

Abstract
After decades of believing the heart loses the ability to regenerate soon after birth, numerous studies are now reporting that the adult heart may indeed be capable of regeneration, although the magnitude of new cardiac myocyte formation varies greatly. While this debate has energized the field of cardiac regeneration and led to a dramatic increase in our understanding of cardiac growth and repair, it has left much confusion in the field as to the prospects of regenerating the heart. Studies applying modern techniques of genetic lineage tracing and carbon-14 dating have begun to establish limits on the amount of endogenous regeneration after cardiac injury, but the underlying cellular mechanisms of this regeneration remained unclear. These same studies have also revealed an astonishing capacity for cardiac repair early in life that is largely lost with adult differentiation and maturation. Regardless, this renewed focus on cardiac regeneration as a therapeutic goal holds great promise as a novel strategy to address the leading cause of death in the developed world.
I. INTRODUCTION
For decades the common dogma was that the adult heart is incapable of regenerating lost myocardium after injury. Achieving cardiac regeneration or stimulating endogenous repair mechanisms to restore cardiac function after injury has been a goal of countless investigators. The longstanding belief that the adult heart has lost its capacity for self-renewal was a result of two simple observations. First, after myocardial infarction, there does not appear to be significant self-healing; instead, the primary repair mechanism is scar formation. Second, primary cardiac cancers are exceedingly rare, and cardiac rhabdomyosarcomas arising from cardiac myocytes are even more so. Furthermore, cardiac rhabdomyosarcomas are primarily felt to be embryonal in origin, not from mature adult cardiac myocytes, consistent with an extremely limited ability of cardiomyocytes to reenter the cell cycle.
In the last decade scientists have questioned whether the mature heart truly lacks the ability to create new myocardium after injury and instead have proposed that there may be significant endogenous regenerative capacity. Numerous reports of both adult cardiac myocyte proliferation and cardiomyogenesis by various endogenous progenitors have been published (Figure 1) (53). These analyses are particularly challenging as the outcome of cell cycle activity is not necessarily cardiac division but instead can be one of many possibilities (Figure 2). Assessing and integrating these often conflicting research reports that both support and alternatively refute the regenerative capacity of the adult mammalian heart has become increasingly difficult. While the debate has certainly fueled renewed interest in the field of cardiac regeneration and expanded our understanding of cardiac growth and repair dramatically, it has left many researchers uncertain of the prospects of regenerating the heart, a therapeutic goal that investigators have pursued for over half a century. We will critically review the data that support both sides of this field of cardiac regeneration and the data that have attempted to quantify cardiomyogenesis using modern approaches. Likewise, we will briefly review the strategies presently being pursued to regenerate the heart after injury including the use of stem cells, which are already being used in clinical trials.
FIGURE 1.

Potential sources of new cardiomyocytes in the adult heart. Schematic diagram summarizing the potential sources of new cardiac myocytes that have been proposed to contribute to myocyte turnover in the adult heart.
FIGURE 2.
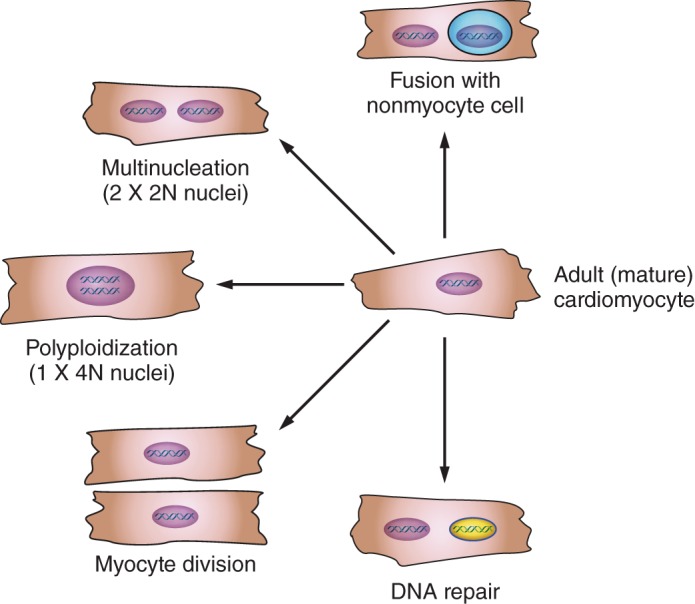
Multiple cell fates are associated with DNA synthesis and reexpression of cell cycle proteins. Multinucleation (DNA replication with karyokinesis but no cytokinesis), polyploidization (DNA replication without karyokinesis or cytokinesis), fusion of nonmyocytes with cardiac myocytes, or DNA repair can all be associated with DNA synthesis and/or reexpression of cell cycle proteins but does not necessarily represent true cardiac myocyte division.
II. HISTORICAL PERSPECTIVE
A. Cardiac Myocyte Proliferation in Lower Vertebrates
Unlike mammals, lower vertebrates are well known to retain a robust potential to regenerate organs after injury including the heart. Cardiomyocytes isolated from newts reenter the cell cycle when stimulated with mitogens, with half these cardiac myocytes becoming multinucleated while the other half undergo division. To divide, newt cardiomyocytes need to partially dissembled their sarcomeric structures and dedifferentiate (phenotypically regress from a differentiated cardiac myocyte into more primitive cell state) (113). Mature zebrafish or newts have substantial cardiac regenerative capacity, being able to restore myocardial structure even after removal of a large portion of the heart apex (40, 47, 54). To identify the origin of the new cardiac myocytes in the regenerated myocardium, investigators created transgenic zebrafish with a cardiac myocyte-specific genetic tracking reporter system (40, 47, 56). In this transgenic model, Cre recombinase-mediated recombination resulted in permanent green fluorescent protein (GFP) labeling of mature cardiac myocytes after exposure to tamoxifen while all the nonmyocytes (including progenitor cells) are GFP negative (Figure 3). When the apex of hearts from these transgenic fish was amputated, preexisting GFP-positive cardiomyocytes dedifferentiate, reenter the cell cycle, then redifferentiate into mature tissue, restoring normal architecture of the heart. There was no contribution from progenitor cells to the regenerated myocardium (40, 47). These dedifferentiated cardiac myocytes originated from both ventricular and atrial cardiomyocytes, but both sources differentiated into mature ventricular cardiomyocytes (133). The basis for the robust regenerative potential of lower vertebrates may be due to the plasticity of their cells. Newt cardiomyocytes implanted into regenerating limbs lost their cardiac phenotype and were able to transdifferentiate into skeletal muscle or chondrocytes. This reprogramming of cardiomyocytes required contact with the limb blastema, prompting the authors to postulate that signals from the limb blastema mediated dedifferentiation of cardiomyocytes, cell proliferation, and redifferentiation into cells of a different fate. Though a “lesser” vertebrate, 70% of the 26,000 zebrafish genes have human counterparts (humans have an estimated 20,000 genes). Therefore, dissecting cellular and molecular mechanisms of heart regeneration in these model organisms may help reshape our approaches in human heart regeneration.
FIGURE 3.
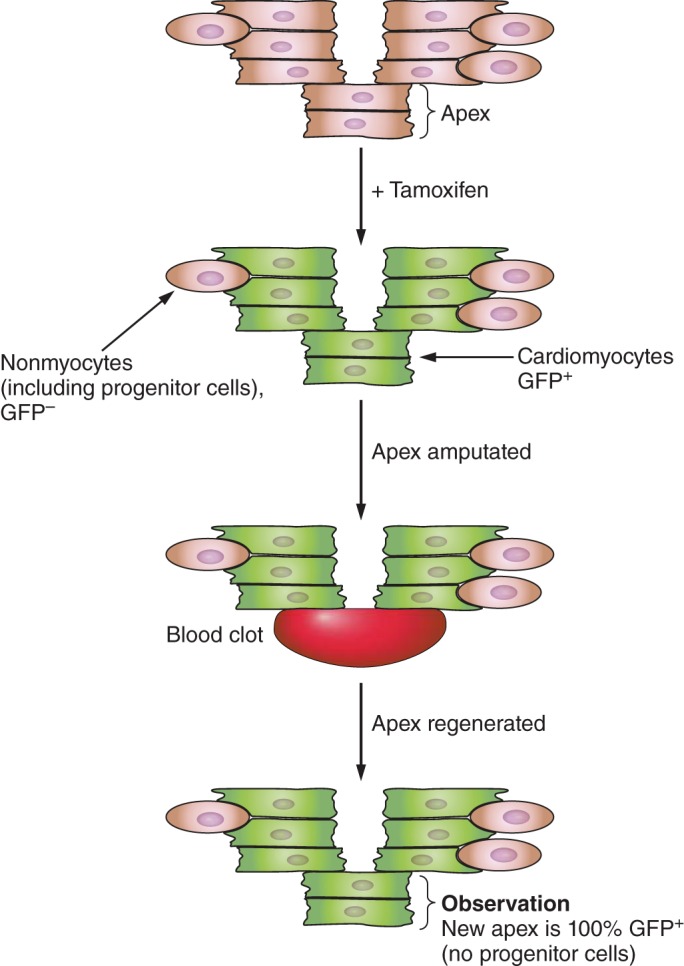
Regenerated cardiomyocytes are derived from preexisting cardiomyocytes not progenitor cells. Schematic diagram of genetic lineage tracing strategy used in zebrafish to identify the source of new cardiac myocytes in the regenerated heart. Cardiomyocytes in transgenic zebrafish were genetically labeled by inducing Cre activity with tamoxifen. These embryos were then grown to adulthood when the heart was amputated and allowed to regenerate. Preexisting cardiomyocytes (green: GFP+) in the injure borderzone dedifferentiate and proliferate to regenerate the resected apex over a period of 30 days.
B. Origins of the Dogma That the Mammalian Heart Is a Nonregenerative Organ
Until recently, our understanding of growth in the postnatal heart and its limited regenerative capacity was based primarily on studies performed in the 1970s–1980s (132). The common dogma was that during mammalian heart development, fetal cardiomyocytes divide robustly, but after birth cardiac myocytes withdraw from the cell cycle and become the highly structured, postmitotic cells that characterize the adult heart (25). Unlike skeletal muscle cells that proliferate as undifferentiated myoblasts, proliferating cardiomyoblasts express a full panel of sarcomeric proteins and contract in the fetal heart. When these cardiac myocytes divide, the sarcomeres disassemble to allow for cytokinesis and then reassemble in the two contractile daughter cells similar to newt cardiomyocytes (113). A detailed analysis of cardiac myocyte size and number in adult human hearts concluded that while the mass of the heart may change through adolescence and in different disease conditions, the number of cardiomyocytes does not (131), although this has recently been challenged (68, 73). Thus it was thought that while adult cardiac myocytes in lower vertebrates retain proliferative potential, the ability for such division dramatically decreases soon after birth in mammals, and the number of cardiomyocytes remains fairly stable through life (68, 104). Cardiomyocytes appear to grow to a finite size and length, which is conserved across mammals, independent of body mass (131). It has been speculated that this stable cell cycle exit was an evolutionary advance in response to the highly structured morphology of the contractile proteins in the adult heart, which allows cardiac myocytes to generate the contractile force needed to sustain higher blood pressure in mammals (132). Regardless, at this time it was felt that most adult mammalian cardiomyocytes enter a stable state of cell cycle senescence, and further postnatal growth of cardiomyocytes and increase in heart mass was almost entirely due to hypertrophy of existing myocytes. This lack of cardiac myocyte cell cycle reentry in mice even after growth stimuli was supported by detailed in vivo analysis using genetic tracking systems (102). Most of the cell cycle activity reported in the adult heart was presumed to arise from nonmyocytes or very limited cell cycle reentry in adult cardiac myocytes that resulted in multinucleation or increased ploidy without proliferation. Although cardiomyocytes account for 90% of the mature heart mass, they make up only 40% of the total cells by number (71). The remaining balance of cells in adult hearts is fibroblasts, endothelial cells, or vascular smooth muscles cells. These noncardiomyocyte cells retain the capacity to divide particularly with stress such as myocardial infarction complicating studies attempting to assess heart regeneration simply by looking at cells reentering the cell cycle.
The advent of radioactive probes allowed a reexamination of the regenerative capacity of rodent hearts. Radiolabeled thymidine should only be incorporated into DNA during S phase of the cell cycle (or to a much lesser extent during DNA repair). In cells capable of replication, exogenously delivered tritiated thymidine ([3H]thymidine) is incorporated into the newly replicated DNA stably marking the cell. This method allowed scientists to identify cell types that are actively dividing in vivo during normal and disease states. These studies demonstrated that <0.0005% adult cardiac myocytes incorporated thymidine over a 24-h period, suggesting a very low capacity for cardiac myocytes to reenter the cell cycle (103). More recent measurements using heavy isotope in a myocyte-specific genetic cell fate mapping mouse model demonstrated a similar rate, which is equivalent to a yearly turnover rate of ∼1% (30, 104). A confounding variable in all studies of cardiac cell cycle activity in vivo is that there are marked differences in rates of ploidy and nuclei number per cardiac myocyte between species (Table 1) (53). Approximately 25% of human cardiomyocytes are binucleated, whereas 75% are binucleated in mice (57, 79). In contrast to normal DNA copy number in mice, many human cardiac myocyte nuclei show increased ploidy (53). In fact, much of the cardiac myocyte cell cycle activity during both normal aging and after myocardial injury lead to an increase in myocyte polyploidy and multinucleation as opposed to new diploid, mononucleated myocytes. This highlights the complex outcomes possible when adult cardiomyocytes enter the cell cycle and difficulty when trying to accurately assess cardiac myocyte proliferation in vivo (96, 104). Even demonstrating mitotic figures could simply represent nuclear division resulting in more nuclei per cardiac myocytes as opposed to true myocyte cytokinesis (8).
Table 1.
Recent studies of cardiomyocyte cell cycle activity in normal and injured hearts
| Age | Per-Day Cycling Rate,* % | Nucleation (n) or Ploidy (N) of New (Cycling) Myocytes | Techniques and Conditions | Reference Nos. |
|---|---|---|---|---|
| Mouse | ||||
| Neonate | 1.00 | 2N ∼66%; 4N ∼29%+ | αMHC-MCM;Z/EG & 15N labeling; MIMS imaging | 96 |
| Young (2–4 mo old) | Sham 0.011 | 1n >2n; no change in ploidy per nucleus; 2N >4N per cell | αMHC-MCM;Z/EG; BrdU labeling 5 wk; isolated cells; flow cytometry | 66 |
| MI 0.021 | ||||
| Young adult | Baseline 0.012 | Baseline: 1n, 49%; 1n/2N, 17% | αMHC-MCM;Z/EG & 15N labeling; MIMS imaging | 1 |
| Sham 0.019 | ||||
| MI 0.411 | MI: 1n/2N, 14%; | |||
| Old (22 mo) | 0.007 | NR | ||
| P7-P8.5 | 0.026 | NR | MADM transgenic model | 1 |
| Young adult | 0.013 | |||
| Human | ||||
| 20 yr | 0.0027 | 2N ∼30%; 4N ∼65%; 8N ∼5%; equal renewal rates in diploid or polyploidy cells | Retrospective 14C birth dating; nuclei; FACS | 9 |
| 70 yr | 0.0012 | |||
| 20 yr | 0.019 | Mostly 1n/2N; polyploidy <1% | Histology; H3pS10 or Ki67; sex chromosome for ploidy | 42 |
| 60 yr | 0.033 | 1n/2N ∼97%; polyploidy ∼3%NR | ||
| 100 yr | 0.088 | |||
| Infant (≤1 yr) | 0.04 | 1n/2N | H3P staining in isolated myocytes | 68 |
| 10–20 yr | 0.009 | |||
Per-day percentage is calculated based on the averaged renewal rate during the survey periods, or as reported in original studies, and according to the used technique identifying cycling cells. +Based on Walsh S et al. Cardiovasc Res 86: 365–373, 2010. NR, not reported.
These issues, namely, that evidence of DNA synthesis, reexpression of cell cycle proteins, or even nuclear division in cardiac myocytes does not necessarily represent true cardiac myocyte division, have been the major hurdles in the field when interpreting results (8). Cell cycle activity in adult cardiac myocytes can represent multinucleation (DNA replication with karyokinesis but no cytokinesis), polyploidization (DNA replication without karyokinesis or cytokinesis), fusion of nonmyocytes with cardiac myocytes, or DNA repair in addition to true proliferative activity (Figure 2) (93). It is potentially even more problematic in diseased hearts such as in heart failure patients where cardiac myocyte ploidy increases dramatically as a patient's heart dilates. The physiological significance of the increased DNA content per myocyte whether through increased nuclei or ploidy is unknown, but after unloading the heart and reducing left ventricular (LV) wall strain, ploidy reverts back towards a normal DNA compliment (123). While these studies support the idea that adult cardiac myocytes can reenter the cell cycle and synthesize DNA, definitive proof for adult cardiac myocyte division in vivo is difficult to document.
C. Controversies in Heart Regeneration Research
Not all investigators believe the heart is postmitotic, with some claiming the heart demonstrates robust self-renewal with cardiac myocyte proliferation and niches of endogenous stem cells. New myocyte formation was claimed to be necessary to balance the equally low rate of apoptosis reported in normal adult hearts (78). The controversies surrounding the presence of an endogenous cardiac stem cell are reviewed below, but with respect to cardiac myocyte division, studies often present images of cardiomyocytes with condensed chromatin that appeared to be in the prophase or metaphase stage of mitosis as proof of dividing cardiac myocytes (8). However, as stated above, these studies could not differentiate between a myocyte undergoing cell division from a more limited nuclear division that would result in increased nuclei per myocyte, which has been well documented to occur in diseased cardiac myocytes. This is particularly true for human cardiac myocytes where 25% are binucleated and 75% of the cardiomyocytes contain nuclei that are polyploidy (containing more than the two homologous copies of chromosomes) (131). In hypertrophied human hearts, this ploidy can increase such that in a moderate to severely hypertrophied heart only 10% of the cardiac myocytes have the normal 2N copies of DNA and 15–30% of the myocytes have as many as 16 copies of the chromosomal complement (132). Studies examining the hearts of cancer patients who received iododeoxyuridine (IdU) therapy, a thymidine analog, as a radiosensitizer for radiation therapy demonstrated the fraction of myocytes labeled by iododeoxyuridine ranged from 2.5 to 46% (42). Similar to thymidine, IdU is incorporated into new DNA of cells during S phase or the repair process. The authors concluded that 0.02–0.14% of cardiac myocytes regenerate each day, which would equate to 22% of the heart regenerating each year, but issues such as increases in ploidy were not addressed (42). These studies are in dramatic contrast to studies performed in mice using a radioactive thymidine analog which estimated much lower rate and recent studies using stable isotope labeling and multi-isotope imaging mass spectrometry (MIMS) allowing irrefutable identification of cardiac myocytes. The investigators demonstrated a low rate of cardiac myocyte DNA synthesis in normal hearts predominantly resulting in changes in ploidy but also infrequent de novo cardiomyocyte formation at a very low rate (<1%/year) through division of preexisting cardiomyocytes during normal ageing. This cardiac myocyte proliferation increased in areas adjacent to myocardial injury but still remained low (96). This conclusion is supported using a completely independent technology of in vivo clonal analysis dubbed “mosaic analysis with double markers” (MADM) in mouse hearts (1). In the MADM assay, the two daughter cells of a dividing cell are indelibly and uniquely “single-labeled” either GFP+ or RFP+ because of interchromosomal Cre-loxP recombination after S phase (137). The results of this analysis in adult mouse hearts demonstrated very low rates of new myocyte formation. While this group did not find difference of cardiac myogenesis between postinfarct or sham hearts, it may be related to the shorter follow-up period as well the intrinsic inefficiency of the recombination-based MADM system (35). Thus accumulating data, at least in mice, seem to strongly support the notion that adult cardiac myocyte cell cycle reentry occurs at a very low rate and myocyte division is rare.
It seems unlikely that technological issues alone would explain the discrepancy between reported regenerative potential of the human (42) compared with mouse, but species specific differences in ability to reenter the cell cycle could. To address this, investigators analyzed human heart samples that took advantage of the fact that prior to the Nuclear Test Ban Treaty of 1963, nearly two decades of nuclear bomb tests occurred above ground resulting in relatively high atmospheric levels of carbon-14. During this period, carbon-14 emitted from the fallout quickly equilibrated throughout the world as 14CO2 and when inhaled or ingested was incorporated into the DNA of newly forming cells. Once the test ban was in effect, levels of 14CO2 quickly decreased so that any new cells generated after the mid 1960s would no longer incorporate 14CO2 at the same rate and cells that were proliferating would dilute the levels of this isotope within their DNA. Through examination of a series of myocardial samples obtained from autopsies spanning this period, investigators demonstrated that at the age of 25, ∼1% per year of the cardiomyocytes were regenerated but by age 75; this number had fallen to 0.45% per year seemingly supporting the concept of very low rates of myocyte turnover in normal human hearts as well (9). Thus, while most studies indicate that the rate of postnatal cardiac myogenesis is very low and declines with age, there is no closure regarding this subject. A conflicting study also based on 14C incorporation in human cardiac myocytes and using a different model to interpret the results reported cardiomyocyte turnover in humans was 7–23% per year (41). The authors concluded that the adult human heart entirely replaces its myocyte population eight times in an average life span; however, this paper was subsequently retracted because of concerns over compromised data, although the specific details have not been made public (41).
So what conclusions can we make from these seemingly discrepant data? We believe that cardiac myocyte cell cycle reentry in normal hearts, whether mouse or human, after adolescence is low and rarely results in new myocyte formation (68, 73). Whether the marked changes in ploidy in human diseased hearts (12) represents a species specific difference in potential to reenter the cell cycle, or is more likely to result in new myocyte formation will require examination of 14C levels in hypertrophied or failing human hearts or prospective heavy isotope labeling experiments similar to mice as has been done to accurately calculate cycling rates in other human tissues (14). Regardless, any study in this area needs to be cognizant of the challenges in accurately measuring myocyte division and acknowledge the limitations of the methodologies used (3). To deal with this complexity, we believe histological examination of myogenesis should be complemented with assays on dissociated single cells including myocytes and nonmyocytes whenever possible. Studies that have used time-lapsed video microscopy to assess cycling in isolated cardiac myocytes have shown virtually no acute cell cycling or division in normal adult cardiac myocytes (10, 95). Likewise, cardiac-specific transgenic models allowing fate tracking that support objective, high-throughput analysis in dissociated single myocytes should be the gold standard when studying heart regeneration in animals. Furthermore, because new myocyte formation is much lower than in other cell types and spans a longer cell cycle progression period, cumulative labeling of proliferative cells with markers such as heavy isotopes or thymidine analogs provides more accurate quantification for myocardial regeneration, even though other cell cycling markers such as Ki67, PCNA, aurora kinase B, phosphorylated histone H3 at serine-10 or -780 (H3pS10, H3pS780) can reveal cell cycle activity at the specific time point being examined. Finally, recognition that markers of DNA synthesis or cycling do not equate with division in cardiac myocytes would dramatically reduce claims of cardiac myocyte proliferation and regeneration.
Although we have believed for decades that cardiomyocytes permanently exit the cell cycle after birth effectively preventing a regenerative response, new data suggest that there may be a persistent, but low, rate of new cardiac myocyte formation even in adult hearts. What is less certain is whether this is a general property of all cardiac myocytes and occurs through dedifferentiation similar to lower vertebrates or whether there is a subset of proliferation competent cardiac myocytes in the adult heart that accounts for this activity. In fact, there are data that indirectly support both of these models. Several investigators have claimed that mononuclear cardiac myocytes in mammals, similar to newt cardiomyocytes, account for the cell activity seen in stimulated hearts (10). Similarly, the ability of dedifferentiation cardiac myocytes to reenter the cell cycle has been known for some time, but this has now been suggested to occur in vivo as well (134). It may be that both processes are occurring as one group has suggested the low rates of myocyte turnover in the adult heart originate from both a pool of persistent cardioblasts (65) and very low rates of dedifferentiation of existing cardiac myocytes after injury (134). They also suggested the regenerative effects of cell therapy could be explained by stimulation of these endogenous cells (65, 66). Differentiating among these alternate models will require novel fate mapping approaches but regardless argues for optimism that approaches to stimulate endogenous cardiac myocyte proliferation is a viable therapeutic approach.
D. Do Endogenous Progenitors Give Rise to New Cardiac Myocytes in the Adult Heart?
Numerous groups have suggested the presence of endogenous cardiac progenitors that have been identified with a series of different cell surface markers. While c-kit+ cardiac stem cells have been promoted extensively as a potential source for endogenous cardiac stem cells, other markers have also been proposed to identify endogenous cardiac stem cells including the SCA-1 (Ly6A) (77) and ABC transporter cells (83) (side population cells that efflux Hoechst dye on flow cytometry) (Figure 1). Cardiospheres, a three-dimensional culture system, have been proposed as an alternative method to expand endogenous cardiac progenitors in vitro (67) even if the identity of the progenitor cell is unknown (100). While cardiospheres appear to contain a rare cardiac progenitor population that has limited cardiac myocyte differentiation potential (29), whether it represents an endogenous cardiac stem cell or is generated ex vivo by the cardiosphere process will await definitive markers that allow its precise identification. There is a vast amount of data demonstrating differentiation of isolated cardiac stem cells in culture or alternatively reinjecting cells expanded in vitro into hearts after injury, which has been reviewed by others (43). However, there are much less data supporting their presence in vivo and a paucity documenting a functional role for endogenous cardiac stem cells. In fact, the direct visualization of endogenous stem cells through coexpression of c-kit and Nkx 2.5 has been shown by a limited number of investigators in mouse or human hearts (7, 119).
To define the functional role of these cardiac stem cells in vivo, investigators have utilized genetic tracking systems to examine their contribution to new cardiomyocyte formation after myocardial infarction. With the use of a common LacZ-GFP labeling model along with an inducible, cardiac-specific Cre recombinase, it is possible to label ∼75% of the existing adult cardiac myocytes with GFP while the nonmyocyte population, presumably including the cardiac stem cell population, remains GFP-negative but LacZ positive. The initial report using this model concluded there was a significant contribution of endogenous progenitors to new myocyte formation after myocardial infarction when they observed a reduction of the percent of GFP-positive myocytes (36). This was expanded further in a subsequent study claiming that cell therapy could further enhance the myogenesis from an endogenous progenitor pool after injury (62). However, in response to criticisms that these results could be also secondary to differential sensitivity to cell death of cardiac myocytes expressing different tracking proteins (GFP versus LacZ), the authors developed a pulse-chase approach with 15N-labeled thymidine and use MIMS to determine new myocyte formation. In contrast to their earlier studies, this approach revealed no contribution of progenitor cells but seemed to support the concept that their earlier results reflected differential death rates as opposed to new myocyte formation from a progenitor cell pool. They demonstrated that the genesis of cardiomyocytes occurs at a very low rate (<1%/year) through division of preexisting cardiomyocytes during normal ageing, a process that increases adjacent to areas of myocardial injury (96). While there continue to be conflicting studies claiming endogenous progenitors are necessary and sufficient for the regeneration and repair of myocardial damage, these studies continue to use the problematic LacZ-GFP genetic tracking model described above that gave inconclusive results (24). The strongest evidence questioning the role of endogenous c-kit cardiac stem cells comes from a detailed genetic lineage mapping study that directly labeled endogenous c-kit cells within the heart and concluded that they contributed an insignificantly small number of cardiomyocytes in the mature adult heart even after injury disputing previous studies that claimed a critical role for c-kit cells in endogenous repair (120). Thus, although there appear to be cells that when isolated and propagated ex vivo have limited capacity to differentiate into cardiovascular cell types, definitive proof for a role for them in normal homeostasis or repair after injury is lacking.
III. STRATEGIES TO REGENERATE THE HEART
A. Stimulating Cardiac Myocyte Proliferation
While cardiac myocytes from lower vertebrates have a significant ability to dedifferentiate, proliferate, and then redifferentiate allowing reconstitution of damaged myocardium, this ability was thought to be lost in mammals. However, experiments published in 2011 suggest that we may be born with this capacity but lose it soon after birth (87). The apex of a newborn mouse heart can be resected, and by 21 days postinjury, the heart regenerates completely without evidence of scarring (87). Analysis of the regenerative process in these neonatal mice demonstrated that it is the cardiomyocytes themselves that dedifferentiate, reenter the cell cycle, and divide to regrow a normal heart. In contrast, if the mouse apex is removed 7 days post birth, the heart will undergo a fibrosis response and result in heavy scarring akin to what would be expected in the adult heart (87). A similar regenerative capacity was seen when newborn mice were subjected to a myocardial infarction (86). This finding is consistent with recent data suggesting there is persistence of cardiomyocyte proliferative capacity well beyond the perinatal period into preadolescence (73). While exciting and suggesting there may be developmental pathways that can be exploited for therapeutic purposes, at least one group has been unable to repeat these findings (2). The reason for this divergent regenerative response remained elusive; however, many investigators have since confirmed the original findings and capacity of the neonatal heart to regenerate itself (92).
Clearly, in disease states, adult mammalian cardiomyocytes have an ability to replicate DNA, but they rarely advance to the step of cytokinesis. Why mature mammalian cardiac myocytes lose their ability to undergo cell division remains unclear, but it correlates with a permanent silencing of G2M cell cycle genes similar to senescent cells. Also similar to senescent cells, the genes responsible for cell division become condensed and wrapped up into transcriptionally inactive heterochromatin in adult cardiac myocytes (95). When heterochromatin formation was disrupted allowing reexpression of cell cycle genes, the adult cardiac myocytes regained the ability to reenter the cell cycle. Of concern to those hoping to use cardiac myocyte proliferation as a therapeutic strategy, these hearts also became dilated and developed systolic dysfunction (95). Whether this dysfunction is specific to the model or a result of the transient disassembly of sarcomeres leading the loss of contractile function is unclear but may provide insight into why mammals have lost the ability for adult cardiac myocytes to reenter the cycle. Another recent report focusing on this perinatal period from day 3 to day 7 when the cardiomyocytes lose their ability to divide demonstrated that Meis1 (myeloid ecotropic viral integration site 1 homolog) is upregulated in myocardium in the early postnatal period and may mediate cardiomyocyte cell cycle exit. When Meis1 protein levels were reduced, there was a 10-fold increase in proliferation of cardiomyocytes within 1 wk (63). How these distinct mechanisms interact is unknown, but both are amenable to molecular manipulation and thus could be regulated to promote cardiac myocyte proliferation in vivo.
Another pathway implicated in regulating cardiac myocyte proliferation is the Hippo-YAP pathway. Hippo is a kinase that functions together with Yes-associated protein (YAP) to regulate cellular growth and survival in multiple organs, including the heart (34, 126). Hippo inactivates YAP, a transcriptional coactivator for TEAD sequence specific DNA-binding proteins, and thereby Hippo can suppress cell cycling. YAP can also stimulate cardiomyocyte proliferation and survival at least in part through Pik3cb by activating the PI3K-AKT pathway (61). Hippo through Yap impedes neonatal heart regeneration, resulting in increased scar formation (33, 125). Cardiac-specific YAP activation after myocardial infarction improved cardiac function, and reduced infarct size by stimulating cardiac myocyte proliferation (60, 125). Interestingly, activating YAP and cardiac myocyte proliferation did not seem to deleteriously affect heart function in adult myocardium (60), suggesting the results seen in earlier studies based on a strategy of deleting the retinoblastoma gene product (Rb) may be specific to that model (95). Together, these findings suggest that activating YAP or its downstream targets after myocardial injury may be a useful strategy to promote regeneration and improve cardiac function. However, a major limitation of this field is an understanding of how these various pathways implicated in cardiac myocyte proliferation interact and are related to each other.
Attempts to directly reactivate adult cardiomyocyte proliferation with cell cycle proteins after injury have proven remarkably difficult (Figure 4). Early studies overexpressed viral oncoproteins such as adenovirus E1A and SV40 large T antigen (SV40), which are well known for their ability in overriding cell cycle checkpoints. Transgenic mice expressing SV40 large-T antigen specifically in the ventricles developed ventricular hyperplasia or cardiomyopathy depending on the developmental timing of expression (23, 44) but also tumors when expressed in the atria (27). Likewise, overexpression of E1A or its downstream effector, E2F-1, activated DNA synthesis but resulted in widespread apoptosis that limits its usefulness as a regeneration strategy (48, 49). Since proliferating cardiac myocytes express high levels of cell cycle activators and low levels of cell cycle inhibitors (13, 84, 85), attempts to induce cardiomyocyte proliferation have often focused on overexpressing cell cycle activators or deleting the inhibitors in the adult heart. Constitutive expression of cyclin D1, D2, or D3 in transgenic mice stimulates DNA synthesis in adult myocardium at very low levels, but only cyclin D2 overexpressing mice demonstrated a reduced infarct size after injury, suggesting limited regeneration had occurred (82). Similar results were also seen in cyclin A2 overexpressing transgenic mice including reduced scar and improved LV function after injury (17, 20). These investigators attempted to translate this into a more clinically relevant model by delivering cyclin A2 via adenovirus to infarcted porcine heart. Cyclin A2 overexpression postinfarct improves cardiac function, which was attributed, at least in part, to enhanced myocyte proliferation (98). A major limitation of studies analyzing the effects of constitutively active transgenes is to differentiate mechanisms that may include dedifferentiation and proliferation of adult cardiac myocytes, stimulation of cardioblasts as suggested above, enhanced proliferation (and differentiation) of endogenous cardiac progenitor cells, or that a subset of cardiac myocytes in the transgenic hearts never underwent terminal differentiation because of the persistent expression of a positive cell cycle activator. Even studies that used adenoviral delivery of cyclin A2 into postinfarct hearts resulting in enhanced cardiac function associated with cardiac myocyte mitosis do not distinguish between these cellular mechanisms (98, 124).
FIGURE 4.
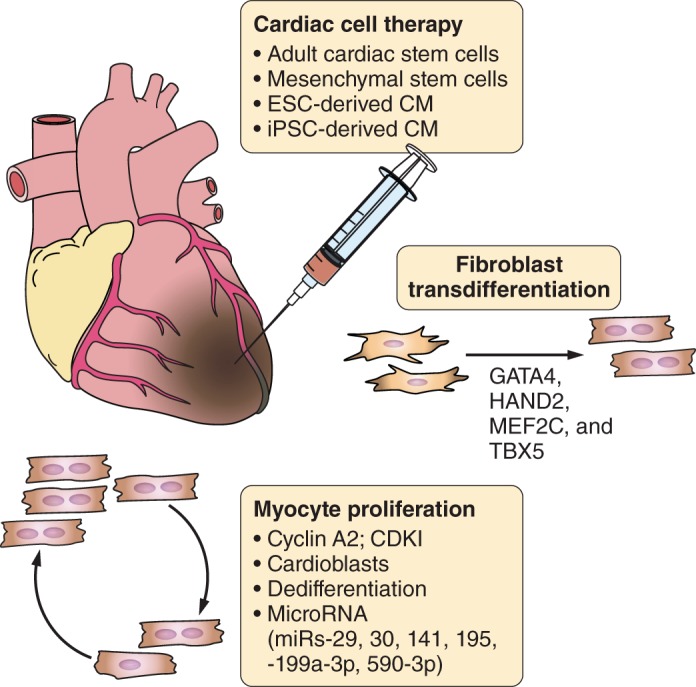
Strategies to regenerate adult heart after injury. Schematic diagram depicting strategies, outlined in the text, for regenerating new heart muscle after cardiac injury.
Cell cycle activity is dependent on the balance between active and negative regulators. These cell cycle inhibitors, which form complexes with cyclin-dependent kinases (CDKs), are dramatically upregulated in adult ventricular myocytes (85). Compared with ventricular myocytes, atrial myocytes express much lower level of CDK inhibitors p21, p53, and 14-3-3, which may explain why they retain the ability to renter the cell cycle even in adult hearts (134). Targeting inhibitory cell cycle molecules in neonatal or adult ventricular myocytes by knocking down CDK inhibitors p21(Waf1), p27(Kip1), and p57(Kip2) by siRNAs induced cell cycle reentry and cytokinesis. Triple knockdown promoted higher myocyte proliferation compared with targeting single or double CDK inhibitors. Whether this approach can stimulate true cardiac regeneration after injury and whether it can be translated to humans remains to be determined, but as of yet, no therapy targeting cell cycle regulators in the heart has yet reach clinical trials in humans.
Recently it was suggested that the signal that provokes cell-cycle arrest of neonatal CM is the perinatal surge in reactive oxygen species (ROS) that causes oxidative DNA damage eliciting a DNA damage response (DDR) in the heart during the first postnatal week (89). The DDR activates pathways through ATM/ATR and Chk1/2 kinases that block CDK activity, causing hypophosphorylation of Rb leading to cell cycle arrest followed by DNA repair or cell arrest with a senescence-like phenotype. While ROS may induce cell cycle exit, the end effectors were not identified (89). Interestingly, it has been reported that HP1 and H3K9me are crucial for DDR and prevent uncontrolled proliferation seen in carcinogenesis (55), which would link this pathway to the heterochromatin formation that silences cell cycle genes in adult cardiac myocytes. Reprogramming of the DNA methylome in postnatal cardiac myocytes may also confer another layer of epigenetic regulation that coordinates the balance of cardiac maturation and cell cycle activity (31). Targeting specific epigenetic changes to induce heart regeneration while promising remains largely untested (81).
A number of investigators have investigated the role of microRNAs (miRNAs) in cardiac myocyte cell cycle control (Figure 4). miRNAs regulate many regulatory pathways in cardiac development including cardiac myocyte proliferation and cell cycle exit. To identify miRNAs that might promote cardiac myocyte proliferation, investigators performed a high-throughput screen using a whole genome miRNA library in neonatal cardiomyocytes. Of the 875 miRNA mimics examined, 40 induced cell cycle progression including miR-590-3p and miR-199a-3p (26). Overexpression of miR-590-3p and miR-199a-3p in neonatal mice increased the proliferation of cardiomyocytes but not cardiac fibroblasts as well as reduced scar formation and improved cardiac function post-MI (26). Although the definitive targets of these miRNAs are unknown, it does suggest that they could be used as therapeutic reagents to promote myocardial regeneration following cardiac injury. With the use of a different approach based on developmental expression, miR-195 (a member of the miR-15 family) was identified as the most highly upregulated miRNA during the perinatal period. Knockdown of the miR-15 family in neonatal mice was associated with an increased number of mitotic cardiomyocytes (86), extending the regenerative window and improving LV function after myocardial infarction (88). Targeting additional miRNAs (miR-29, miR-30, and miR-141) that are upregulated during postnatal maturation but downregulated during dedifferentiation and proliferation of adult cardiac myocytes promotes myocyte cell cycle and proliferation (15, 135). Regardless of the approach, there are now numerous molecular targets that can be exploited to promote cardiac myocyte proliferation; however, this therapy is not without risks. Cardiac myocyte proliferation and dedifferentiation may induce heart failure if cardiac myocyte cell cycle reentry is associated with reduced contractile function (50, 95).
B. Cell Transplantation With Adult and Cardiac Stem Cells
1. Cardiac cell therapy with adult stem cells
Cardiac cell transplantation was first described in the mid-1990s by several groups using various cell types (70, 115). Transplanting cells directly into and around the injured myocardium reduced scar formation and improved LV function postmyocardial infarction (70, 114). Although there were initial reports that the mechanism of benefit of the transplanted cells was due to transdifferentiation into cardiac myocytes, this was subsequently proven not to be the case (91). Instead, it is now felt that the majority of the beneficial effects of these cells are related to the release of paracrine and autocrine factors that modulate apoptosis, inflammation, vascularity, and healing of the infarcted tissue (43). Thus, when investigators reported that a bone marrow-derived cell, expressing the cytokine receptor c-kit, could differentiate into cardiomyocytes and regenerate injured myocardium, there was intense interest. It was reported that these mobilized c-kit-positive cells could regenerate 68% of the infarcted left ventricle wall and improve cardiac function when directly injected into infarcted heart tissue (80). While this study garnered great excitement, two independent groups, using genetic lineage tracing and parabiotic systems, demonstrated that endogenous c-kit expressing cells from the bone marrow could not differentiate into cardiomyocytes and regenerate heart tissue. The bone marrow c-kit-positive cells did not demonstrate any significant conversion into cardiomyocytes, nor was there any significant capacity of these cells to restore heart function (6, 69). Despite questions as to the reproducibility of these findings, cell therapy with bone marrow-derived cells was translated into human subjects almost immediately (5, 106). However, after over a decade of use and thousands of patients receiving cell therapy, both the magnitude and mechanism of any benefits remain uncertain. Although investigators initially believed that the benefits of these bone marrow-derived cell arose from their ability to form cardiomyocytes by transdifferentiation similar to the claims in the 1990s, there is now general agreement that long-term engraftment of transplanted cells and differentiation of the injected cells to cardiomyocytes is very limited, if it occurs at all. Instead, investigators now believe the benefits of cardiac cell therapy are independent of engraftment and differentiation and that cells delivered to the heart release soluble factors that mediate cardiac repair, neovascularization, and cytoprotection in a paracrine manner (112). It has been proposed that transplanted cells activate endogenous regenerative mechanisms leading to cardiac myogenesis, but consensus on this new hypothesis will await more definitive lineage tracking experiments and confirmation by independent labs. Although large phase III trials that are adequately powered to determine the benefits of cell therapy are still lacking, recent meta-analysis of published trials comparing bone marrow-derived cells to conventional therapy concluded there was about a 3% absolute improvement in LV ejection fraction (28). Of concern with this conclusion is that there was a strong correlation between the number of discrepancies in a study and the reported increment in ejection fraction. In the five trials without discrepancies, there was no effect of bone marrow stem cell therapy on ejection fraction (74). While future clinical trials of bone marrow cell therapy may demonstrate that this is a promising approach after myocardial injury, there are little data to support that this benefit is secondary to new myogenesis.
2. Endogenous cardiac stem and progenitor cells
Given that investigators have reported endogenous cardiac stem cells it was assumed they might have added regenerative properties compared with other cell sources. To test this hypothesis, these studies have primarily concentrated on two sources of cells. The most work has focused on c-kit expressing cells that reside in murine and human hearts. Numerous animal studies have demonstrated that these cardiac c-kit cells could be isolated and transplanted into recipient animals reconstituting the infarcted ventricular wall, reducing infarct size and restoring LV function (7). On the basis of this promising preclinical work, a human clinical trial was begun in which c-kit+ cells from left atrial appendage at the time of bypass surgery were isolated and reinjected into infarcted tissue (SCIPIO trial) (11). An interim analysis suggested that intracoronary infusion of autologous cardiac c-kit cells is effective in improving LV systolic function and reducing infarct size in patients with heart failure after myocardial infarction. While these results are encouraging, there was an expression of concern after issues were raised over data presented in two supplemental figures (116). It is not clear how this expression of concern impacts the overall conclusions of the study, but hopefully ongoing studies will provide a definitive answer on the efficacy of cardiac c-kit cells.
Cardiosphere transplantation has also been proposed as a means of regenerating the heart after myocardial infarction. While cardiospheres contain a rare cardiac progenitor population that has limited cardiac myocyte differentiation potential (29), their therapeutic benefit in vivo is likely independent of this progenitor cell and has been proposed to be secondary to stimulating endogenous repair mechanisms including new myogenesis (59, 65). Although the active cell in cardiospheres is unknown, CD90-negative population tended to perform better in terms of cardiac differentiation potential and therapeutic benefit in injured hearts (19, 29). The first phase I trial in humans using cardiospheres, CADUCEUS, did not demonstrate a significant difference in LV ejection fraction or volumes between groups; however, compared with the control group, CDC-treated patients showed an increase in viable myocardium and reductions in scar mass at the 6-month time point (64). Although exciting, these results need to be viewed with caution as the numbers of patients were small and follow-up was not uniform. Clinical trials with larger numbers of patients and longer follow-up times are necessary to establish efficacy of endogenous cardiac cells in ischemic cardiomyopathy.
C. Pluripotent Stem Cells
1. Embryonic stem cells
The heart's limited regenerative ability has driven scientists to look for other sources of renewable cardiomyocytes. Human embryonic stem cells (ESC), first isolated by James Thomson in 1998 (117), offered a potential source. Embryonic stem cells are cultured cells isolated from the inner cell mass of the blastocyst in the early stages of embryogenesis. These cells retain the potential to differentiate into any cell type if given the appropriate growth factors. Initial attempts to culture cardiomyocytes relied on embryoid body formation, which gave poor yields of contractile cells, often less than 10% of the total population (45). There was also a belief that the heart milieu itself could provide either critical cell-cell interactions or growth factors to direct embryonic stem cells to a cardiac phenotype and integrate into surrounding myocardium. This notion was quickly dispelled as injected ESCs into mouse myocardium formed large teratomas rather than mature cardiomyocytes (75, 109). These studies also demonstrated that the differentiated products of ESCs are not “immune-privileged” as had been suggested for the parental cells (58) and were quickly rejected by recipient animals unless these animals were immunosuppressed (108). Since the cardiac milieu is incapable of directing cardiomyocyte specific differentiation, several groups focused on dissecting the signaling pathways that direct a primordial cell towards the cardiovascular lineage. Research performed in Xenopus models demonstrated that transforming growth factor (TGF)-β superfamily members play a central role in directing a cardiac fate. Initial signaling with activin induces mesoderm formation (51) followed by bone morphogenic proteins (BMPs) to direct ESCs into immature myocytes (107). Timed delivery of activin A and BMP-4 to human ESCs generates cultures where 30–80% of the cells are cardiomyocytes. These cardiac myocytes, while contractile, likely represent an immature developmental stage as they could be passaged and expanded in culture for some time (52, 127). However, these cells can be induced to mature in vitro if cultured in three-dimensional matrices under load-bearing or electrical stimulation conditions, which results in increased sarcomere banding, myofibril formation, and spontaneous contractions (97, 118). Human ESC-derived cardiac myocytes (ESC-CM) also matured into typical, mature rod-shaped myocytes when transplanted into primate hearts in vivo (21).
Human ESC-CM transplanted into the hearts of athymic rats (39) or immunocompetent guinea pigs treated with cyclosporine (99) have been shown to survive for up to 4 wk. ESC-CM also engraft in the pig heart and electrically couple to the host myocardium (46). Transplanted human ESC-CM postinfarct have been shown to improve fractional shortening or ejection fraction by 20–40% in rats (16, 52). In rodent postinfarct models, ESC-CM have also been shown to reduce spontaneous or induced ventricular arrhythmias (99). While exciting, there has been discussion of whether these improvements are durable since by 12 wk the enhanced cardiac function was no longer statistically different from the control animals (121). More recently, human ESC-CMs were shown to survive, engraft, and electrically couple to host myocardium in a non-human primate model (21). This is the first report of significant remuscularization by exogenous cells in a large animal model. However, the ESC-CM transplants demonstrated increased ventricular arrhythmias not seen in earlier experiments in rodents. The mechanism of these arrhythmias is unclear but may be due to the size of the grafts, developmental stage of the injected cardiac myocytes, or slower heart rate of primate host allowing reentry circuits. Clearly this issue, along with immunosuppression and potential for teratoma formation, will need to be addressed before this can be translated into humans, but this remains a very promising approach to remuscularize infarcted hearts.
2. Induced pluripotent stem cells
Soon after the fusion of sex specific germ cells to create the zygote, a wave of genetic modifications starts to silence genes and limit the cell's ability from becoming any cell type as the process of differentiation to a more specified cell type begins. Although mature somatic cells still contain all the genetic information of that first totipotent zygote, it no longer retains an ability to revert back to an earlier state. In the 1950s botanists famously isolated a somatic cell from a carrot and were able to culture and grow a new carrot demonstrating, at least in plants, an ability for somatic cells to overcome their lineage restrictions and revert back to a totipotent state (105). This was followed rapidly by cloning of Xenopus in 1958 (32) and later of larger mammals cloned by transferring the nucleus from a somatic cell to an unfertilized egg (122). This demonstrated a long-held belief that somatic cells retained all the genetic information required to recapitulate a totipotent cell. The method, namely, somatic cell nuclear transfer (SCNT), by which a somatic cell nucleus could be reprogrammed was not unique to the unfertilized egg as researchers discovered that fusion with ESCs could also induce a similar change (22). These pioneering studies laid the foundation for the landmark study published in 2006 demonstrating that a cocktail of four genes (c-Myc, Oct3/4, Sox2, and Klf4) known to maintain a pluripotent state in stem cells, when expressed in mouse fibroblasts using retroviruses, could convert these somatic cells into cells with capabilities similar to ESCs. These cells were called “induced pluripotent stem” (iPS) cells (111). One year later, fibroblasts from humans were reprogrammed back to a pluripotent stem cell state using similar cocktails of genes: one set with c-MYC, OCT3/4, SOX2 and the other set with KLF4, or OCT3/4, SOX2, NANOG, LIN28 (110, 130). Since these discoveries, many papers have emerged demonstrating novel methods for achieving similar results using fewer genes (37, 72) or nonviral methodologies that aimed to avoid potentially mutagenic effects of integrating viral delivery methods (76). Investigators have also been able to substitute many of the original factors with the addition of small molecules (37) or miRNAs(4) or by modifying culture conditions to promote pluripotency (129). These methods have made generating iPS cell lines faster and cheaper, but the significance of residual epigenetic and transcriptional aberrations after reprogramming on their safety in clinical settings will need to be determined before they replace more convention pluripotent stem cell sources. Careful analysis of pluripotent stem cells derived via SCNT or by iPSC factors revealed comparable levels of genomic aberrations, suggesting neither reprogramming method is superior and that genomic instability may be inherent to the reprogramming process (39). Therefore, ESC will probably remain the first choice for human cell therapy until the functional consequences of these aberrations are known or new reprogramming techniques are developed that prevents them from accumulating.
Injection of iPS-derived cardiac myocytes, smooth muscle cells, and endothelial cells demonstrated engraftment of all cell types (128). Interestingly, there were significant improvements in LV function despite relatively low rates of engraftment compared with earlier studies using ES cell-derived cardiac myocytes in nonhuman primates (21). The authors suggest that the benefits might be multifaceted including reductions in apoptosis, angiogenesis, and improved metabolism. This highlights the fact that cardiac cell therapy may work by multiple mechanisms distinct from remuscularization, and whether regenerating significant amounts of new myocardium has added benefit needs to be determined.
Numerous investigators have demonstrated that iPS cells can be differentiated into cardiac myocytes (94) and that these cells demonstrate properties comparable to cardiomyocytes derived from ESCs (94). Armed with the knowledge that somatic cells could be reprogrammed using combinations of transcription factors, researchers have attempted to reprogram fibroblast directly into differentiated cells (Figure 4). A combination of three transcription factors was used to convert exocrine pancreatic cells to endocrine cells or neurons (136). In 2010, a trio of transcription factors (Gata4, Mefc2, and Tbx5) were identified after screening a pool of 14 candidate genes involved in cardiomyocyte development that when infected into mouse fibroblasts could induce 20% of cells to become cardiomyocyte-like cells, with the first signs of cardiomyocyte specific protein expression after only 3 days (38). However, other investigators have not been able to reproduce this efficiency using the three transcription factor cocktail (18), and a later study suggested that four transcription factors, GATA4, HAND2, MEF2C, and TBX5, were required to efficiently transform fibroblasts into beating cardiac-like myocytes in vitro (101). Infecting the postinfarct heart with these transcription factors (90) or combining them with HAND2(101) resulted in 4–35% of the cardiomyocytes in the region bordering the infarct zone being derived from the infected fibroblasts, with a reduction in scar area and improved LV function. However, these studies are in their infancy, and it is likely as the field matures that issues will also arise from this approach. Obviously, if these findings prove reproducible, it could provide another therapy to generate de novo cardiac myocytes in vivo. The robustness of the conversion of fibroblasts is poor, and these studies have primarily been performed with rodent cells. Whether similar approaches will work in humans needs to be explored. Retroviral infection of fibroblasts has many caveats when considering human applications, namely, concern for infection frequency, random genomic insertion sites, tissue specific infection, and inflammatory reaction. Furthermore, while the results are indeed exciting, the frequency of fibroblast-derived cardiomyocytes in the infarct border region was small despite significant improvements in heart function and decreased scar size. The push to achieve more efficient cardiac transdifferentiation will need to be balanced by the potential negative consequences of depleting endogenous fibroblasts and endothelial cells, since these cell types also play a critical role in postinfarct repair.
IV. CONCLUSION AND FUTURE PROSPECTS
While there is growing consensus that the mammalian heart is capable of some self-renewal, it appears quite limited and insufficient to address the loss of cardiac myocytes after injury such as myocardial infarction. Despite numerous studies to the contrary, there is mounting evidence that the prospects of substantial myocardial regeneration outside of the early perinatal period without genetic manipulation or without transplanting exogenous myocytes is very unlikely. However, a number of exciting new technologies are being tested in preclinical through phase I human studies (Figure 4).
Despite its rapid translation into clinical studies, the field of cardiac cell therapy is in flux. Whether the beneficial effects attributed to adult stem cell therapy in ischemic heart disease are paracrine in nature or are related to the activation of a persistent endogenous repair system that is dormant under normal circumstances remains to be determined. In addition, whether the robust remuscularization feasible with pluripotent stem cells has additive benefits will need to be determined. This highlights the critical need to explore the additional benefit that ESC- and iPSC-based cardiac myocyte transplantation can provide over that by other cell sources incapable of cardiac differentiation. Regardless, the renewed interest in this field has stimulated multiple parallel fields of study that are likely to result in new potential therapies in treating cardiovascular disease.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01 HL70748 and P01 HL094374 (to W. R. MacLellan) and by American Heart Association Award 15BGIA22410026 (to Y. Zhang). Y. Zhang and W. R. MacLellan were in part supported by the Research Award from the John Locke Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
We thank the many post-docs and students that have contributed to this work and Kyohei Oyama and Daniel El-Nachef specifically for their helpful suggestions and comments on the manuscript.
Address for reprint requests and other correspondence: W. R. MacLellan, 1959 NE Pacific St., Box 356422, Seattle, WA 98195 (e-mail: wrmaclellan@cardiology.washington.edu).
REFERENCES
- 1.Ali SR, Hippenmeyer S, Saadat LV, Luo L, Weissman IL, Ardehali R. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc Natl Acad Sci USA 111: 8850–8855, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersen DC, Ganesalingam S, Jensen CH, Sheikh SP. Do neonatal mouse hearts regenerate following heart apex resection? Stem Cell Reports 2: 406–413, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ang KL, Shenje LT, Reuter S, Soonpaa MH, Rubart M, Field LJ, Galinanes M. Limitations of conventional approaches to identify myocyte nuclei in histologic sections of the heart. Am J Physiol Cell Physiol 298: C1603–C1609, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anokye-Danso F. Reprogramming somatic cells into pluripotent stem cells using miRNAs. Methods Mol Biol 1150: 273–281, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Assmus B, Schachinger V, Teupe C, Britten M, Lehmann R, Dobert N, Grunwald F, Aicher A, Urbich C, Martin H, Hoelzer D, Dimmeler S, Zeiher AM. Transplantation of Progenitor Cells and Regeneration Enhancement in Acute Myocardial Infarction (TOPCARE-AMI). Circulation 106: 3009–3017, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Balsam LB, Wagers AJ, Christensen JL, Kofidis T, Weissman IL, Robbins RC. Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature 428: 668–673, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114: 763–776, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P. Evidence that human cardiac myocytes divide after myocardial infarction. N Engl J Med 344: 1750–1757, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science 324: 98–102, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138: 257–270, 2009. [DOI] [PubMed] [Google Scholar]
- 11.Bolli R, Chugh AR, D'Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, Sanada F, Elmore JB, Goichberg P, Cappetta D, Solankhi NK, Fahsah I, Rokosh DG, Slaughter MS, Kajstura J, Anversa P. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet 378: 1847–1857, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Brodsky VY, Sarkisov DS, Arefyeva AM, Panova NW, Gvasava IG. Polyploidy in cardiac myocytes of normal and hypertrophic human hearts; range of values. Virchows Arch 424: 429–435, 1994. [DOI] [PubMed] [Google Scholar]
- 13.Brooks G, Poolman RA, McGill CJ, Li JM. Expression and activities of cyclins and cyclin-dependent kinases in developing rat ventricular myocytes. J Mol Cell Cardiol 29: 2261–2271, 1997. [DOI] [PubMed] [Google Scholar]
- 14.Busch R, Neese RA, Awada M, Hayes GM, Hellerstein MK. Measurement of cell proliferation by heavy water labeling. Nat Protoc 2: 3045–3057, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Cao X, Wang J, Wang Z, Du J, Yuan X, Huang W, Meng J, Gu H, Nie Y, Ji B, Hu S, Zheng Z. MicroRNA profiling during rat ventricular maturation: a role for miR-29a in regulating cardiomyocyte cell cycle re-entry. FEBS Lett 587: 1548–1555, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Caspi O, Huber I, Kehat I, Habib M, Arbel G, Gepstein A, Yankelson L, Aronson D, Beyar R, Gepstein L. Transplantation of human embryonic stem cell-derived cardiomyocytes improves myocardial performance in infarcted rat hearts. J Am Coll Cardiol 50: 1884–1893, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Chaudhry HW, Dashoush NH, Tang H, Zhang L, Wang X, Wu EX, Wolgemuth DJ. Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J Biol Chem 279: 35858–35866, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Chen JX, Krane M, Deutsch MA, Wang L, Rav-Acha M, Gregoire S, Engels MC, Rajarajan K, Karra R, Abel ED, Wu JC, Milan D, Wu SM. Inefficient reprogramming of fibroblasts into cardiomyocytes using Gata4, Mef2c, and Tbx5. Circ Res 111: 50–55, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng K, Ibrahim A, Hensley MT, Shen D, Sun B, Middleton R, Liu W, Smith RR, Marban E. Relative roles of CD90 and c-kit to the regenerative efficacy of cardiosphere-derived cells in humans and in a mouse model of myocardial infarction. J Am Heart Assoc 3: e001260, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng RK, Asai T, Tang H, Dashoush NH, Kara RJ, Costa KD, Naka Y, Wu EX, Wolgemuth DJ, Chaudhry HW. Cyclin A2 induces cardiac regeneration after myocardial infarction and prevents heart failure. Circ Res 100: 1741–1748, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Chong JJ, Yang X, Don CW, Minami E, Liu YW, Weyers JJ, Mahoney WM, Van BB, Palpant NJ, Gantz JA, Fugate JA, Muskheli V, Gough GM, Vogel KW, Astley CA, Hotchkiss CE, Baldessari A, Pabon L, Reinecke H, Gill EA, Nelson V, Kiem HP, Laflamme MA, Murry CE. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 510: 273–277, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cowan CA, Atienza J, Melton DA, Eggan K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science 309: 1369–1373, 2005. [DOI] [PubMed] [Google Scholar]
- 23.DeLeon J, Federoff HJ, Dickson DW, Vikstrom KL, Fishman GI. Cardiac and skeletal myopathy in beta myosin heavy-chain simian virus 40 tsA58 transgenic mice. Proc Natl Acad Sci USA 91: 519–523, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellison GM, Vicinanza C, Smith AJ, Aquila I, Leone A, Waring CD, Henning BJ, Stirparo GG, Papait R, Scarfo M, Agosti V, Viglietto G, Condorelli G, Indolfi C, Ottolenghi S, Torella D, Nadal-Ginard B. Adult c-kit(pos) cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell 154: 827–842, 2013. [DOI] [PubMed] [Google Scholar]
- 25.Erokhina IL, Rumyantsev PP. Ultrastructure of DNA-synthesizing and mitotically dividing myocytes in sinoatrial node of mouse embryonal heart. J Mol Cell Cardiol 18: 1219–1231, 1986. [DOI] [PubMed] [Google Scholar]
- 26.Eulalio A, Mano M, Dal FM, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492: 376–381, 2012. [DOI] [PubMed] [Google Scholar]
- 27.Field LJ. Atrial natriuretic factor-SV40 T antigen transgenes produce tumors and cardiac arrhythmias in mice. Science 239: 1029–1033, 1988. [DOI] [PubMed] [Google Scholar]
- 28.Fisher SA, Brunskill SJ, Doree C, Mathur A, Taggart DP, Martin-Rendon E. Stem cell therapy for chronic ischaemic heart disease and congestive heart failure. Cochrane Database Syst Rev 4: CD007888, 2014. [DOI] [PubMed] [Google Scholar]
- 29.Gago-Lopez N, Awaji O, Zhang Y, Ko C, Nsair A, Liem D, Stempien-Otero A, MacLellan WR. THY-1 receptor expression differentiates cardiosphere-derived cells with divergent cardiogenic differentiation potential. Stem Cell Reports 2: 576–591, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garbern JC, Lee RT. Cardiac stem cell therapy and the promise of heart regeneration. Cell Stem Cell 12: 689–698, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilsbach R, Preissl S, Gruning BA, Schnick T, Burger L, Benes V, Wurch A, Bonisch U, Gunther S, Backofen R, Fleischmann BK, Schubeler D, Hein L. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat Commun 5: 5288, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gurdon JB, Elsdalet R, Fischberg M. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature 182: 64–65, 1958. [DOI] [PubMed] [Google Scholar]
- 33.Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL, Martin JF. Hippo signaling impedes adult heart regeneration. Development 140: 4683–4690, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332: 458–461, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henner A, Ventura PB, Jiang Y, Zong H. MADM-ML, a mouse genetic mosaic system with increased clonal efficiency. PLoS One 8: e77672, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med 13: 970–974, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, Melton DA. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol 26: 795–797, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142: 375–386, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johannesson B, Sagi I, Gore A, Paull D, Yamada M, Golan-Lev T, Li Z, LeDuc C, Shen Y, Stern S, Xu N, Ma H, Kang E, Mitalipov S, Sauer MV, Zhang K, Benvenisty N, Egli D. Comparable frequencies of coding mutations and loss of imprinting in human pluripotent cells derived by nuclear transfer and defined factors. Cell Stem Cell 15: 634–642, 2014. [DOI] [PubMed] [Google Scholar]
- 40.Jopling C, Sleep E, Raya M, Marti M, Raya A, Izpisua Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 464: 606–609, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kajstura J, Rota M, Cappetta D, Ogorek B, Arranto C, Bai Y, Ferreira-Martins J, Signore S, Sanada F, Matsuda A, Kostyla J, Caballero MV, Fiorini C, D'Alessandro DA, Michler RE, Del MF, Hosoda T, Perrella MA, Leri A, Buchholz BA, Loscalzo J, Anversa P. Cardiomyogenesis in the aging and failing human heart. Circulation 126: 1869–1881, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Kajstura J, Urbanek K, Perl S, Hosoda T, Zheng H, Ogorek B, Ferreira-Martins J, Goichberg P, Rondon-Clavo C, Sanada F, D'Amario D, Rota M, Del MF, Orlic D, Tisdale J, Leri A, Anversa P. Cardiomyogenesis in the adult human heart. Circ Res 107: 305–315, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Karantalis V, Balkan W, Schulman IH, Hatzistergos KE, Hare JM. Cell-based therapy for prevention and reversal of myocardial remodeling. Am J Physiol Heart Circ Physiol 303: H256–H270, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katz EB, Steinhelper ME, Delcarpio JB, Daud AI, Claycomb WC, Field LJ. Cardiomyocyte proliferation in mice expressing alpha-cardiac myosin heavy chain-SV40 T-antigen transgenes. Am J Physiol Heart Circ Physiol 262: H1867–H1876, 1992. [DOI] [PubMed] [Google Scholar]
- 45.Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, Livne E, Binah O, Itskovitz-Eldor J, Gepstein L. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest 108: 407–414, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kehat I, Khimovich L, Caspi O, Gepstein A, Shofti R, Arbel G, Huber I, Satin J, Itskovitz-Eldor J, Gepstein L. Electromechanical integration of cardiomyocytes derived from human embryonic stem cells. Nat Biotechnol 22: 1282–1289, 2004. [DOI] [PubMed] [Google Scholar]
- 47.Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, Evans T, Macrae CA, Stainier DY, Poss KD. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 464: 601–605, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirshenbaum LA, Chakraborty S, Schneider MD. Human E2F-1 reactivates cell cycle progression in ventricular myocytes and represses cardiac gene transcription. Dev Biol 179: 402–411, 1996. [DOI] [PubMed] [Google Scholar]
- 49.Kirshenbaum LA, Schneider MD. Adenovirus E1A represses cardiac gene transcription and reactivates DNA synthesis in ventricular myocytes, via alternative pocket protein- and p300-binding domains. J Biol Chem 270: 7791–7794, 1995. [DOI] [PubMed] [Google Scholar]
- 50.Kubin T, Poling J, Kostin S, Gajawada P, Hein S, Rees W, Wietelmann A, Tanaka M, Lorchner H, Schimanski S, Szibor M, Warnecke H, Braun T. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell 9: 420–432, 2011. [DOI] [PubMed] [Google Scholar]
- 51.Ladd AN, Yatskievych TA, Antin PB. Regulation of avian cardiac myogenesis by activin/TGFbeta and bone morphogenetic proteins. Dev Biol 204: 407–419, 1998. [DOI] [PubMed] [Google Scholar]
- 52.Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, Police S, O'Sullivan C, Collins L, Chen Y, Minami E, Gill EA, Ueno S, Yuan C, Gold J, Murry CE. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol 25: 1015–1024, 2007. [DOI] [PubMed] [Google Scholar]
- 53.Laflamme MA, Murry CE. Heart regeneration. Nature 473: 326–335, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laube F, Heister M, Scholz C, Borchardt T, Braun T. Re-programming of newt cardiomyocytes is induced by tissue regeneration. J Cell Sci 119: 4719–4729, 2006. [DOI] [PubMed] [Google Scholar]
- 55.Lemaitre C, Soutoglou E. Double strand break (DSB) repair in heterochromatin and heterochromatin proteins in DSB repair. DNA Repair 19: 163–168, 2014. [DOI] [PubMed] [Google Scholar]
- 56.Lepilina A, Coon AN, Kikuchi K, Holdway JE, Roberts RW, Burns CG, Poss KD. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 127: 607–619, 2006. [DOI] [PubMed] [Google Scholar]
- 57.Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 28: 1737–1746, 1996. [DOI] [PubMed] [Google Scholar]
- 58.Li L, Baroja ML, Majumdar A, Chadwick K, Rouleau A, Gallacher L, Ferber I, Lebkowski J, Martin T, Madrenas J, Bhatia M. Human embryonic stem cells possess immune-privileged properties. Stem Cells 22: 448–456, 2004. [DOI] [PubMed] [Google Scholar]
- 59.Li TS, Cheng K, Malliaras K, Smith RR, Zhang Y, Sun B, Matsushita N, Blusztajn A, Terrovitis J, Kusuoka H, Marban L, Marban E. Direct comparison of different stem cell types and subpopulations reveals superior paracrine potency and myocardial repair efficacy with cardiosphere-derived cells. J Am Coll Cardiol 59: 942–953, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, Yau AL, Buck JN, Gouin KA, van Gorp PR, Zhou B, Chen J, Seidman JG, Wang DZ, Pu WT. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res 115: 354–363, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin Z, Zhou P, von Gise A, Gu F, Ma Q, Chen J, Guo H, van Gorp PR, Wang DZ, Pu WT. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ Res 116: 35–45, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Loffredo FS, Steinhauser ML, Gannon J, Lee RT. Bone marrow-derived cell therapy stimulates endogenous cardiomyocyte progenitors and promotes cardiac repair. Cell Stem Cell 8: 389–398, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, Sadek HA. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 497: 249–253, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, Czer LS, Marban L, Mendizabal A, Johnston PV, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marban E. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet 379: 895–904, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Malliaras K, Ibrahim A, Tseliou E, Liu W, Sun B, Middleton RC, Seinfeld J, Wang L, Sharifi BG, Marban E. Stimulation of endogenous cardioblasts by exogenous cell therapy after myocardial infarction. EMBO Mol Med 6: 760–777, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malliaras K, Zhang Y, Seinfeld J, Galang G, Tseliou E, Cheng K, Sun B, Aminzadeh M, Marban E. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol Med 5: 191–209, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Messina E, De AL, Frati G, Morrone S, Chimenti S, Fiordaliso F, Salio M, Battaglia M, Latronico MV, Coletta M, Vivarelli E, Frati L, Cossu G, Giacomello A. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res 95: 911–921, 2004. [DOI] [PubMed] [Google Scholar]
- 68.Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, Silberstein LE, Dos Remedios CG, Graham D, Colan S, Kuhn B. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci USA 110: 1446–1451, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, Pasumarthi KB, Virag JI, Bartelmez SH, Poppa V, Bradford G, Dowell JD, Williams DA, Field LJ. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature 428: 664–668, 2004. [DOI] [PubMed] [Google Scholar]
- 70.Murry CE, Wiseman RW, Schwartz SM, Hauschka SD. Skeletal myoblast transplantation for repair of myocardial necrosis. J Clin Invest 98: 2512–2523, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nag AC. Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios 28: 41–61, 1980. [PubMed] [Google Scholar]
- 72.Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 26: 101–106, 2008. [DOI] [PubMed] [Google Scholar]
- 73.Naqvi N, Li M, Calvert JW, Tejada T, Lambert JP, Wu J, Kesteven SH, Holman SR, Matsuda T, Lovelock JD, Howard WW, Iismaa SE, Chan AY, Crawford BH, Wagner MB, Martin DI, Lefer DJ, Graham RM, Husain A. A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell 157: 795–807, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nowbar AN, Mielewczik M, Karavassilis M, Dehbi HM, Shun-Shin MJ, Jones S, Howard JP, Cole GD, Francis DP. Discrepancies in autologous bone marrow stem cell trials and enhancement of ejection fraction (DAMASCENE): weighted regression and meta-analysis. BMJ 348: g2688, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nussbaum J, Minami E, Laflamme MA, Virag JA, Ware CB, Masino A, Muskheli V, Pabon L, Reinecke H, Murry CE. Transplantation of undifferentiated murine embryonic stem cells in the heart: teratoma formation and immune response. FASEB J 21: 1345–1357, 2007. [DOI] [PubMed] [Google Scholar]
- 76.O'Doherty R, Greiser U, Wang W. Nonviral methods for inducing pluripotency to cells. Biomed Res Int 2013: 705902, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, Entman ML, Schneider MD. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA 100: 12313–12318, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med 336: 1131–1141, 1997. [DOI] [PubMed] [Google Scholar]
- 79.Olivetti G, Cigola E, Maestri R, Corradi D, Lagrasta C, Gambert SR, Anversa P. Aging, cardiac hypertrophy and ischemic cardiomyopathy do not affect the proportion of mononucleated and multinucleated myocytes in the human heart. J Mol Cell Cardiol 28: 1463–1477, 1996. [DOI] [PubMed] [Google Scholar]
- 80.Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Bone marrow cells regenerate infarcted myocardium. Nature 410: 701–705, 2001. [DOI] [PubMed] [Google Scholar]
- 81.Oyama K, El-Nachef D, Zhang Y, Sdek P, MacLellan WR. Epigenetic regulation of cardiac myocyte differentiation. Front Genet 5: 375, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pasumarthi KB, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res 96: 110–118, 2005. [DOI] [PubMed] [Google Scholar]
- 83.Pfister O, Oikonomopoulos A, Sereti KI, Sohn RL, Cullen D, Fine GC, Mouquet F, Westerman K, Liao R. Role of the ATP-binding cassette transporter Abcg2 in the phenotype and function of cardiac side population cells. Circ Res 103: 825–835, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Poolman RA, Brooks G. Expressions and activities of cell cycle regulatory molecules during the transition from myocyte hyperplasia to hypertrophy. J Mol Cell Cardiol 30: 2121–2135, 1998. [DOI] [PubMed] [Google Scholar]
- 85.Poolman RA, Gilchrist R, Brooks G. Cell cycle profiles and expressions of p21CIP1 and P27KIP1 during myocyte development. Int J Cardiol 67: 133–142, 1998. [DOI] [PubMed] [Google Scholar]
- 86.Porrello ER, Johnson BA, Aurora AB, Simpson E, Nam YJ, Matkovich SJ, Dorn GW, van RE, Olson EN. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res 109: 670–679, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science 331: 1078–1080, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci USA 110: 187–192, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI, Sadek HA. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157: 565–579, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485: 593–598, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reinecke H, Poppa V, Murry CE. Skeletal muscle stem cells do not transdifferentiate into cardiomyocytes after grafting. J Mol Cell Cardiol 34: 241–249, 2002. [DOI] [PubMed] [Google Scholar]
- 92.Sadek HA, Martin JF, Takeuchi JK, Leor J, Nei Y, Giacca M, Lee RT. Multi-investigator letter on reproducibility of neonatal heart regeneration following apical resection. Stem Cell Reports 3: 1, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sandritter W, Adler CP. Numerical hyperplasia in human heart hypertrophy. Experientia 27: 1435–1437, 1971. [DOI] [PubMed] [Google Scholar]
- 94.Schenke-Layland K, Rhodes KE, Angelis E, Butylkova Y, Heydarkhan-Hagvall S, Gekas C, Zhang R, Goldhaber JI, Mikkola HK, Plath K, MacLellan WR. Reprogrammed mouse fibroblasts differentiate into cells of the cardiovascular and hematopoietic lineages. Stem Cells 26: 1537–1546, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sdek P, Zhao P, Wang Y, Huang CJ, Ko CY, Butler PC, Weiss JN, MacLellan WR. Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes. J Cell Biol 194: 407–423, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu TD, Guerquin-Kern JL, Lechene CP, Lee RT. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 493: 433–436, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Serena E, Figallo E, Tandon N, Cannizzaro C, Gerecht S, Elvassore N, Vunjak-Novakovic G. Electrical stimulation of human embryonic stem cells: cardiac differentiation and the generation of reactive oxygen species. Exp Cell Res 315: 3611–3619, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shapiro SD, Ranjan AK, Kawase Y, Cheng RK, Kara RJ, Bhattacharya R, Guzman-Martinez G, Sanz J, Garcia MJ, Chaudhry HW. Cyclin A2 induces cardiac regeneration after myocardial infarction through cytokinesis of adult cardiomyocytes. Sci Transl Med 6: 224ra27, 2014. [DOI] [PubMed] [Google Scholar]
- 99.Shiba Y, Fernandes S, Zhu WZ, Filice D, Muskheli V, Kim J, Palpant NJ, Gantz J, Moyes KW, Reinecke H, Van BB, Dardas T, Mignone JL, Izawa A, Hanna R, Viswanathan M, Gold JD, Kotlikoff MI, Sarvazyan N, Kay MW, Murry CE, Laflamme MA. Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature 489: 322–325, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marban E. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 115: 896–908, 2007. [DOI] [PubMed] [Google Scholar]
- 101.Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, Hill JA, Bassel-Duby R, Olson EN. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485: 599–604, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis during hypertrophy in adult mice. Am J Physiol Heart Circ Physiol 266: H1439–H1445, 1994. [DOI] [PubMed] [Google Scholar]
- 103.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol Heart Circ Physiol 272: H220–H226, 1997. [DOI] [PubMed] [Google Scholar]
- 104.Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol Heart Circ Physiol 271: H2183–H2189, 1996. [DOI] [PubMed] [Google Scholar]
- 105.Steward FC, Mapes MO, Kent AE, Holsten RD. Growth and development of cultured plant cells. Science 143: 20–27, 1964. [DOI] [PubMed] [Google Scholar]
- 106.Strauer BE, Brehm M, Zeus T, Kostering M, Hernandez A, Sorg RV, Kogler G, Wernet P. Repair of infarcted myocardium by autologous intracoronary mononuclear bone marrow cell transplantation in humans. Circulation 106: 1913–1918, 2002. [DOI] [PubMed] [Google Scholar]
- 107.Sugi Y, Lough J. Activin-A and FGF-2 mimic the inductive effects of anterior endoderm on terminal cardiac myogenesis in vitro. Dev Biol 168: 567–574, 1995. [DOI] [PubMed] [Google Scholar]
- 108.Swijnenburg RJ, Schrepfer S, Govaert JA, Cao F, Ransohoff K, Sheikh AY, Haddad M, Connolly AJ, Davis MM, Robbins RC, Wu JC. Immunosuppressive therapy mitigates immunological rejection of human embryonic stem cell xenografts. Proc Natl Acad Sci USA 105: 12991–12996, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Swijnenburg RJ, Tanaka M, Vogel H, Baker J, Kofidis T, Gunawan F, Lebl DR, Caffarelli AD, de Bruin JL, Fedoseyeva EV, Robbins RC. Embryonic stem cell immunogenicity increases upon differentiation after transplantation into ischemic myocardium. Circulation 112: I166–I172, 2005. [DOI] [PubMed] [Google Scholar]
- 110.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861–872, 2007. [DOI] [PubMed] [Google Scholar]
- 111.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663–676, 2006. [DOI] [PubMed] [Google Scholar]
- 112.Tang XL, Rokosh G, Sanganalmath SK, Yuan F, Sato H, Mu J, Dai S, Li C, Chen N, Peng Y, Dawn B, Hunt G, Leri A, Kajstura J, Tiwari S, Shirk G, Anversa P, Bolli R. Intracoronary administration of cardiac progenitor cells alleviates left ventricular dysfunction in rats with a 30-day-old infarction. Circulation 121: 293–305, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tate JM, Oberpriller JO, Oberpriller JC. Analysis of DNA synthesis in cell cultures of the adult newt cardiac myocyte. Tissue Cell 21: 335–342, 1989. [DOI] [PubMed] [Google Scholar]
- 114.Taylor DA, Atkins BZ, Hungspreugs P, Jones TR, Reedy MC, Hutcheson KA, Glower DD, Kraus WE. Regenerating functional myocardium: improved performance after skeletal myoblast transplantation. Nat Med 4: 929–933, 1998. [DOI] [PubMed] [Google Scholar]
- 115.Taylor DA, Silvestry SC, Bishop SP, Annex BH, Lilly RE, Glower DD, Kraus WE. Delivery of primary autologous skeletal myoblasts into rabbit heart by coronary infusion: a potential approach to myocardial repair. Proc Assoc Am Physicians 109: 245–253, 1997. [PubMed] [Google Scholar]
- 116.The LE. Expression of concern: the SCIPIO trial. Lancet 383: 1279, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science 282: 1145–1147, 1998. [DOI] [PubMed] [Google Scholar]
- 118.Tulloch NL, Muskheli V, Razumova MV, Korte FS, Regnier M, Hauch KD, Pabon L, Reinecke H, Murry CE. Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ Res 109: 47–59, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Urbanek K, Quaini F, Tasca G, Torella D, Castaldo C, Nadal-Ginard B, Leri A, Kajstura J, Quaini E, Anversa P. Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy. Proc Natl Acad Sci USA 100: 10440–10445, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, Middleton RC, Marban E, Molkentin JD. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 509: 337–341, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Van Laake LW, Passier R, Monshouwer-Kloots J, Verkleij AJ, Lips DJ, Freund C, den OK, Ward-van OD, Korving J, Tertoolen LG, van Echteld CJ, Doevendans PA, Mummery CL. Human embryonic stem cell-derived cardiomyocytes survive and mature in the mouse heart and transiently improve function after myocardial infarction. Stem Cell Res 1: 9–24, 2007. [DOI] [PubMed] [Google Scholar]
- 122.Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KH. Viable offspring derived from fetal and adult mammalian cells. Nature 385: 810–813, 1997. [DOI] [PubMed] [Google Scholar]
- 123.Wohlschlaeger J, Schmitz KJ, Takeda A, Takeda N, Vahlhaus C, Stypmann J, Schmid C, Baba HA. Reversible regulation of the retinoblastoma protein/E2F-1 pathway during “reverse cardiac remodelling” after ventricular unloading. J Heart Lung Transplant 29: 117–124, 2010. [DOI] [PubMed] [Google Scholar]
- 124.Woo YJ, Panlilio CM, Cheng RK, Liao GP, Suarez EE, Atluri P, Chaudhry HW. Myocardial regeneration therapy for ischemic cardiomyopathy with cyclin A2. J Thorac Cardiovasc Surg 133: 927–933, 2007. [DOI] [PubMed] [Google Scholar]
- 125.Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R, Olson EN. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci USA 110: 13839–13844, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, Richardson JA, Bassel-Duby R, Olson EN. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal 4: ra70, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, Henckaerts E, Bonham K, Abbott GW, Linden RM, Field LJ, Keller GM. Human cardiovascular progenitor cells develop from a KDR(+) embryonic-stem-cell-derived population. Nature 453: 524–528, 2008. [DOI] [PubMed] [Google Scholar]
- 128.Ye L, Chang YH, Xiong Q, Zhang P, Zhang L, Somasundaram P, Lepley M, Swingen C, Su L, Wendel J, Guo J, Jang A, Rosenbush D, Greder L, Dutton J, Zhang J, Kamp T, Kaufman D, Ge Y, Zhang J. Cardiac repair in a porcine model of acute myocardial infarction with human induced pluripotent stem cell-derived cardiovascular cells. Cell Stem Cell 15: 750–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yoshida Y, Takahashi K, Okita K, Ichisaka T, Yamanaka S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 5: 237–241, 2009. [DOI] [PubMed] [Google Scholar]
- 130.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science 318: 1917–1920, 2007. [DOI] [PubMed] [Google Scholar]
- 131.Zak R. Cell proliferation during cardiac growth. Am J Cardiol 31: 211–219, 1973. [DOI] [PubMed] [Google Scholar]
- 132.Zak R. Development and proliferative capacity of cardiac muscle cells. Circ Res 35 Suppl: 26, 1974. [PubMed] [Google Scholar]
- 133.Zhang R, Han P, Yang H, Ouyang K, Lee D, Lin YF, Ocorr K, Kang G, Chen J, Stainier DY, Yelon D, Chi NC. In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature 498: 497–501, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang Y, Li TS, Lee ST, Wawrowsky KA, Cheng K, Galang G, Malliaras K, Abraham MR, Wang C, Marban E. Dedifferentiation and proliferation of mammalian cardiomyocytes. PLoS One 5: e12559, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang Y, Matsushita N, Eigler T, Marban E. Targeted MicroRNA interference promotes postnatal cardiac cell cycle re-entry. J Regen Med 2: 2, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455: 627–632, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L. Mosaic analysis with double markers in mice. Cell 121: 479–492, 2005. [DOI] [PubMed] [Google Scholar]