

Abstract
Genetic heterogeneity has been recognised in Peutz-Jeghers syndrome (PJS) (over 230 STK11 gene mutations reported). We report a rare PJS phenotype with early extensive gastrointestinal (GI) presentation and a new genetic variant. The case presented as haematochezia and mucocutaneous pigmentation (the patient was 3 years of age). Endoscopy showed several polyps throughout the stomach/colon (PJ-type hamartomas); the larger polyps were resected. Small bowel imaging detected multiple jejunum/ileum small polyps. During 8 years of follow-up of this asymptomatic patient, an increasing number of diffusely distributed polyps was observed and polypectomies were performed. Subsequently, the patient failed consultations; when the patient was 13 years of age, emergency surgery was required due to small bowel intussusception (ileal polyp). A STK11 gene study identified two missense variants in heterozygous (yet unknown significance but probably pathogenic): c.854T>A (exon 6) and c.446C>T* (exon 2) (*not previously reported). We report two STK11 gene variants (one not previously described) of yet undetermined causality in a paediatric patient presenting with extensive GI involvement at a very early age, with no family medical history. Structural and functional repercussion of the newly described variants should be further investigated.
Background
Peutz-Jeghers syndrome (PJS) is an autosomal dominant condition characterised by mucocutaneous pigmentation, gastrointestinal (GI) hamartomatous polyposis and multisystemic oncogenic predisposition.1–4 Its estimated incidence ranges from 1/200 000 to 1/50 000 births, with no sex, racial or ethnic predominance.3 4 At paediatric age, clinical manifestations may include subocclusive events caused by GI polyps, requiring regular clinical, imaging and endoscopic surveillance.2 4 5
Although a number of STK11 gene mutations (serine/threonine-protein kinase 11) have been increasingly identified in 50–90% of sporadic and in almost 100% of familial cases,3 6 7 wide genetic heterogeneity is recognised and genotype–phenotype associations are not yet clearly established.8–10
In addition to reporting a new PJS genetic variant, the authors further intended to contribute to characterisation of genotype–phenotype associations, highlighting the clinical and molecular peculiarities of an illustrative PJS case (fully detailed), with very early presentation, and extensive/progressive proximal and distal GI involvement (previous reports of such large sized gastric and colonic polyps at an early age are rare). Interestingly, considering the unremarkable family history, it might possibly represent a ‘de novo’ variant. Further elucidation of genotype–phenotype associations and of long-term outcome (including oncogenic risk) will certainly improve current clinical care, quality of life and life expectancy of these patients.10–12
Case presentation
A 14-year-old boy presented with intermittent lower GI tract bleeding and anal mass exteriorisation when he was 2.5 years old. At the time, melanic pigmentation was noticed in the buccal mucosa and perioral region, and iron-deficiency anaemia requiring ambulatory iron therapy was confirmed. The patient was then submitted to surgical excision of two pediculate rectal polyps (1 and 1.5 cm in diameter). Histological examination revealed PJ-type hamartomatous polyps, thus confirming the diagnosis of PJS.
The boy is the youngest third child of non-consanguineous parents, with no relevant medical history, apart from a discrete language disorder, which evolved favourably with language therapy support. He was born under unfavourable sociodemographic circumstances, to severely illiterate parents. None of his first generation relatives had GI problems or mucocutaneous pigmentation. His paternal grandmother was described as presenting discrete facial pigmentation, which could not be confirmed in medical visits.
After diagnosis, initial endoscopy at 3 years of age and subsequent annual endoscopic evaluations of upper and lower GI tract disclosed the presence of multiple diffuse small polyps as well as a few large-sized polyps distributed throughout the stomach, colon and rectum (1.5–4.5 cm in diameter). Sequential endoscopic excision was performed, according to the polyps’ location and size, and all were histologically suggestive of PJ-type hamartomas, without dysplasia (figure 1). No polyps had been initially identified through small bowel imaging. Follow-up diagnostic work up included clinical and analytical evaluations, periodic upper and lower GI tract endoscopies as well as small bowel imaging studies (radiology and magnetic enteroresonance) and extra-intestinal screening (namely testicular ultrasound), on an individualised basis.
Figure 1.
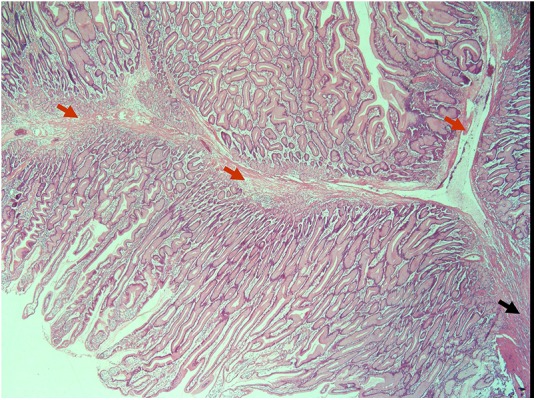
Histological features of a large gastric Peutz-Jeghers-type polyp resected in a patient 3 years of age. The pedicle (black arrow) and the arborescent central bundle of smooth muscle fibres (red arrows) surrounded by normal mucosa (H&E) are shown.
Meanwhile, despite family awareness of the disease implications, the patient missed several appointments due to sociodemographic constraints, which prevented regular and adequate surveillance from 10 to 13 years of age. During the 8-year follow-up period, he had no more GI bleeding or other significant GI symptoms.
When the patient was 13 years of age, MR enterography was performed, which detected multiple small polyps throughout the jejunum and ileum, some larger small bowel polyps (<20 mm) and an asymptomatic small bowel intussusception in relation to a 22 mm polyp (white arrow) (figure 2). Soon after, he was submitted to surgical intervention due to small bowel intussusception. Laparotomy was performed, with manual intussusception resolution, excision of a 2 cm ileal polyp and resection of the first jejunum loop (33 cm), which was filled with large sessile and pediculate polyps (0.5–4 cm). There were no further complications since then.
Figure 2.
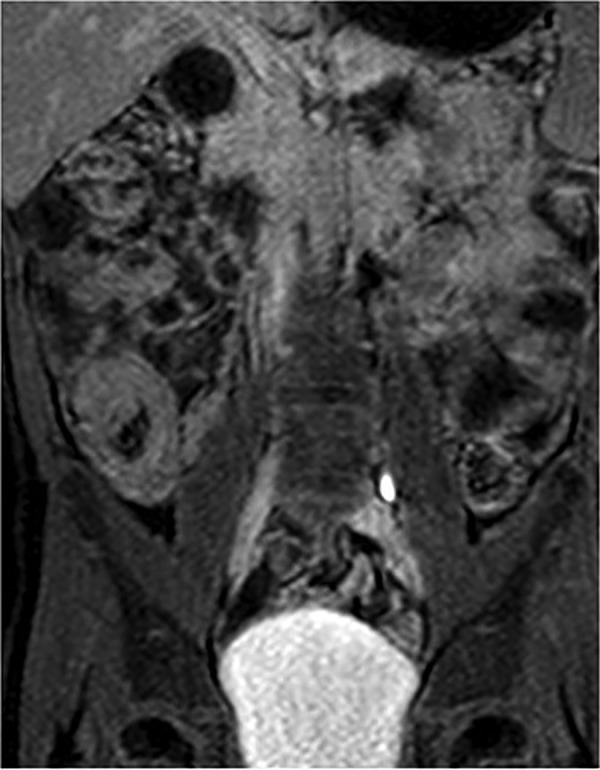
MR enterography performed in the patient at 13 years of age detected multiple small polyps throughout the jejunum and ileum, some larger small bowel polyps (<20 mm) and an asymptomatic small bowel intussusception in relation to a 22 mm polyp (white arrow).
Currently, with the patient 14 years of age, the latest endoscopic evaluation confirmed extensive GI involvement, with diffuse and multiple small gastroduodenal (over 100; 2–3 mm in diameter), colorectal polyps (over 30; 2–3 mm in diameter) and two large gastric polyps (2 cm), which required polypectomy (figure 3). A hamartomatous histological pattern was patent, with no dysplasia. The patient has been proposed to undergo diagnostic/therapeutic balloon enteroscopy preceded by videocapsule evaluation. Additional surveillance with abdominal and testicular ultrasound was normal.
Figure 3.
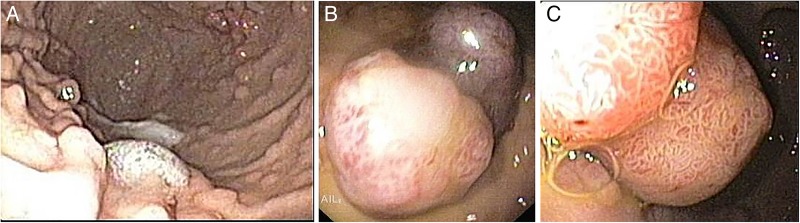
Upper gastrointestinal tract endoscopy (14 years of age) showing multiple small polyps throughout the stomach (approximately 2 mm diameter) and two large-sized pediculated polyps in the small curvature (1×1 cm and 2×1 cm diameter).
Finally, molecular study of the STK11 gene, performed by direct sequencing of its codifying regions (NM_000455.4), identified two missense variants in heterozygosis: c.854T>A at exon 6, previously described as having an unknown significance, and the variant c.446C>T at exon 2, not previously reported. The two variants resulted in substitution of leucine to glutamine at the 285 codon (p.Leu285Gln) and from proline to leucine at the 149 codon (p.Pro149Leu) of serine/threonine-protein kinase 11, respectively. Both of the aminoacids are preserved and their modification seems not to be tolerated. The in silico bioinformatic predictions of the first variant suggested a high probability of a pathogenic effect, and the analysis of the latter was not conclusive. Family molecular study is underway.
Discussion
PJS diagnosis is based on clinical findings and variable expressivity is common.2–4 13 In the case described, melanotic lesions in the oral mucosa were present in an early phase. However, GI bleeding was the first noticed symptom, accompanied by iron deficiency anaemia due to the presence of two large rectal polyps. Once its hamartomatous PJ-type intestinal nature was confirmed, the diagnosis was established, according to current consensus statements.2 3
All the resected polyps consisted of typical PJ-type hamartomas, the hallmark of the syndrome (as described in figure 1).2–4 Polyps usually predominate in the small bowel,2–4 13 but may also occur with variable frequency and in large number in the large bowel, stomach and duodenum. Their rapid and diffuse growth pattern was clearly evident in our patient from a very early age (3 years) through the years of follow-up. Interestingly, the early occurrence of predominantly large-sized proximal (stomach) and distal (colon, rectum) polyps, has been rarely reported. So far, no extra-intestinal organs seem to be involved, as expected at a young age.2–4 Complications usually occur before the age of 10 years in about 40% of patients.1 3 4 Morbidity and mortality are mainly caused by small bowel intussusception,1 3 4 13 which can first present between 5 and 16 years of age.1 3 4
There is an increased oncogenic risk to affected young adults, including in the GI tract and extra-intestinal organs.3 4 11 12 The risk is lower but not negligible at paediatric age, including pre-pubertal testicular tumours.12 Life expectancy of patients with PJS is significantly shorter, with a median age of death at 51 years, in relation to GI complications before 30 years of age, and due to malignancy thereafter.3 11 12 Current surveillance protocols are controversial and not yet evidence based, due to the rarity of the disorder.2 13 14 Routine endoscopic surveillance with polypectomy decreases the frequency of emergency laparotomy and bowel loss resulting from intussusception.2 14 MR enterography and, recently, videocapsule endoscopy, have improved diagnosis and management of small bowel polyps.14 Balloon-assisted enteroscopy allows for removal of deep small-bowel polyps, representing a therapeutic technique with short-term and long-term benefits.2 14
Specifically in this case, very early extensive presentation with large-sized polyps and the occurrence of small bowel intussusception, may predict a more severe phenotype and clinical outcome. According to current recommendations, the latest that endoluminal periodic evaluation should begin is at 8–10 years of age (upper and lower GI tract endoscopy and small bowel imaging or videocapsule),2 14 although significant lesions may develop earlier, as in the reported patient. Pancreas and testes should also be monitored no later than from the second decade on, with annual ultrasound evaluation.2 14 In fact, recently, Goldstein and Hoffenberg14 have suggested initial screening at age 4–5 years with capsule endoscopy and upper and lower endoscopy, as well as evaluation for testicular tumours.
Currently, only mutations in the STK11 gene (small arm of chromosome 19, position 19p13.3) have been described as causing PJS. The gene STK11 includes 22637 base pairs and 10 exons, 9 of which contain a coding sequence of 48.6 kDa protein, which fulfils the function of serine-threonine kinase.3 6 It is a suppressor gene, and the kinase encoded by it plays an important role in cell metabolism regulation and proliferation, cell polarity, p53-mediated apoptosis and in other signalling pathways.3 6 9
Until now, over 230 distinct STK11 gene mutations with a heterogeneous nature have been identified in the Human Gene Mutation Database: 72 point missense/nonsense mutations and 26 mutations in splicing sites, as well as 102 small insertions, deletions and indel-type mutations.2 6 The remaining 31 mutations described concern rearrangements of larger fragments of the STK11 sequence, including 25 large deletions, 3 large insertions and 3 combined mutations.2 6 Interestingly, a notable proportion of PJS samples do not carry any mutation in STK11, suggesting possible genetic heterogeneity in the disease and the potential existence of another causative genetic locus, yet to be identified.3 6 7
Previous reports reveal that molecular testing of this gene identifies disease-causing mutations in 80–94% of affected individuals.5 6 There is no family history of PJS in approximately 45% of patients,3 6 but the exact proportion of de novo gene mutations is still unknown.3 6 9 Thus family testing is important. The presence of these genetic variants in healthy parents would make these variants benign, as this is an autosomal-dominant condition. However, it is not possible to exclude PJS with certainty in a child’s parents and siblings simply on a clinical basis, because it may occur without pigmented lesions or GI problems.3 6 If neither parent is a carrier, the possibility of being de novo mutations would be more likely, although other possibilities should be excluded (alternate paternity or maternity).
Despite the high detection rate of direct sequencing of the STK11 gene, enabling detection of more than 95% of the most prevalent mutations (reduced to 70% in the absence of family history),3 6 larger rearrangements and longer deletions or duplications detectable by other methods may have been missed.3 6 Most of the described variants in patients with PJS seem to affect the catalytic kinase domain of the protein (encoded by aminoacids 49–309), resulting in absence of kinase activity and disruption of the STK11 functions.3 6 Despite the presence of two variants in STK11 gene in the reported case, it is not yet possible to establish their causality. If both of these mutations are confirmed to be pathogenic, one may speculate that the function of STK11 gene and its protein might even be more profoundly affected, therefore determining a more severe phenotype, with diffuse and continuous growing of large polyps, and a potentially greater risk of oncogenic transformation.
Results from genotype–phenotype studies have so far been controversial.10 Lim et al12 associated the presence of a STK11 mutation to greater oncogenic risk, differently from other authors, considering that its type or site would not influence cancer risk. On the contrary, nonsense mutations seem to predispose to more GI surgeries, a higher polyp count, an earlier age at first polypectomy8 10 15 and higher frequency of cancers.8 Also, Amos et al10 reported that missense mutations were associated with later onset of symptoms. Recently, Fu et al15 reported two new nonsense STK11 mutations in two boys undergoing first surgery at very young ages (2 and 4 years).
Little is known about the genetic implications of specific STK11 mutations with regard to their role in dysplastic and malignant transformation of GI polyps. Wang et al16 investigated STK11 mutations and genotype–phenotype correlations in a study involving 116 Chinese patients with PJS from 52 unrelated families. The mutation detection rate was 67.3% and, interestingly, 29.6% of all mutations were in exon 7, the shortest out of the nine exons. Strikingly, mutations affecting protein kinase domain XI, encoded in part by exon 7, correlated with a 90% (9/10) incidence of GI polyp dysplasia.
In another relevant study from Mehenni et al,17 the presence of pathogenic mutations in the LKB1/STK11 gene in 46 unrelated PJS families was assessed and genotype–phenotype correlation searched, concerning the development of cancer in 170 PJS family members. In 59% (27/46) of unrelated PJS cases, pathogenic mutations in the LKB1/STK11 gene, including nine novel mutations, were identified. The new mutations were two splice site deletion-insertions, two missenses, one nonsense and four abnormal splice sites. Genotype–phenotype analysis did not yield any significant differences between patients carrying mutations in LKB1/STK11 versus those without mutations, even with respect to primary biliary adenocarcinoma. These studies increased the mutational spectrum of LKB1/STK11 allelic variants.
Another interesting feature deserving further elucidation is the occurrence of individuals with PJS-like pigmentation but no polyposis, designated as isolated mucocutaneous melanotic pigmentation (IMMP). Boardman et al18 identified a significantly increased relative risk for breast and gynaecologic cancer in 26 women with IMMP, although LKB1 mutations were not detected in the patients tested.
Conclusion
The reported case illustrates the typical course of PJS (continuous growing of hamartomatous polyps with a diffuse GI distribution), as well as the rare exuberant GI expression with significant clinical consequences at a very young age. Despite the paradigmatic phenotype, the whole clinical impact of the identified genetic variants remains undetermined, particularly in relation to polyp development, long term outcome and oncogenic risk. Further molecular study, extensive to family members, might elucidate if a de novo mutation has occurred. Finally, contribution of novel diagnostic (MR enterography, videocapsule) and therapeutic approaches, such as double-balloon enteroscopy, enabling accurate GI evaluation and excision of lesions with more remote intestinal location, will help in improving quality of life and life expectancy of these patients. In conclusion, two STK11 gene variants (one not previously reported) were found in a paediatric patient presenting with extensive GI involvement at a very early age, with no family medical history. The present report widened the spectrum of STK11 variants in patients with PJS. Further genotype–phenotype accurate studies should elucidate structural and functional repercussion of newly described variants.
Learning points.
The present report should alert clinicians to Peutz-Jeghers syndrome (PJS) diagnostic criteria and its typical course, emphasising a rare exuberant GI expression with clinical significance at a very young age.
Novel diagnostic and therapeutic approaches enable accurate GI evaluation and intervention, improving patients’ morbidity and mortality.
Two STK11 gene variants are reported (one not previously described), of yet undetermined causality; molecular study including family studies is of paramount importance, as it can further contribute to elucidating genotype–phenotype associations.
Individualised surveillance strategies are crucial in all patients with PJS at the paediatric age.
Acknowledgments
Dr Ana Maria Palha, Pathology Department, University Hospital Santa Maria (histopathological evaluation); Dr Luisa Lobo, Imagiology Department, University Hospital Santa Maria (Imagiology evaluation).
Footnotes
Contributors: All the authors collaborated to manuscript writing; additionally, AIL was responsible for supervision and overall review of the manuscript, in charge of the patient and endoscopy performance; JD performed the molecular study.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.El Tayeb AA, Ibrahim NH, El Tayeb AA et al. Peutz-Jeghers syndrome in children and adolescents. Ann Pediatr Surg 2008;4:37–41. [Google Scholar]
- 2.Beggs AD, Latchford AR, Vasen HF et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut 2010;59:975–86. 10.1136/gut.2009.198499 [DOI] [PubMed] [Google Scholar]
- 3.McGarrity TJ, Amos CI, Frazier ML et al. Peutz-Jeghers syndrome. GeneReviews™. Seattle: University of Washington, 1993–2015. (Updated 25 July 2013). http://www.ncbi.nlm.nih.gov/books/NBK1266/ (accessed 18 Jan 2015). [Google Scholar]
- 4.Andrade AC, Carvalho Júnior EC, Dantas KS et al. Síndrome de Peutz-Jeghers, Relato de caso. Rev Col Bras Cir 2008;35:210–11. 10.1590/S0100-69912008000300015 [DOI] [Google Scholar]
- 5.Tchekmedyian A, Amos CI, Bale SJ et al. Findings from the Peutz-Jeghers syndrome Registry of Uruguay. PLoS ONE 2013;8:e79639 doi:10.1371/journal.pone.0079639 (accessed 18 Jan 2015). 10.1371/journal.pone.0079639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borun P, Bartkowiak A, Banasiewicz T et al. High resolution melting analysis as a rapid and efficient method of screening for small mutation in the STK11 gene in patients with Peutz-Jeghers syndrome. BMC Med Genet 2013;14:58 http://www.biomedcentral.com/1471-2350/14/58 (accessed 18 Jan 2015). 10.1186/1471-2350-14-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu D, Guo H, Xu X et al. Two variants in STK11 gene in Chinese patients with Peutz-Jeghers syndrome. J Genet 2012;91:205–8. 10.1007/s12041-012-0152-8 [DOI] [PubMed] [Google Scholar]
- 8.Salloch H, Reinacher-Schick A, Schulmann K et al. Truncating mutations in Peutz-Jeghers syndrome are associated with more polyps, surgical interventions and cancers. Int J Colorectal Dis 2010;25:97–107. 10.1007/s00384-009-0793-0 [DOI] [PubMed] [Google Scholar]
- 9.Papp J, Kovacs ME, Solyom S et al. High prevalence of germline STK11 mutations in Hungarian Peutz-Jeghers syndrome patients. BMC Med Genet 2010;11:169 http://www.biomedcentral.com/1471-2350/11/169 (accessed 18 Jan 2015). 10.1186/1471-2350-11-169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amos CI, Keitheri-Cheteri MB, Sabripour M et al. Genotype-phenotype correlations in Peutz-Jeghers syndrome. J Med Genet 2004;41:327–33. 10.1136/jmg.2003.010900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olschwang S, Boisson C, Thomas G. Peutz-Jeghers families unlinked to STK11/LKB1 gene mutations are highly predisposed to primitive biliary adenocarcinoma. J Med Genet 2001;38:356–60. 10.1136/jmg.38.6.356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim W, Hearle N, Shah B et al. Further observations on LKB1/STK11 status and cancer risk in Peutz-Jeghers syndrome. Br J Cancer 2003;89:308–13. 10.1038/sj.bjc.6601030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopes AI, Gonçalves J, Palha AM et al. Síndrome de Peutz-Jeghers. Diversidade de Expressão Gastrointestinal em Idade Pediátrica e Considerações sobre a sua Abordagem Clínica. Acta Med Port 2004;17:445–50. [PubMed] [Google Scholar]
- 14.Goldstein SA, Hoffenberg EJ. Peutz-Jegher syndrome in childhood: need for updated recommendations? J Pediatr Gastroenterol Nutr 2013;56:191–5. 10.1097/MPG.0b013e318271643c [DOI] [PubMed] [Google Scholar]
- 15.Fu J, Wen Z, Wang F et al. Genetic and Clinical Analyses of Southern C Chinese children with Peutz-Jeghers syndrome. Genet Test Mol Biomarkers 2015;19:528–31. 10.1089/gtmb.2015.0109 [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Wu B, Mosig RA et al. STK11 domain XI mutations: candidate genetic drivers leading to the development of dysplastic polyps in Peutz-Jeghers syndrome. Hum Mutat 2014;35:851–8. 10.1002/humu.22549 [DOI] [PubMed] [Google Scholar]
- 17.Mehenni H, Resta N, Guanti G et al. Molecular and clinical characteristics in 46 families affected with Peutz-Jeghers syndrome. Dig Dis Sci 2007;52:1924–33. 10.1007/s10620-006-9435-3 [DOI] [PubMed] [Google Scholar]
- 18.Boardman LA, Pittelkow MR, Couch FJ et al. Association of Peutz-Jeghers-like mucocutaneous pigmentation with breast and gynecologic carcinomas in women. Medicine (Baltimore) 2000;79:293–8. 10.1097/00005792-200009000-00002 [DOI] [PubMed] [Google Scholar]