be released from cells either actively in a regulated process, or passively during necrosis [41, 42]. Our previous work showed that over 94 % of cells remained viable after treatment with PPC (5 μM) and SIN-1 (1.0 mM) as used in the present experiments [32]. These results suggest that prooxidant-induced HMGB1 release from HEK-Blue mTLR4 cells is not caused by cell death, but is likely due to an active process regulated by the levels of iROS. Consistent with our data, exogenous hydrogen peroxide was also recently shown to induce HMGB1 release from hepatocytes [43] and macrophages [44]. Indeed, antioxidants prevented HMGB1 release in hypoxic hepatocytes in vitro, and in liver ischemia/reperfusion (I/R) (trauma) model [43].
Structurally, HMGB1 is composed of three domains: two positively charged tandem DNA-binding HMGB1 box domains (A and B) linked by a short flexible basic linker that allows the two domains to behave independently as free protein moieties, and a negatively charged carboxyl terminus (the acidic tail) [55]. Analysis of the structure-function of HMGB1 has revealed that the pro-inflammatory properties of HMGB1 reside in Box B domain of the molecule, whereas the DNA-binding BoxA acts antagonistically to inhibit the activities mediated by the full-length HMGB1 [46]. BoxA is the most highly conserved DNA binding domain of HMGB1 protein, and acts as an antagonist for HMGB1 binding to TLR4, and inhibits HMGB1 cytokine effects in vivo. HMGB1 (BoxA), when isolated from the rest of the molecule, acts as a specific antagonist of HMGB1. Thus, HMGB1 itself can act as a proinflammatory stimulus to activate monocytes and endothelial cells to release proinflammatory cytokines [47,48]. Stimulation of endothelial cells with HMGB1 resulted in the secretion of diverse proinflammatory cytokines, including G-CSF, IL-8, TNF-α and MCP1 [49], which contributes to inflammatory responses. Furthermore, stimulation by HMGB1 increased TLR4-mediated NF-κB activation in human neutrophils [50], which is confirmed here with HEK-Blue mTLR4 cells. Interestingly, anti-TLR4 antibody inhibited HMGB1-induced IL-8 and TNF-α secretion in several cell lines [51], which is consistent with our present results. Consistent with this, our results showed that TLR4 neutralizing antibody inhibited SEAP and TNF-α release, markers of NF-κB activation in TLR4 signaling pathway. HMGB1 signaling appears capable of maintaining either an autocrine or a paracrine positive feed forward or feedback loops. HMGB1-induced stimulation can lead to TNF-α secretion [10], which in turn can increase HMGB1 secretion from macrophages and endothelial cells [10] by activating TLR4 that resulted in NF-κB activation (Fig. 11).
Fig. 11. A simplified schematic representation of a putative mechanism for injury-induced exogenous prooxidant-coupled HMGB1 stimulation of TLR4 signaling through NFκB activation.
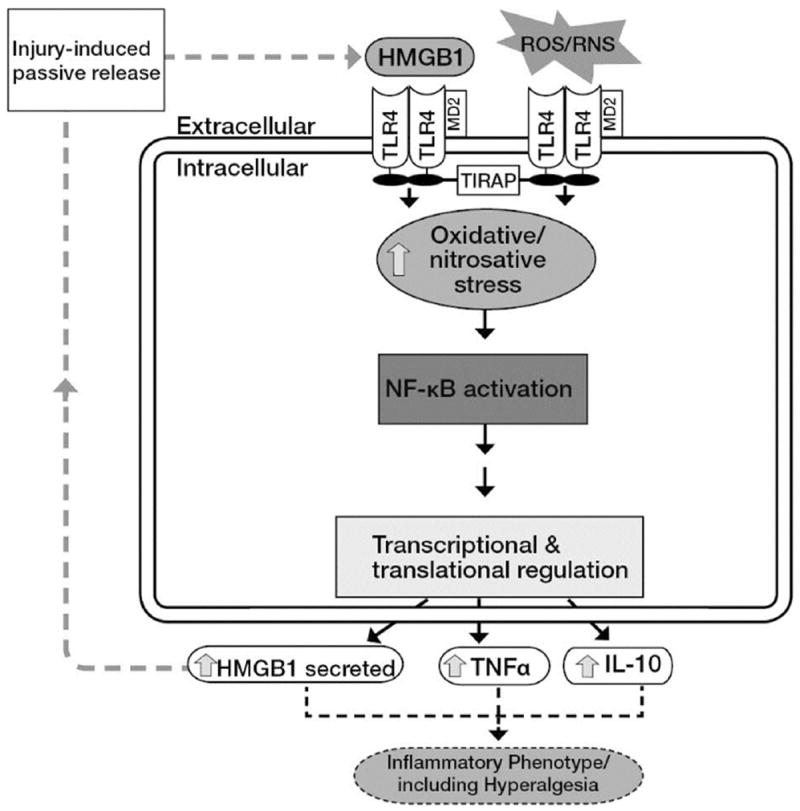
Prooxidants and HMGB1 act in concert to stimulate TLR4. Extracellularly released HMGB1 due to injury can potentially activate TLR4 in an autocrine or paracrine fashion. TLR4 is stimulated in response to exogenous oxidants resulting in the activation of c-Src, which interacts with TLR4 [32]. Increased formation of TLR4/c-Src complex leads to enhanced recruitment of different cytosolic adaptor proteins including toll-interleukin 1 receptor adaptor protein (TIRAP) that results in c-Src/NFκB/IκBα coupled activation.
Extracellular and cytoplasmic expressions of HMGB1 have been observed in tissue sections from synovitis in both human arthritis and in experimental arthritic models [51]. After therapeutic application in collagen-induced arthritis, HMGB1 (BoxA) markedly decreased arthritic score, reduced synovitis, and decreased IL-1β expression, in the synovia [52]. Consistent with this finding, HMGB1 (BoxA) fragment in the present study decreased HMGB1-induced but not prooxidant-mediated NF-κB activity confirming the specificity of BoxA in blocking the effects of HMGB1. Results in vitro showed that the neutralizing antibody effectively blocked rHMGB1-induced TNF-α production in RAW264.7 cells [53]. Pre-treatment of animals with HMGB1 neutralizing antibody also significantly reduced the expression of TNF-α in the kidneys after I/R insult [53]. It has been suggested that a novel way to therapeutically target HMGB1 could be to induce its nuclear retention in order to inhibit its extracellular release [54].
Recently, Evankovich et al [55] showed that hepatocytes actively release HMGB1 in response to oxidative stress, which suggests that these parenchymal cells can provide danger signals to neighboring immune cells in the liver to promote inflammation and organ damage. Although HMGB1 is passively released following necrosis, the findings demonstrated that oxidative stress in hepatocytes could lead to early shuttling from the nucleus to cytoplasm, followed by its subsequent release in the absence of cell death. This suggests that HMGB1 mobilization in hepatocytes is an active, regulated process. The pathways that govern HMGB1 release from hepatocytes are still not clear; nonetheless, the proximal events appear to involve TLR4 activation and calcium signaling through calcium/calmodulin-dependent protein kinases (CaMKs) [56]. In other cell types, downstream events governing HMGB1 release have also been linked to oxidation/reduction [57] and post-translational modifications that include phosphorylation [58] and acetylation [59]. In the present study, we have not examined the mechanism of oxidant-induced HMGB1 release. This is an ongoing study in the laboratory.
After NF-κB activation, various cytokines including TNF-α and chemokines are produced [60]. With a correlation of NF-κB activation with inflammatory disease in animal models of arthritic pain, and allergic airway disease [61], but association of NF-κB activity and inflammatory disease is not easy to interpret. This is because both pro- and anti-inflammatory mediators are produced during inflammation, and the balance between these factors is likely to dictate disease progression and outcome [62]. Consistent with this hypothesis, stimulation of TLR4 signaling pathway by LPS, HMGB1 and prooxidants induced increases in both TNF-α and IL-10 production, as representative pro- and anti-inflammatory, respectively. Adib-Conguy et al [63] reported that TLR4 mediates the production of TNF-α and IL-10 during systemic inflammation. TNF-α not only regulates the expression of a variety of other pro-inflammatory cytokines including IL-1, IL-6, PDGF, TGF-β but also enhances chemotaxis of macrophages and neutrophils at the site of inflammation [64,65]. However, IL-10 has multifaceted anti-inflammatory properties, including inhibition of the prototypic inflammatory transcription factor NF-κB, leading to suppression of cytokine production [66]. IL-10 may increase during inflammation to supersede pro-inflammatory cytokine signaling cascade [67]. In an effort to provide a mechanistic explanation for the prooxidant-induced increases in both TNFα (proinflammatory) and IL-10 (an antiinflammatory), we determined the ratios of the expression levels of TNF-α to IL-10. We surmised that the ratios would represent a balance between the two types of cytokines and their biologically relevant functions. Indeed, the extent of inflammation could possibly depend on high ratio of TNF-α to IL-10 [68]. Surprisingly, TNF-α/IL-10 ratio was higher in HMGB1- and PPC-treated cells than cells treated with LPS-EK. Moreover, co-treatment of PPC and HMGB1 resulted in a higher TNF-α/IL-10 ratio than either PPC or HMGB1 alone, which unequivocally confirmed that prooxidants can promote exHMGB1-induced inflammatory response. This conclusion directly supports our present data on ROS/HMGB1-induced synergistic release of TNFα and IL-6 from primary murine macrophages derived from TLR4-WT compared with those from TLR4-KO mice.
In conclusion, our data provide a unifying mechanism for initiating and maintaining nonbacterial systemic inflammatory responses in a wide range of human disease states in which ONS plays a role. Intriguingly, an evolutionary remnant of the host defense against exogenous pathogens and/or our microbiota (TLRs as a member of PRR family) is “co-opted” by molecules produced by the host such as HMGB1 and ROS, which are released during inflammation and in normal cellular processes. It is also noteworthy that in vitro and in vivo a member of the TLR family, TLR4, is especially promiscuous with respect to potential ligands [69,70]. Both the “danger model” and the “oxygen paradox” hypotheses as proposed by Matzinger [71] and Davies [1], respectively, are in line with our reasoning here. Thus, the extent of HMGB1/ROS/TLR4-coupled activation of NF-κB signaling pathway has the potential to serve as a mechanistic link for inflammatory pathophysiology of diverse disease states in any organ system [72].
Acknowledgments
We thank Dr. Ankit B. Shah for providing us the initial training on the operations of flow cytometer.
The project described was supported by Award Number DE021888 from the National Institute of Dental & Craniofacial Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Dental & Craniofacial Research or the National Institutes of Health.
Abbreviations
- ONS
oxidative/nitrosative stress
- TLR
toll-like receptor
- HEK
human embryonic kidney
- MD
myeloid of differentiation
- CD
cluster of differentiation
- pAb
polyclonal antibody
- LPS-EK (Ultrapure)
lipopolysaccharide from E. coli K12
- TLR4-KO macrophages
macrophages derived from complete TLR4 knockout mice
- TLR4-WT macrophages
macrophages derived from wild-type mice
- PPC
potassium peroxychromate
- SEAP
secreted embryonic alkaline phosphatase
- FBS
DMEM, Dulbecco’s modified Eagle’s medium
- ANOVA
analysis of variance
- MTT
(3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- TBARS
thiobarbituric acid reacting substances
- MDA
malonyldialdehyde
- NF-κB
nuclear factor kappa B
- iROS
intracellular reactive oxygen species
- TNF-α
tumor necrosis factor alpha
- ELISA
enzyme-linked immuno-sorbent assay
Footnotes
The authors have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davies KJ. Oxidative stress: the paradox of aerobic life. Biochem Soc Symp. 1995;61:1–31. doi: 10.1042/bss0610001. [DOI] [PubMed] [Google Scholar]
- 2.Giustarini D, Dalle-Donne I, Tsikas D, Rossi R. Oxidative stress and human diseases: Origin, link, measurement, mechanisms, and biomarkers. Crit Rev Clin Lab Sci. 2009;46(5-6):241–281. doi: 10.3109/10408360903142326. [DOI] [PubMed] [Google Scholar]
- 3.Davies KJ. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life. 2000;50(4-5):279–289. doi: 10.1080/713803728. Review. [DOI] [PubMed] [Google Scholar]
- 4.Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic Biol Med. 2010;48(9):1121–1132. doi: 10.1016/j.freeradbiomed.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiang M, Fan J, Fan J. Association of Toll-like receptor signaling and reactive oxygen species: a potential therapeutic target for post-trauma acute lung injury. Mediators Inflamm. 2010 doi: 10.1155/2010/916425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 7.Ve T, Gay NJ, Mansell A, Kobe B, Kellie S. Adaptors in toll-like receptor signaling and their potential as therapeutic targets. Curr Drug Targets. 2012;3:1360–1374. doi: 10.2174/138945012803530260. [DOI] [PubMed] [Google Scholar]
- 8.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010 doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 10.Andersson U, Erlandsson-Harris H, Yang H, Tracey KJ. HMGB1 as a DNA-binding cytokine. J Leukoc Biol. 2002;72:1084–1091. [PubMed] [Google Scholar]
- 11.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26(2):174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 12.Mosquera JA. Role of the receptor for advanced glycation end products (RAGE) in inflammation. Invest Clin. 2010;51(2):257–268. [PubMed] [Google Scholar]
- 13.Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, Hirashima M, Uede T, Takaoka A, Yagita H, Jinushi M. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13(9):832–842. doi: 10.1038/ni.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, Shams SS, Yang H, Varani L, Andersson U, Tracey KJ, Bachi A, Uguccioni M, Bianchi ME. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209(9):1519–1528. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wähämaa H, Schierbeck H, Hreggvidsdottir HS, Palmblad K, Aveberger AC, Andersson U, Harris HE. High mobility group box protein 1 in complex with lipopolysaccharide or IL-1 promotes an increased inflammatory phenotype in synovial fibroblasts. Arthritis Res Ther. 2011;13(4):R136. doi: 10.1186/ar3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009;86(3):573–576. doi: 10.1189/jlb.1008585. [DOI] [PubMed] [Google Scholar]
- 17.Hreggvidsdottir HS, Ostberg T, Wähämaa H, Schierbeck H, Aveberger AC, Klevenvall L, Palmblad K, Ottosson L, Andersson U, Harris HE. The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J Leukoc Biol. 2009;86(3):655–662. doi: 10.1189/jlb.0908548. [DOI] [PubMed] [Google Scholar]
- 18.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 20.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 21.Harris HE, Raucci A. Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep. 2006;7:774–778. doi: 10.1038/sj.embor.7400759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qin YH, Dai SM, Tang GS, Zhang J, Ren D, Wang ZW, Shen Q. HMGB1 enhances proinflammatory activity of lipopolysaccharide by promoting the phosphorylation of MAPK p38 through receptor for advanced glycation end products. J Immunol. 2009;183:6244–6250. doi: 10.4049/jimmunol.0900390. [DOI] [PubMed] [Google Scholar]
- 23.Karki R, Igwe OJ. Toll-like receptor 4-mediated nuclear factor kappa B activation is essential for sensing exogenous oxidants to propagate and maintain oxidative/nitrosative cellular stress. PLoS ONE. 2013;8(9):e73840. doi: 10.1371/journal.pone.0073840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antoine DJ, Harris HE, Andersson U, Tracey KJ, Bianchi ME. A systematic nomenclature for the redox states of high mobility group box (HMGB) proteins. Mol Med. 2014;20:135–137. doi: 10.2119/molmed.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baird MB, Massie HR, Piekielniak MJ. Formation of lipid peroxides in isolated rat liver microsomes by singlet molecular oxygen. Chem Biol Interact. 1977;16(2):145–153. doi: 10.1016/0009-2797(77)90124-7. [DOI] [PubMed] [Google Scholar]
- 26.Miesel R, Kroger H, Kurpisz M, Weser U. Induction of arthritis in mice and rats by potassium peroxochromate and assessment of disease activity by whole blood chemiluminescence and 99mpertechnetate-imaging. Free Radic Res. 1995;23:213–227. doi: 10.3109/10715769509064035. [DOI] [PubMed] [Google Scholar]
- 27.Trackey JL, Uliasz TF, Hewett SJ. SIN-1-induced cytotoxicity in mixed cortical cell culture: peroxynitrite-dependent and -independent induction of excitotoxic cell death. J Neurochem. 2001;79(2):445–455. doi: 10.1046/j.1471-4159.2001.00584.x. [DOI] [PubMed] [Google Scholar]
- 28.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls: The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266(7):4244–4250. [PubMed] [Google Scholar]
- 29.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14(Unit 14.1) doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karki R, Zhang Y, Igwe OJ. Activation of c-Src: a hub for exogenous pro-oxidant-mediated activation of Toll-like receptor 4 signaling. Free Radic Biol Med. 2014;71:256–269. doi: 10.1016/j.freeradbiomed.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang H, Wang H, Ju Z, Ragab AA, Lundbäck P, Long W, Valdes-Ferrer SI, He M, Pribis JP, Li J, Lu B, Gero D, Szabo C, Antoine DJ, Harris HE, Golenbock DT, Meng J, Roth J, Chavan SS, Andersson U, Billiar TR, Tracey KJ, Al-Abed Y. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med. 2015;212(1):5–14. doi: 10.1084/jem.20141318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. 2013;93(6):865–873. doi: 10.1189/jlb.1212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meng J, Lien E, Golenbock DT. MD-2-mediated ionic interactions between lipid A and TLR4 are essential for receptor activation. J Biol Chem. 2010;285(12):8695–8702. doi: 10.1074/jbc.M109.075127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nathan C. Specificity of a third kind: reactive oxygen and nitrogen intermediates in cell signaling. J Clin Invest. 2003;111(6):769–778. doi: 10.1172/JCI18174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287(2):C246–C256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 38.Daringer NM, Schwarz KA, Leonard JN. Contributions of unique intracellular domains to switchlike biosensing by Toll-like receptor 4. J Biol Chem. 2015;290(14):8764–8777. doi: 10.1074/jbc.M114.610063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalyanaraman B, DarleyUsmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, 2nd, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med. 2112;52(1):1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMGB-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 41.Wang H, Vishnubhakat JM, Bloom O, Zhang M, Ombrellino M, Sama A, Tracey KJ. Proinflammatory cytokines (tumor necrosis factor and interleukin 1) stimulate release of high mobility group protein-1 by pituicytes. Surgery. 1999;126:389–392. [PubMed] [Google Scholar]
- 42.Bell CW, Jiang W, Reich CF, 3rd, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol. 2006;291:C1318–C1325. doi: 10.1152/ajpcell.00616.2005. [DOI] [PubMed] [Google Scholar]
- 43.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR, Billiar TR. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–2923. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang D, Shi Y, Kang R, Li T, Xiao W, Wang H, Xiao X. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol. 2006;81:741–747. doi: 10.1189/jlb.0806540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stott K, Tang GS, Lee KB, Thomas JO. Structure of a complex of tandem HMG boxes and DNA. J Mol Biol. 2010;360(1):90–104. doi: 10.1016/j.jmb.2006.04.059. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, Harris HE, Czura CJ, Wang H, Ulloa L, Wang H, Warren HS, Moldawer LL, Fink MP, Andersson U, Tracey KJ, Yang H. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med. 2003;9:37–45. [PMC free article] [PubMed] [Google Scholar]
- 47.Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 48.Treutiger CJ, Mullins GE, Johansson AS, Rouhiainen A, Rauvala HM, Erlandsson-Harris H, Andersson U, Yang H, Tracey KJ, Andersson J, Palmblad JE. High mobility group 1 B-box mediates activation of human endothelium. J Intern Med. 2003;254:375–385. doi: 10.1046/j.1365-2796.2003.01204.x. [DOI] [PubMed] [Google Scholar]
- 49.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMGB-1 as a mediator of acute lung inflammation. J Immunol. 2000;165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 50.Park JS, Arcaroli J, Yum HK, Yang H, Wang H, Yang KY, Choe KH, Strassheim D, Pitts TM, Tracey KJ, Abraham E. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am J Physiol Cell Physiol. 2003;284:C870–C879. doi: 10.1152/ajpcell.00322.2002. [DOI] [PubMed] [Google Scholar]
- 51.Kokkola R, Sundberg E, Ulfgren AK, Palmblad K, Li J, Wang H, Ulloa L, Yang H, Yan XJ, Furie R, Chiorazzi N, Tracey KJ, Andersson U, Harris HE. High mobility group box chromosomal protein 1: a novel proinflammatory mediator in synovitis. Arthritis Rheum. 2002;46:2598–2603. doi: 10.1002/art.10540. [DOI] [PubMed] [Google Scholar]
- 52.Kokkola R, Li J, Sundberg E, Aveberger AC, Palmblad K, Yang H, Tracey KJ, Andersson U, Harris HE. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 2003;48:2052–2058. doi: 10.1002/art.11161. [DOI] [PubMed] [Google Scholar]
- 53.Li J, Gong Q, Zhong S, Wang L, Guo H, Xiang Y, Ichim TE, Wang CY, Chen S, Gong F, Chen G. Neutralization of the extracellular HMGB1 released by ischaemic damaged renal cells protects against ischaemia-reperfusion injury. Nephrol Dial Transplant. 2011;26:469–478. doi: 10.1093/ndt/gfq466. [DOI] [PubMed] [Google Scholar]
- 54.Chorny A, Anderson P, Gonzalez-Rey E, Delgado M. Ghrelin protects against experimental sepsis by inhibiting high-mobility group box 1 release and by killing bacteria. J Immunol. 2008;180:8369–8377. doi: 10.4049/jimmunol.180.12.8369. [DOI] [PubMed] [Google Scholar]
- 55.Evankovich J, Cho SW, Zhang R, Cardinal J, Dhupar R, Zhang L, Klune JR, Zlotnicki J, Billiar T, Tsung A. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem. 2010;285(51):39888–39897. doi: 10.1074/jbc.M110.128348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR, Billiar TR. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204(12):2913–2923. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoppe G, Talcott KE, Bhattacharya SK, Crabb JW, Sears JE. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res. 2006;312(18):3526–3538. doi: 10.1016/j.yexcr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 58.Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol. 2006;177(11):7889–7897. doi: 10.4049/jimmunol.177.11.7889. [DOI] [PubMed] [Google Scholar]
- 59.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22(20):5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simmonds RE, Foxwell BM. Signaling, inflammation and arthritis: NF-κB and its relevance to arthritis and inflammation. Rheumatology. 2008;47:584–590. doi: 10.1093/rheumatology/kem298. [DOI] [PubMed] [Google Scholar]
- 61.Lawrence T. The nuclear factor NFκB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;(1):a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lawrence T, Gilroy DW. Chronic inflammation: A failure of resolution? Int J Exp Pathol. 2007;88:85–94. doi: 10.1111/j.1365-2613.2006.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adib-Conguy M, Moine P, Asehnoune K, Edouard A, Espevik T, Miyake K, Werts C, Cavaillon JM. Toll-like receptor-mediated tumor necrosis factor and interleukin-10 production differ during systemic inflammation. Am J Respir Crit Care Med. 2003;168:158–164. doi: 10.1164/rccm.200209-1077OC. [DOI] [PubMed] [Google Scholar]
- 64.Larrick JW, Wright SC. Cytotoxic mechanism of tumour necrosis factor-alpha. FASEB J. 1990;4:3215–3223. doi: 10.1096/fasebj.4.14.2172061. [DOI] [PubMed] [Google Scholar]
- 65.Feldman AM, Combes A, Wagner D, Kadakomi T, Kuboka T, Li YY, McTrernan C. The role of tumor necrosis factor in the pathophysiology of heart failure. J Am Coll Cardiol. 2000;35:537–544. doi: 10.1016/s0735-1097(99)00600-2. [DOI] [PubMed] [Google Scholar]
- 66.Wang P, Wu P, Siegel MI. Interleukin (IL)-10 inhibits nuclear factor kappa B (NF kappa B) activation in human monocytes: IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. J Biol Chem. 1995;270:9558–9563. doi: 10.1074/jbc.270.16.9558. [DOI] [PubMed] [Google Scholar]
- 67.Elenkov IJ, Chrousos GP. Stress hormones, proinflammatory and antiinflammatory cytokines, and autoimmunity. Ann N Y Acad Sci. 2002;966:290–303. doi: 10.1111/j.1749-6632.2002.tb04229.x. [DOI] [PubMed] [Google Scholar]
- 68.You Z, Luo C, Zhang W, Chen Y, He J, Zhao Q, Zuo R, Wu Y. Pro- and anti-inflammatory cytokines expression in rat’s brain and spleen exposed to chronic mild stress: involvement in depression. Behav Brain Res. 2011;225:135–141. doi: 10.1016/j.bbr.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 69.Goligorsky MS. TLR4 and HMGB1: partners in crime? Kidney Int. 2011;80(5):450–452. doi: 10.1038/ki.2011.170. [DOI] [PubMed] [Google Scholar]
- 70.Lucas K, Maes M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Mol Neurobiol. 2013;48(1):190–204. doi: 10.1007/s12035-013-8425-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 72.Tang D, Kang R, Zeh HJ, 3rd, Lotze MT. High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal. 2011;14(7):1315–1335. doi: 10.1089/ars.2010.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]