

Abstract
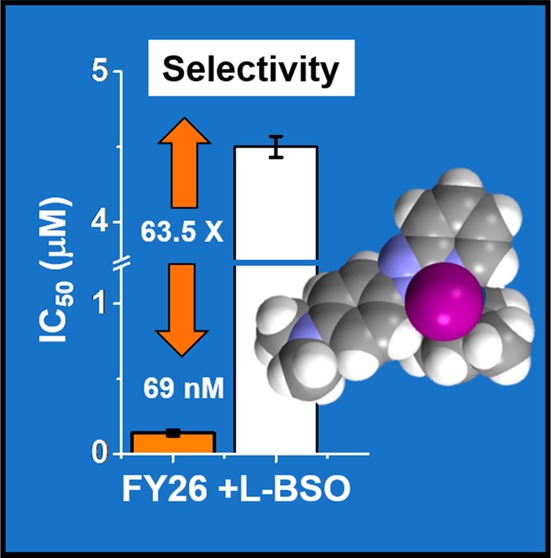
Combination with redox modulators can potentiate the anticancer activity and maximize the selectivity of organometallic complexes with redox-based mechanisms of action. We show that nontoxic doses of l-buthionine sulfoximine increase the selectivity of organo-Os complex FY26 for human ovarian cancer cells versus normal lung fibroblasts to 63-fold. This increase is not due to changes in the mechanism of action of FY26 but to the decreased response of cancer cells to oxidative stress.
Introduction
The development of resistance is a major clinical problem with current anticancer drugs. Multitargeting by using a combination of drugs provides a potential strategy to overcome resistance. We are exploring the concept of redox synergism for combination therapy, a strategy that could additionally lower the doses of metallodrugs. Metal complexes based on Ru(II), Os(II), and Ir(III) are being developed as viable alternatives to platinum drugs used in the clinic.1−3
Metal-based anticancer drugs can interfere in cellular redox chemistry in several ways: directly through metal or ligand redox centers, or indirectly by binding to biomolecules involved in cellular redox pathways. This opens the possibility to target the redox balance in cancer cells, which may be a highly effective, multiple site approach.4 In the present work, we show that combining metal-based drugs and redox modulators not only improves potency but also has great repercussions for the selectivity of the complex toward cancer cells.
Conventional platinum drugs target DNA and therefore rely on the high proliferation rate of cancer cells as a basis for selectivity.5 In contrast, organometallic osmium(II) complexes, such as FY26, [Os(η6-p-cym)(p-NMe2-Azpy)I]PF6 (Figure 1a), have a novel mechanism of action centered on increasing the level of reactive oxygen species (ROS) produced in cancer cells.6 Cancer cells are primed for this intervention because they are already redox-hyperactive and in most cases present malfunctioning mitochondria. Several factors are known to contribute to mitochondrial dysfunction in cancer cells, including mtDNA mutations, oncogenic stress, and p53 mutations.7−9 Functional mitochondria are responsible for a plethora of cellular processes, including ATP production, ROS generation, and cell death.10 Dysfunctional mitochondria are unable to control ROS generation efficiently, leading to inherent oxidative stress in cancer cells.11 This allows osmium compounds, such as FY26, to exert selective toxicity toward cancer cells over normal cells.
Figure 1.
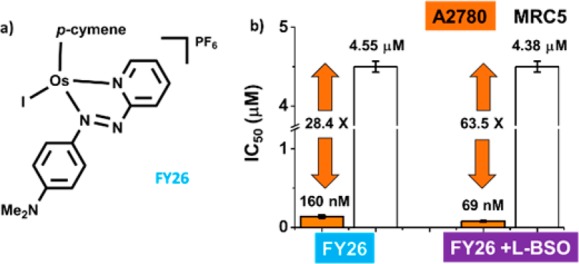
(a) Chemical structure of FY26, (b) antiproliferative activity of FY26 ± 5 μM L-BSO in A2780 ovarian cancer cells (orange) and MRC5 human lung fibroblasts (white). The selectivity factor (IC50-MRC5/IC50-A2780) increases from 28.4× to 63.5×.
Multicomponent therapy, in which the drugs used are synergistic, can provide dose reduction and subsequent minimization of adverse side effects while avoiding the development of resistance.12,13 Some examples of conventional combination therapy for cancer include the use of aphidicolin glycinate and cisplatin (CDDP) in the treatment of melanomas14 and paclitaxel combined with carboplatin for ovarian and NSCL cancers.15
We have shown that a number of active organometallic anticancer agents based on Ru(II), Os(II), and Ir(III) have potential redox components in their mechanisms of action. For such complexes, the possibility arises of using combination therapy together with redox modulators to increase their potency. This is attractive for lowering the doses of metal complexes that need to be administered.4
Osmium complex FY26 is highly active toward several cancer cell lines, in particular, it exhibits submicromolar activity in A2780 ovarian, A549 lung, HCT116 colon, and MCF7 breast cancer cell lines.16,17 FY26 is more potent than cisplatin by at least 2 orders of magnitude in the NCI-60 cell line screen (GI50 FY26 0.28 μM versus 10.3 μM for CDDP) as well as in the 809-cell line screen of the Sanger Institute (mean GI50 FY26 0.75 μM versus 36.7 μM for CDDP).6
We have further investigated the anticancer activity and mechanism of action on FY26 toward ovarian cancer, using A2780 cells as a model. Current statistics, held by CR-UK, indicate that we lose one woman every 2 h due to this disease. Importantly, we have reported that the activity of FY26 can be potentiated by coadministration of a nontoxic dose of the redox modulator l-buthionine sulfoximine, L-BSO, an inhibitor of γ-glutamylcysteine synthetase.4,16 This effect has also been observed for piano-stool complexes based on Ru(II) and Ir(III). In the case of the activity of FY26 toward A2780 ovarian cancer cells, potency improves by 2.3-fold when coadministered with 5 μM of L-BSO, with the IC50 decreasing from 160 ± 10 nM to 69 ± 5 nM.4 In the present work, we explore the implications of this activity improvement at the cellular level. We show that combination of FY26 with nontoxic doses of L-BSO dramatically increases the selectivity of the Os complex for cancer cells over normal fibroblasts.
Results and Discussion
We describe a series of experiments in which we investigated the effect of low, nontoxic doses of the redox modulator l-buthionine sulfoximine, L-BSO, an inhibitor of the enzyme γ-glutamyl cysteine synthetase that catalyzes the first and rate-limiting step in the production of glutathione (GSH). This redox modulator has been in clinical trials for malignant melanoma (NCI-T93-0176O) and refractory progressive neuroblastoma (NCT00002730) in combination with melphalan. The results so far indicate that it can be safely used to generate a 40% GSH depletion in patients.18,19
In the present work, L-BSO has been used in combination with the organo-Os(II) complex FY26 in studies on the antiproliferative activity in A2780 ovarian cancer cells and MRC5 human fibroblasts, effects on GSH levels, on the induction of ROS and superoxide, the involvement of apoptosis in cell death, and on mitochondrial membrane potentials and cell-cycle profiles.
Antiproliferative Activity
The antiproliferative activity of FY26 has previously been reported in the A2780 ovarian cell line (IC50 = 160 nM) as well as the possibility of enhancing its activity by coadministration with 5 μM L-BSO.4 We have investigated whether this potentiation of anticancer activity is also observed for inactive Os(II) piano-stool complexes. For this, we determined the IC50 of FY77 ([Os(η6-bip)(Cl-Azpy)Cl]PF6 IC50 > 100 μM) and FY122 ([Os(η6-p-cym) (OH-Impy)I]PF6 IC50 = 30 ± 2 μM) which are structurally related organometallic complexes but have much lower potencies than FY26. We then determined the percentages of cell survival when different concentrations of FY77 and FY122 are coadministered with 5 μM of L-BSO (Supporting Information (SI) Figure 1). The biphenyl complex FY77 remains inactive, with percentages of cell survival above 93%. Only partial enhancement of activity was observed for FY122. This indicates that L-BSO does not confer antiproliferative activity per se and can only enhance the performance of a complex that is already biologically active.
We hypothesized that the cellular effect of L-BSO in reducing GSH levels is directly related to the enhancement of antiproliferative activity. To confirm this, we investigated whether the effect of coincubation with L-BSO could be reversed by also coadministering GSH. A2780 ovarian cancer cells exposed to a fixed concentration of FY26 (0.10 or 0.30 μM) were coincubated with (a) 5 μM L-BSO, (b) 5 μM GSH, (c) 5 μM L-BSO and 5 μM GSH, (d) 50 μM GSH, or (e) 5 μM L-BSO and 50 μM GSH (Figure 2 and SI Figure 2).
Figure 2.
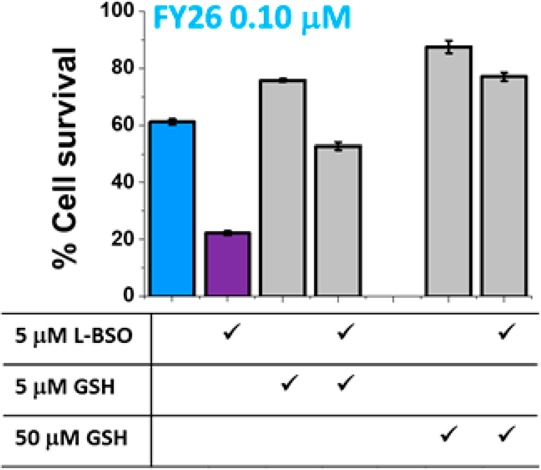
Effect of coadministration of 0.10 μM FY26 with 5 μM L-BSO in the presence/absence of GSH (5 and 50 μM) on the percentage of cell survival of A2780 ovarian cancer cells.
The administration of 0.10 μM of FY26 and 5 μM L-BSO, as expected, reduced the percentage of cell survival compared to the administration of the complex on its own. This effect was partially reversed in the presence of GSH as well as L-BSO (Figure 2). In fact, when cells were coincubated with only FY26 and GSH, the percentage of cell survival increased in comparison to the osmium drug alone. This suggests that the effect of L-BSO may be related to modulating the cellular response to the osmium drug. More specifically, the role of L-BSO is to reduce the level of GSH as a cellular detoxification agent.
The anticancer activity of metal-based drugs may well involve both cytostatic and cytotoxic effects. To investigate the contribution of these two effects, the percentages of survival of cells exposed to the drug when allowed or not allowed to recover in drug free medium were compared. This provides an indication of cytostasis and cytotoxicity, respectively.
We carried out this experiment on A2780 ovarian cancer cells exposed for 24 h to various concentrations of FY26. These values were then compared to cells that were drug-exposed under similar conditions but were then allowed to recover for further 72h in drug-free medium (SI Figure 3a). At concentrations between 75 and 0.3 μM FY26 the percentage of cell survival was always significantly lower for cells that were allowed to recover, which highlights the cytostatic effect of FY26. Meanwhile at the lowest concentration tested (30 nM), no differences were observed and the percentage of cell survival was very similar to that of untreated controls. We also determined the percentages of cell survival when A2780 cells were exposed to 5 μM L-BSO in coadministration with various concentrations of FY26 (SI Figure 3b). The use of the redox modulator did not change the trends observed, and once more the percentage of cells in the experiment that included 72 h recovery in drug-free medium was lower. This suggests that in the presence and absence of L-BSO the behavior of FY26 is mostly cytostatic.
Cancer Cell Selectivity
The selectivity factor for a given compound can be defined as the ratio between its activity (IC50) in normal cells (MRC5 fibroblasts in the present case) compared to cancer cells. The selectivity of FY26 (IC50-MRC5/IC50-A2780) is 28, while the corresponding factor for CDDP is only 9.5.20 We studied the effect on the selectivity factor of combined administration of a 5 μM nontoxic dose of L-BSO together with FY26 to A2780 human ovarian cancer cells compared to MRC5 fibroblasts. Remarkably, the selectivity factor increased to 63.5× (Figure 1b, SI Table 1). The activity in the fibroblasts remains unchanged, but the potency in the ovarian cancer cells increases. Such a differential activity might allow the use of combination therapy for dose reduction and translate into reduction of unwanted side effects.
Cellular GSH Levels
We investigated the effect of L-BSO on the GSH levels in A2780 ovarian cancer cells. We treated A2780 cells with three different concentrations of L-BSO: 1, 5, and 50 μM. Compared to untreated controls, 1 μM L-BSO treatment did not cause significant variations in the GSH levels (to 95%), but 5 μM L-BSO induced an approximate 50% drop (to 56%), and the highest concentration (50 μM) caused a 63% reduction (to 37%; Figure 3a, SI Table 2).
Figure 3.
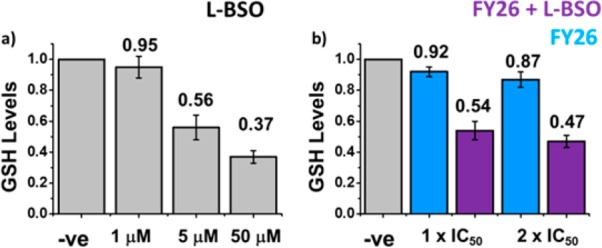
GSH levels in A2780 cells exposed for 24 h to (a) L-BSO (1, 5, and 50 μM), (b) FY26 (IC50 and 2× IC50) ± 5 μM L-BSO. In all cases, data have been normalized against negative untreated controls.
We chose 5 μM L-BSO to carry out experiments on coadministration with FY26. In this case, the levels of GSH were reduced, as expected, to ca. 54% of those of the untreated controls. The variation caused by a single dose of FY26 at IC50 concentration was a decrease to 92%. At 2× IC50 concentration, a statistically significant reduction to 87% was observed. It is possible that at high concentrations of the complex, GSH is used as a cellular strategy for drug detoxification (Figure 3b, SI Table 2). This effect has been observed before, for example, reduced levels of GSH (caused by L-BSO administration) can restore cellular sensitivity to the Ru(III) anticancer drug KP1019.21
ROS/Superoxide Induction
We then determined the induction of ROS and superoxide in A2780 ovarian cancer cells exposed for 24 h to complex FY26 at the IC50 concentration (160 nM) using flow cytometry and compared this to the ROS and superoxide levels observed in MRC5 fibroblasts under similar conditions. The experiment allowed the simultaneous determination of total oxidative stress (e.g., peroxides, peroxynitrite, and hydroxyl radicals) using the FL-1 green channel and the generation of superoxide using FL-2 orange channel. Ovarian cancer cells exposed to FY26 showed high levels of both total ROS and superoxide, with most of the cell population in the FL-1+/FL-2+ upper right quadrant of a dot plot. Meanwhile, untreated negative controls remained in the lower left quadrant, with low levels in both channels (Figure 4a,b, SI Table 3). In contrast, MRC5 fibroblasts exposed to FY26 exhibited elevated levels of total ROS but showed low induction of superoxide, with most of the population in the lower half of the FL-2 orange channel (Figure 4c).
Figure 4.

Flow cytometry analysis of the induction of total ROS and superoxide: (a) positive and negative controls in A2780 ovarian cancer cells, (b) FY26 (IC50) ± 5 μM L-BSO in A2780 cells, and (c) MRC5 normal fibroblasts exposed to FY26 (IC50). In all cases, the experiments included 24 h of drug exposure and no-recovery time.
These observations are consistent with the proposed mechanism of action for FY26, which is based on the disruption of the cellular redox balance.6 We hypothesize that FY26 exerts its anticancer activity by generating a burst of superoxide, hence exploiting the weaknesses of defective mitochondria in cancer cells. This could explain the selective toxicity observed between cancer cells and normal fibroblasts. The MRC5 fibroblasts which contain normal functioning mitochondria are capable of rebalancing the redox state of the cell efficiently, while the A2780 progress toward cell death.
A similar experiment was carried out using a combination of FY26 and 5 μM of L-BSO in A2780 ovarian cancer cells. We again determined the levels of total ROS and superoxide induction. Both of these readings remained unchanged compared to the administration of the osmium complex alone, showing the majority of the cell population in the upper right quadrant FL-1+/FL-2+ (Figure 4b SI Table 4). Remarkably, no further increase in the levels of superoxide is observed in the presence of FY26 and L-BSO, which indicates that the enhancement in anticancer activity is the result of a weakened cell response to the osmium complex due to the lower GSH levels. As a second set of negative controls (apart from the untreated cells), A2780 cells exposed to 5 μM of L-BSO showed only population in the FL-1–/FL-2– lower left quadrant, indicating low levels of both superoxide and total ROS.
We have reported that the levels of superoxide induction in A2780 ovarian cancer cells can be directly related to the antiproliferative activity of Os(II) piano-stool complexes, in particular to the activity of FY26, FY77, and FY122.6 There is no production of superoxide in cells exposed the inactive complex FY77, and no improvement in anticancer activity when the complex is coadministered with 5 μM L-BSO. This is consistent with the idea that L-BSO can only enhance the potency of already active complexes, such as FY26, and that it does so by lowering GSH levels, hence weakening the cellular response to the osmium-complex.
Induction of Apoptosis
We have previously reported that IC50 concentrations of FY26 do not cause significant apoptosis in A2780 ovarian cancer cells after 24 h of drug exposure.6 Under these conditions, the majority of the cell population in a flow cytometry dot plot remains in the lower left quadrant with FL-1 measuring Annexin V fluorescence intensity and FL-2 reading propidium iodide fluorescence. Apoptosis, as programmed cell death, starts with the loss of symmetry of the phosphatidylserine membrane, which allows Annexin binding (early apoptosis), followed by loss of the membrane integrity. At this point, cells become permeant to propidium iodide and generate high fluorescence in the FL-2 channel after DNA intercalation (late apoptosis). FY26 has been shown to cause apoptotic cell death in A549 lung cancer cells only at concentrations 10× IC50, nonetheless at a dose of 0.5 μM the apoptotic population can still be disregarded.22
We exposed A2780 ovarian cancer cells to FY26 under similar conditions (24 h drug exposure at IC50 concentration), this time coadministered with 5 μM L-BSO with the aim of determining whether the use of the redox modulator would induce higher levels of programmed cell death. Figure 5a (SI Table 5) shows that combination with the redox modulator does not cause significant changes to the apoptotic response. Most of the cell population showed only low levels of Annexin binding and propidium iodide intercalation, hence they are located in the FL-1–/FL-2– lower left quadrant of the dot plot. There are, nonetheless, a statistically relevant (p < 0.001) number of nonviable cells located in the upper left quadrant FL-1–/FL-2+, which only exhibit high propidium iodide fluorescence; this is consistent with a nonapoptotic mechanism of cell death. The experiment was also carried out using a single dose of 5 μM L-BSO as a second set of negative controls. There were no statistically significant differences between the cells exposed to the redox modulator and the untreated controls, as all the population remained in the lower left quadrant, with low Annexin V and low propidium iodide fluorescence.
Figure 5.
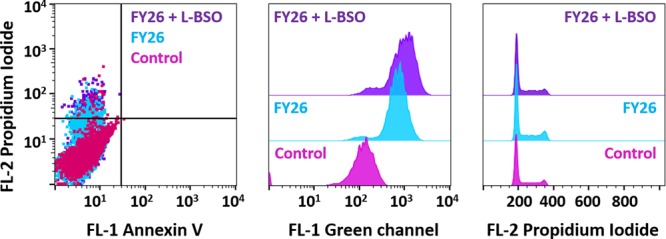
Flow cytometry analysis of A2780 cells exposed for 24 h to FY26 (IC50) ± 5 μM L-BSO: (a) induction of apoptosis, (b) changes in the mitochondrial membrane potential, and (c) cell cycle analysis.
A nonapoptotic mechanism of cell death does not rule out the involvement of mitochondrial dysfunction or redox disruption. In fact, it has been reported that pathogenic mitochondrial oxidation23 and even autophagic cell death24 can be linked to cellular redox disruption. There is also a need to consider that the mechanism of cell death caused by this type of multitargeted metal-based complex may be novel and difficult to map onto known mechanisms.
Mitochondrial Membrane Potential
We also investigated changes in the mitochondrial membrane potential of A2780 cells exposed to FY26 ± 5 μM L-BSO using flow cytometry. First, we used the Os(II) complex on its own, with a drug exposure of 24 h at IC50 (160 nM) concentration. The experiment relies on the JC-10 mitochondrial stain which exists as a red aggregate inside the mitochondria, however, upon changes in the membrane potential, the dye is released into the cytosol and converted into its monomeric green form. Following the increase of intensity in the FL-1 green channel, it is possible to quantify the release of the stain and hence gain information on changes in the mitochondrial membrane potential. A FL-1 histogram revealed that FY26 is capable of changing the potential of the mitochondrial membrane as indicated by a gain in the fluorescence intensity. Similarly, we carried out the experiment with coadministration with 5 μM L-BSO. This combination did not affect markedly the results of this experiment, and changes in the mitochondrial membrane potential were still observed (Figure 5b, SI Table 6).
Effects on A2780 Cell Cycle
Cell cycle profiles of A2780 cells exposed for 24 h to FY26 at IC50 concentration (160 nM) ± 5 μM L-BSO were obtained by flow cytometry using propidium iodide as a DNA stain. In both cases, with and without the redox modulator, it was observed that after the drug exposure period and no recovery time, A2780 cells were arrested in G1 phase and there was no significant population in a sub-G1 phase (Figure 5c). These results highlight the cytostatic effect of the Os(II) complex and are consistent with the above studies of apoptosis in which 24 h of drug exposure does not lead to a majority population of nonviable cells. It is also consistent with the results obtained when cells exposed to FY26 are allowed or not to recover in drug-free medium before determining the percentages of cell survival (vide supra).
Conclusions
It is well-known that L-BSO can deplete GSH levels in cells by inhibiting the enzyme γ-glutamylcysteine synthetase.21 Furthermore, several previous studies have related intracellular levels of GSH with resistance to metal-based anticancer agents. Metal–GSH adducts can be recognized by ATP-mediated efflux transporters and MDR proteins, which translates into reduced cell accumulation of the drugs.25,26 Most importantly, cellular GSH is a scavenger of reactive oxygen species.
FY26, a potent Os(II) anticancer drug candidate, exerts its activity by generating reactive oxygen species and disrupting the redox balance in cancer cells.4,6,22 We have shown here that coadministration of FY26 and nontoxic doses of L-BSO allows the potentiation of its anticancer activity, and most remarkably, it improves the selectivity for cancer cells versus normal fibroblasts. Reduced GSH levels (caused by the L-BSO) not only undermine cellular detoxification pathways but most importantly diminish the cell’s ability to respond to oxidative stress.
Co-administration of FY26 with the redox modulator L-BSO does not alter the levels of ROS/superoxide produced in the cancer cells nor does it enhance the effect of the Os(II) complex on the mitochondrial membrane potential. However, such a combination plays a key role in the cellular response to this damage. Cell responses to redox disruption differ greatly from cancer to normal cells. The vulnerability of the former arises from elevated energy requirements and the presence of dysfunctional mitochondria, while in the latter, balancing mechanisms are effective and redox variations, such as those induced by FY26, are readily reversed. Most notably, and in contrast to the cancer cells, treatment of normal fibroblasts with FY26 does not induce high levels of superoxide (Figure 4).
Because metal-based drugs are often multitargeted, a “systems pharmacology” approach which considers their effects on feedback loops from interlinked pathways is likely to be beneficial for understanding their mechanisms of action and improving their design. “Smart” and “synergic” combinations can be exploited to maximize the selectivity of metal-based anticancer drug candidates. The increase in selectivity of FY26 to a 63-fold difference between MRC5 fibroblasts and A2780 ovarian cancer cells is quite dramatic and would have a significant impact on therapeutic regimes if it could be translated into clinical practice.
Experimental Section
Materials and Methods
Organometallic Os(II) complexes FY26, FY77, and FY122 were synthesized and characterized as reported previously.16 Staurosporine, propidium iodide (94%) RNase A, pyocyanin and carbonyl cyanide 3-chlorophenylhydrazone, CCCP, L-BSO, and GSH were purchased from Sigma-Aldrich.
Cell Culture
A2780 human ovarian carcinoma and MRC5 human fetal lung fibroblasts were obtained from the European Collection of Cell Cultures (ECACC) and grown in Roswell Park Memorial Institute medium (RPMI-1640), supplemented with 10% of fetal calf serum, 1% of 2 mM glutamine, and 1% penicillin/streptomycin. All cells were grown as adherent monolayers at 310 K in a 5% CO2 humidified atmosphere and passaged at approximately 70–80% confluency.
In Vitro Growth Inhibition Assay
Briefly, 96-well plates were used to seed 5000 cells per well. The plates were left to preincubate in drug-free media at 310 K for 48 h before adding different concentrations of the compounds to be tested. A drug exposure period of 24 h was allowed. After this, supernatants were removed by suction and each well was washed with PBS. A further 72 h was allowed for the cells to recover in drug-free medium at 310 K. The SRB assay was used to determine cell viability. IC50 values, as the concentration which caused 50% of cell death, were determined as duplicates of triplicates in two independent sets of experiments and their standard deviations were calculated. CDDP-exposed and untreated cells were used as positive and negative controls, respectively.
Redox Modulation Assays
These experiments were performed using the protocol previously described for IC50 determination with the following modifications. Briefly, a 96-well plate was seeded with 5000 cells per well. Cells were preincubated in drug-free medium for 48 h at 310 K before adding the metal complex FY26 together with 5 μM L-BSO. To prepare the stock solution of the drug, the solid complex was dissolved first in DMSO and then diluted in a 50:50 v/v mixture of cell culture medium:saline (0.9% w/v NaCl in sterile water) to a maximum DMSO concentration of 5% v/v. This stock was further diluted using RPMI until working concentrations were achieved. Separately, a stock solution of L-BSO was prepared in saline. Both solutions, FY26 and L-BSO, were added to each well independently but within 5 min of each other. Drug exposure and recovery time were 24 and 72 h, respectively. The SRB assay was used to determine cell viability.
These experiments used cells treated with different concentrations of CDDP as positive controls and two sets of negative controls; in the first one, the cells were kept untreated, while in the second set, the cells were exposed to 5 μM L-BSO. The differences in cell survival between the two sets of negative controls were not statistically significant in any experiment.
A similar protocol was used to investigate the effect of coadministration with GSH. A2780 cells were drugged using fixed concentrations of FY26 (0.10 and 0.30 μM) and 5 μM L-BSO in the presence/absence of 5 and 50 μM GSH. Drug exposure and recovery time were 24 and 72 h, respectively. The SRB assay was used to determine cell viability.
Cell Cycle Analysis
Cells were seeded in a 6-well plate using 1.0 × 106 cells per well. They were preincubated in drug-free media at 310 K for 24 h, after which FY26 ± 5 μM L-BSO were added using equipotent concentrations equal to IC50 and 2× IC50 for the Os(II) complex. After 24 h of drug exposure, supernatants were removed by suction and cells were washed with PBS. Finally, cells were harvested using trypsin. DNA staining was achieved by resuspending the cell pellets in PBS containing propidium iodide (PI) and RNase A. Cell pellets were resuspended in PBS before being analyzed by flow cytometry using the maximum excitation of PI-bound DNA at 536 nm and its emission at 617 nm. Data were processed using Flowjo software. These experiments used two sets of negative controls, a first one using untreated cells and a second one with cells treated only with 5 μM L-BSO. These experiments were carried out in duplicates of triplicates in independent experiments; although only selected dot plots are shown, full numerical data and statistical analysis can be found in the Supporting Information.
Induction of Apoptosis
Flow cytometry analysis of apoptotic populations were carried out using the Annexin V-FITC apoptosis detection kit (Sigma-Aldrich) according to the manufacturer’s instructions. Briefly, cells were seeded in 6-well plates (1.0 × 106 cells per well), preincubated for 24 h in drug-free media at 310 K, after which they were exposed to FY26 ± 5 μM L-BSO for further 24 h (equipotent concentrations of FY26 equal to IC50 and 2× IC50). Cells were harvested using trypsin and stained using PI/Annexin V-FITC. After staining in the dark, cell pellets were analyzed in a Becton Dickinson FACScan flow cytometer. For positive-apoptosis controls, A2780 cells were exposed for 2 h to staurosporine (1 μg/mL). Cells for apoptosis studies were used with no previous fixing procedure so as to avoid nonspecific binding of the annexin V-FITC conjugate. Negative controls included untreated cells and cells treated only with 5 μM L-BSO. These experiments were carried out in duplicates of triplicates in independent experiments; although only selected dot plots are shown, full numerical data and statistical analysis can be found in the Supporting Information.
ROS and Superoxide Determination
Flow cytometry analysis of ROS/superoxide generation by exposure to FY26 ± 5 μM L-BSO was carried out using the Total ROS/Superoxide detection kit (Enzo Life Sciences) according to the supplier’s instructions. Briefly, 1.0 × 106 cells per well were seeded in a 6-well plate. Cells were preincubated in drug-free media at 310 K for 24 h in a 5% CO2 humidified atmosphere, and then drugs were added at equipotent concentrations equal to IC50 and 2× IC50 of FY26 and ±5 μM L-BSO. After 24 h of drug exposure, supernatants were removed by suction and cells were washed and harvested. Staining was achieved in the dark by resuspending the cell pellets in buffer containing the orange/green fluorescent reagents. Cells were analyzed in a Becton Dickinson FACScan flow cytometer using Ex/Em 490/525 nm for the oxidative stress and Ex/Em 550/620 nm for superoxide detection. Data were processed using Flowjo software. Negative controls included untreated cells and cells treated only with 5 μM L-BSO. Positive controls were obtained using pyocyanin. These experiments were carried out as duplicates of triplicates in independent experiments; although only selected dot plots are shown, full numerical data and statistical analysis can be found in the Supporting Information.
Mitochondrial Membrane Potential Assay
Analysis of the changes of mitochondrial potential was carried out using the Abcam, JC-10 mitochondrial membrane potential assay kit according to the manufacturer’s instructions. Briefly, 1.0 × 106 cells were seeded in 6-well plates and left to incubate for 24 h in drug-free medium at 310 K in a humidified atmosphere. Solutions of FY26 at IC50 and 2× IC50 concentrations ±5 μM L-BSO were added in triplicate and the cells left to incubate for further 24 h under similar conditions. Supernatants were removed by suction, and each well was washed with PBS before detaching the cells using trypsin-EDTA. Staining of the samples was done in flow cytometry tubes protected from light, incubating 30 min at room temperature. Samples were immediately analyzed on a Beckton Dickinson FACScan with fluorescence detection. Data were processed using Flowjo. Negative controls included untreated cells and cells treated only with 5 μM L-BSO. Positive controls were obtained using CCCP. These experiments were carried out as duplicates of triplicates in independent experiments; although only selected dot plots are shown, full numerical data and statistical analysis can be found in the Supporting Information.
Glutathione Assay
GSH levels in cells exposed to FY26 ± L-BSO were determined using the Glutathione (GSH/GSSG/Total) Assay Kit from BioVision according to the manufacturer’s instructions. Briefly, 1.0 × 106 cells were seeded in 6-well plates and left to incubate for 24 h in drug-free medium at 310 K in a humidified atmosphere. Drug solutions of FY26 at IC50 and 2× IC50 concentration ±5 μM L-BSO were added in triplicate and the cells left to incubate for further 24 h under similar conditions. Supernatants were removed by suction, and each well was washed with PBS before detaching the cells using trypsin-EDTA. Cell pellets were resuspended in 100 μL of assay buffer together with 20 μL of cold 6N perchloric acid (PCA). A 60 μL aliquot was vortexed to a uniform emulsion and kept on ice for 5 min before spinning it at 13000g for 2 min. The supernatant, containing GSH, was collected and the protein pellet discarded. For the fluorescence-based assay, a 40 μL aliquot of each sample was neutralized using 6 M KOH before being kept on ice for min and spun at 13000g for 2 min at 4 °C. The GSH concentration was determined by reading the absorbance of the o-phthalaldehyde (OPA) probe at Ex/Em 340/420 nm after 40 min incubation at room temperature (10 μL of the probe were added to 10 μL each sample and diluted to a final volume of 100 μL). Values were normalized to the protein content of each sample determined by a Bradford assay. These experiments were carried out as duplicates of triplicates in independent experiments.
Statistical Analysis
In all cases, independent two-sample t tests with unequal variances, Welch’s tests, were carried out to establish statistical significance of the variations (p < 0.001 for ***, p < 0.01 for **, and p < 0.05 for *).
Acknowledgments
We thank Miss B. Qamar for technical assistance with cell culture, Russell Needham and Dr. Fu Ying for providing FY26, FY77, and FY122. We also thank Dr. Sanchez-Cano and members of the EU COST Action CM1105 for stimulating discussions. This research was supported by the ERC (Grants 247450 and 324594), AWM/ERDF, and EPSRC IAA.
Glossary
Abbreviations Used
- ROS
reactive oxygen species
- mtDNA
mitochondrial DNA
- CDDP
cisplatin
- NCI
National Cancer Institute
- L-BSO
l-buthionine sulfoximine
- GSH
glutathione
- SRB
sulforhodamine B
- CCCP
carbonyl cyanide 3-chlorophenylhydrazone
- MDR
multidrug resistance
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00655.
Author Contributions
I.R.-C. wrote of the manuscript, M.M. and I.R.-C. carried out the research. I.R.-C. and P.J.S. designed the experiments and analyzed the data.
The authors declare no competing financial interest.
Supplementary Material
References
- Gasser G.; Metzler-Nolte N. The Potential of Organometallic Complexes in Medicinal Chemistry. Curr. Opin. Chem. Biol. 2012, 16, 84–91. 10.1016/j.cbpa.2012.01.013. [DOI] [PubMed] [Google Scholar]
- Van Rijt S. H.; Sadler P. J. Current Applications and Future Potential for Bioinorganic Chemistry in the Development of Anticancer Drugs. Drug Discovery Today 2009, 14, 1089–1097. 10.1016/j.drudis.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry N. P. E.; Sadler P. J. Exploration of the Medical Periodic Table: Towards New Targets. Chem. Commun. 2013, 49, 5106–5131. 10.1039/c3cc41143e. [DOI] [PubMed] [Google Scholar]
- Romero-Canelón I.; Sadler P. J. Next-Generation Metal Anticancer Complexes: Multitargeting via Redox Modulation. Inorg. Chem. 2013, 52, 12276–12291. 10.1021/ic400835n. [DOI] [PubMed] [Google Scholar]
- Tennant D.; Durán R. V.; Gottlieb E. Targeting Metabolic Transformation for Cancer Therapy. Nat. Rev. Cancer 2010, 10, 267–277. 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- Hearn J. M.; Romero-Canelón I.; Munro A. F.; Fu Y.; Pizarro A. M.; Garnett M. J.; McDermott U.; Carragher N. O.; Sadler P. J. Potent Organo-Osmium Complex Shifts Metabolism in Epithelial Ovarian Cancer Cells. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E3800–E3805. 10.1073/pnas.1500925112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L.; Larochette N.; Zamzami N.; Kroemer G. Mitochondria as Therapeutic Targets for Cancer Chemotherapy. Oncogene 2006, 25, 4812–4830. 10.1038/sj.onc.1209598. [DOI] [PubMed] [Google Scholar]
- Kroemer G. Mitochondria in Cancer. Oncogene 2006, 25, 4630–4632. 10.1038/sj.onc.1209589. [DOI] [PubMed] [Google Scholar]
- Pinton P.; Kroemer G. Cancer Therapy: Altering Mitochondrial Properties. Nat. Chem. Biol. 2014, 10, 89–90. 10.1038/nchembio.1440. [DOI] [PubMed] [Google Scholar]
- Collins Y.; Chouchani E. T.; James A. M.; Menger K. E.; Cocheme H. M.; Murphy M. P. Mitochondrial Redox Signalling at a Glance. J. Cell Sci. 2012, 125, 801–806. 10.1242/jcs.098475. [DOI] [PubMed] [Google Scholar]
- Scheffler I. E. A Century of Mitochondrial Research: Achievements and Perspectives. Mitochondrion 2001, 1, 3–31. 10.1016/S1567-7249(00)00002-7. [DOI] [PubMed] [Google Scholar]
- Greco W. R.; Bravo G.; Parsons J. C. The Search for Synergy: A Critical Review from a Response Surface Perspective. Pharmacol. Rev. 1995, 47, 331–385. [PubMed] [Google Scholar]
- Greco W. R.; Faessel H.; Levasseur L. The Search for Cytotoxic Synergy between Anticancer Agents: A Case of Dorothy and the Ruby Slippers?. J. Natl. Cancer Inst. 1996, 88, 699–700. 10.1093/jnci/88.11.699. [DOI] [PubMed] [Google Scholar]
- O’Dwyer P. J.; Moyer J. D.; Suffness M.; Harrison S. D.; Cysyk R.; Hamilton T. C.; Plowman J. Antitumor Activity and Biochemical Effects of Aphidicolin Glycinate (NSC 303812) Alone and in Combination with Cisplatin in Vivo. Cancer Res. 1994, 54, 724–729. [PubMed] [Google Scholar]
- Greco F.; Vicent M. J. Combination Therapy: Opportunities and Challenges for Polymer-Drug Conjugates as Anticancer Nanomedicines. Adv. Drug Delivery Rev. 2009, 61, 1203–1213. 10.1016/j.addr.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Fu Y.; Habtemariam A.; Pizarro A. M.; van Rijt S. H.; Healey D. J.; Cooper P.; Shnyder S. D.; Clarkson G. J.; Sadler P. J. Organometallic Osmium Arene Complexes with Potent Cancer Cell Cytotoxicity. J. Med. Chem. 2010, 53, 8192–8196. 10.1021/jm100560f. [DOI] [PubMed] [Google Scholar]
- Shnyder S. D.; Fu Y.; Habtemariam A.; van Rijt S. H.; Cooper P.; Loadman P. M.; Sadler P. J. Anti-Colorectal Cancer Activity of an Organometallic Osmium Arene Azopyridine Complex. MedChemComm 2011, 2, 666–668. 10.1039/c1md00075f. [DOI] [Google Scholar]
- Bailey H. H.; Mulcahy R. T.; Tutsch K. D.; Arzoomanian R. Z.; Alberti D.; Tombes M. B.; Wilding G.; Pomplun M.; Spriggs D. R. Phase I Clinical Trial of Intravenous l-Buthionine Sulfoximine and Melphalan: An Attempt at Modulation of Glutathione. J. Clin. Oncol. 1994, 12, 194–205. [DOI] [PubMed] [Google Scholar]
- Trachootham D.; Zhang W.; Huang P. In Oxidative Stress and Drug Resistance in Cancer; Siddik Z. H.; Mehta K., Eds.; Springer: New York, 2009. [Google Scholar]
- Romero-Canelón I.; Salassa L.; Sadler P. J. The Contrasting Activity of Iodido versus Chlorido Ruthenium and Osmium Arene Azo- and Imino-Pyridine Anticancer Complexes: Control of Cell Selectivity, Cross-Resistance, p53 Dependence, and Apoptosis Pathway. J. Med. Chem. 2013, 56, 1291–1300. 10.1021/jm3017442. [DOI] [PubMed] [Google Scholar]
- Jungwirth U.; Kowol C. R.; Keppler B. K.; Hartinger C. G.; Berger W.; Heffeter P. Anticancer Activity of Metal Complexes: Involvement of Redox Processes. Antioxid. Redox Signaling 2011, 15, 1085–1127. 10.1089/ars.2010.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rijt S. H.; Romero-Canelón I.; Fu Y.; Shnyder S. D.; Sadler P. J. Potent Organometallic Osmium Compounds Induce Mitochondria-Mediated Apoptosis and S-Phase Cell Cycle Arrest in A549 Non-Small Cell Lung Cancer Cells. Metallomics 2014, 6, 1014–1022. 10.1039/c4mt00034j. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Limphong P.; Pieper J.; Liu Q.; Rodesch C. K.; Christians E.; Benjamin I. J. Glutathione-Dependent Reductive Stress Triggers Mitochondrial Oxidation and Cytotoxicity. FASEB J. 2012, 26, 1442–1451. 10.1096/fj.11-199869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L.; Vitale I.; Abrams J. M.; Alnemri E. S.; Baehrecke E. H.; Blagosklonny M. V.; Dawson T. M.; Dawson V. L.; El-Deiry W. S.; Fulda S.; Gottlieb E.; Green D. R.; Hengartner M. O.; Kepp O.; Knight R.; Kumar S.; Lipton S.; Lu X.; Madeo F.; Malorni W.; Mehlen P.; Nuñez G.; Peter M. E.; Piacentini M.; Rubinsztein D. C.; Shi Y.; Simon H.-U.; Vandenabeele P.; White E.; Yuan J.; Zhivotovsky B.; Melino G.; Kroemer G. Molecular Definitions of Cell Death Subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartalou M.; Essigmann J. M. Mechanisms of Resistance to Cisplatin. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2001, 478, 23–43. 10.1016/S0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- Shen D. W.; Pouliot L. M.; Hall M. D.; Gottesman M. M. Cisplatin Resistance: A Cellular Self-Defense Mechanism Resulting from Multiple Epigenetic and Genetic Changes. Pharmacol. Rev. 2012, 64, 706–721. 10.1124/pr.111.005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.