

Abstract
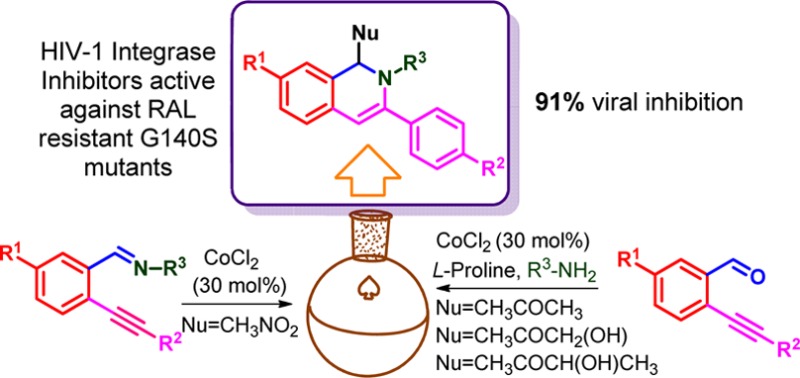
6-Endo-dig-cyclization is an efficient method for the synthesis of 1,2-dihydroisoquinolines. We have synthesized few 1,2-dihydroisoquinolines having different functionality at the C-1, C-3, C-7, and N-2 positions for evaluation against HIV-1 integrase (HIV1-IN) inhibitory activity. A direct nitro-Mannich condensation of o-alkynylaldimines and dual activation of o-alkynyl aldehydes by inexpensive cobalt chloride yielded desired compounds. Out of 24 compounds, 4m and 6c came out as potent integrase inhibitors in in vitro strand transfer (ST) assay, with IC50 value of 0.7 and 0.8 μM, respectively. Molecular docking of these compounds in integrase revealed strong interaction between metal and ligands, which stabilizes the enzyme–inhibitor complex. The ten most active compounds were subjected to antiviral assay. Out of those, 6c reduced the level of p24 viral antigen by 91%, which is comparable to RAL in antiviral assay. Interestingly, these compounds showed similar ST inhibitory activity in G140S mutant, suggesting they can act against resistant strains.
Keywords: Multicomponent reaction, integrase, integrase inhibitors, molecular docking
In the area of medicinal research efforts, the discovery, development, and evolution of novel and potent HIV-1 integrase (IN) inhibitors1 remains a significant scientific endeavor, as the high mutation ability of this virus results in resistance against drugs. HIV-1 IN is a 32 kDa protein that catalyzes the incorporation reaction of a viral genome inside the host cell and can then be taken as an absolute target for treating HIV-1 inhibition.2,3 Highly conserved DDE (D64, D116, and E152 amino acids) motif in catalytic core domain (CCD) of HIV-1 IN bind to divalent metal cofactors (Mg2+ and Mn2+).1,4,5 These metal ions present at the catalytic active site of an enzyme form a ligand–Mg2+–IN complex with the inhibitors. This ligand–Mg2+–IN complex would subsequently blocks the transition state of the IN–DNA complex by competing with the target DNA substrate, acting as an “interfacial inhibitor”.1 Treatment with FDA-approved raltegravir (RAL, Isentress or MK-0518)6 demonstrated significant and sustained suppression of viral RNA levels to less than 50 copies/mL, along with a substantial increase in CD4 immune cell counts.7 RAL has shown failure in the selection of mutations at integrase position Y143, Q148, or N155.8 Elvitegravir (EVG) (GS-9137)9 and dolutegravir (DTG) (S/GSK-1349572)10 are recently approved drugs along with INSTIs, which show improved efficacies against RAL resistant strains (Figure 1).11−13 However, viral strains that are highly resistant against EVG14 and DTG15 have been recently reported demonstrating multiple mutations in integrase.
Figure 1.

Structures of the best known HIV-1 integrase strand transfer inhibitors.
Earlier it was reported that 2-hydroxyisoquinoline-1,3(2H,4H)-dione scaffold chelates Mg2+ causing inhibition of HIV-1 IN and/or the HIV-1 reverse transcriptase ribonuclease H domain.16,17 Billamboz et al. further established that this class of compounds inhibit viral replication of HIV-1 in MT-4 cells.18 In line with continuous search for a better integrase inhibitor, we have selected dihydroisoquinoline as pharmacophore with a combination of nucleophiles having differential ability to chelate with Mg2+ ions. Based on these facts, we designed and synthesized three types of 1,2-dihydroisoquinolines derivatives bearing different functional groups at the C-1, C-3, C-7, and N-2 positions (Figure 2).
Figure 2.
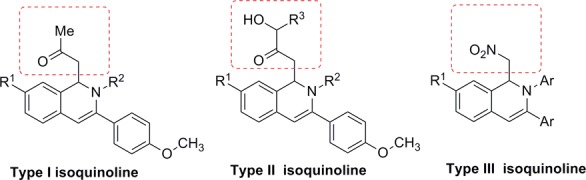
Designed isoquinolines as potential HIV-1 integrase strand transfer inhibitors.
Molecular docking of these compounds to integrase protein showed strong interactions between both. The antiviral and strand transfer inhibition assay suggested good biological and biochemical responses. At last, the combined efforts proved 6c as one of the most potent molecules in the antiviral assay among the ten compounds 4e, 4f, 4m, 4s, 4p, 6a–d.
The type I 1,2-dihydroisoquinolines 4a–h were synthesized by the reaction of o-alkynyl aldehyde 1a–b with ketones 2a–c and amines 3a–f via dual activation process as per the reported literature.19−21 Under the effective catalysis of inexpensive anhydrous CoCl2, intermolecular attack of enamine (generated by the reaction l-proline with ketones) onto o-alkynylaldimines (formed by the reaction of o-alkynylaldehyde with amine) resulted in the desired 6-endo-dig cyclized products 4a–h (Scheme 1). Reactions of 2-alkynylbenzaldehydes 1a–b with propanone (2a) and 4-aminophenol (3a) afforded the desired products 4a and 4h in 65 and 68% yields, respectively, after 12 h; whereas reaction of substrate 1b with propanone and p-toluidine 3e provided the product 4f in 87% yield in less reaction time (4 h). In the case of 4-aminophenol 3a, the presence of a hydroxy group at the p-position of the ring increases the possibility of the intermolecular hydrogen bonding and decreases the nucleophilicity of the NH2 group as well as the formation of imine; however, in the case of p-toluidine 3e the presence of an electron-releasing methyl group at the 4-position of the ring increases the nucleophilicity (+R effect) of the NH2 group, which facilitates the formation of the key intermediate imine.
Scheme 1. Synthesis of 1,2-Dihydroisoquinolines 4a–s.

Reactions were performed using 1.08 mmol of 2-alkynylbenzaldehyde 1, 5.0 equiv of ketone 2, 1 equiv of amine 3, 30 mol % CoCl2, 10 mol % l-proline in 5.0 mL of EtOH at 60 °C for 4 h unless otherwise noted.
Isolated yield.
Reaction time 12 h.
Reaction time 16 h.
It is notable that the electron-withdrawing (−CN, −NO2, −F) groups are present at the p-position of the phenyl ring of amines, providing the desired products 4b–e and 4g in comparatively low yields, which could be due to reduced nucleophilicity (−R effect) of the NH2 group. Dihydroisoquinolines 4i–s with a requisite hydroxyl group at the α-position of the keto group (Figure 2, type II isoquinolines) were synthesized in 28–48% yield by the reaction of substrate 1a with respective amines, hydroxyacetone (2b), and 3-hydroxybutan-2-one (2c). The low yields might be attributed to the low reactivity of ketones (able to form intramolecular hydrogen bond). All of the dihydroisoquinolines were obtained as racemic mixtures (R and S).
Type III, 1,2-dihydroisoquinolines (Figure 2) were prepared by nitro-Mannich condensation by our reported procedure.19 Dihydroisoquinolines 6a–e were synthesized in 58–90% yields via attack of aci-nitromethane on o-alkynylaldimines under cobalt-catalysis (Scheme 2). Substrate 5c and 5d bearing electron-withdrawing groups at the C-5 position provided the desired products in 72 and 70% yields, respectively; however, substrate 5e with an electron-releasing group yielded the desired product 6e in a 58% yield and required a longer reaction time.
Scheme 2. Synthesis of Nitro-Substituted 1,2-Dihydroisoquinolines,

The reaction was carried out using 5a–e (1 equiv) and nitromethane (2 equiv) in the presence of 30 mol % CoCl2 in 1,2-dichloroethane at 80 °C for 6 h.
Isolated yield.
Reaction time 10 h.
The synthesized compounds were examined for anti-integrase activity through in vitro biochemical assays, adapted from previously described methods.22 The strand transfer IC50 value (μM) and cell-based (TZM-bl) cytotoxicity CC50 value (μM) at 48 h were determined for all derivatives (Table 1). It was observed that dihydroisoquinolines having 7-fluoro function 4a–d were found to be less potent while the IC50 and CC50 values of 7-nitro derivatives of 1,2-dihydroisoquinoline 4e–h showed a significant range from 0.4 to 6.6 μM and 7.7 to 23.5 μM, respectively. Furthermore, the 1,2-dihydroisoquinoline bearing hydroxyl acetone 4i–m and hydroxyl butanone 4n–s functionality at the C-1 position showed an IC50 ranging from 0.82 to 22.1 μM and 5.2 to 25.2 μM, respectively. Additionally, the CC50 values were in the range of 8.5 to 50.9 μM for both earlier mentioned derivatives. However, in the case of the 6a–e derivatives (nitromethane group at the C-1 position) IC50 of 0.7 to 3.1 μM and CC50 10.3 to 32.2 μM were observed, which gave a lead to move on to the next step. The inhibitory activity of the compounds was also evaluated on one of the RAL resistant IN mutants G140S.23 Interestingly our compounds showed almost similar activity against mutant strain compared with the WT strain (Table 1), suggesting these compounds can act against RAL resistant IN. Furthermore, these molecules were screened in an antiviral assay using TZM-bl cell (Hela cell clone engineered to express CD4 and CCR5 receptors).24 Compounds 4m and 6c had shown good antiviral activity and a safety index of 4- and 1073-fold, respectively. p24 is a viral protein that is not present in normal uninfected cells. The early diagnosis of HIV infection can be measured by a p24 antigen assay, which is standard screening assay for HIV testing. TZM-bl cells may or may not be correlated with infectivity of other target cell types, e.g., PBMC, whereas p24 directly correlates with infectivity of cells. Hence we subjected ten potent compounds 4e, 4f, 4m, 4s, 4p, and 6a–e to antiviral assays on VSV-G pseudotyped virus-pNL4–3(WT-HIV-1) using a p24 antibody25−27 (Figure 3). At a concentration of 10 μM, compound 6c exhibited 91% reduction in the band intensity of p24 (calculated by densitometry analysis shown in Figure 3 and Figure S1), while for raltegravir, the band was observed to be reduced by 92%, and 4m exhibited 70.9% reduction in band intensity of p24 at 10 μM. Thus, taking into account cytotoxicity and the antiviral properties of 6c, having a nitro group as a metal coordinating unit showed promising potency in antiviral assays compared to corresponding hydroxyacetone bearing dihydroisoquinoline; 4m with a nitro group at the C-7 can also be considered a potent molecule.
Table 1. Inhibition of HIV-1IN, Cytotxicity, and Antviral Activity of Substituted 1,2-Dihydroxyquinolines.
| entry | %STa inhibition at 10 μM dose in WT | IC50 of ST activity in μm | IC50 of STb activity in G140S mutant | EC50 (μM) In TZM-bl cells | CC50 on TZM-bl cells (μm) | TI |
|---|---|---|---|---|---|---|
| RAL | 94.7 | 0.007 | 0.05 | 0.15 | >200 | >1300 |
| 4a | 65.7 | 12.0 | 14.0 | 21.4 | 60.5 | 2.8 |
| 4b | 44.2 | ND | ND | ND | ND | − |
| 4c | 40.2 | ND | ND | ND | 11.2 | − |
| 4d | 4.4 | ND | 20.0 | ND | ND | − |
| 4e | 62.4 | 0.46 | 0.64 | 36.3 | 7.7 | 0.2 |
| 4f | 69.9 | 0.6 | 0.65 | 26.3 | 10.6 | 0.4 |
| 4g | 67.5 | 2.9 | 2.8 | 22.5 | 15.4 | 0.6 |
| 4h | 65.0 | 6.6 | 7.6 | 31.4 | 23.5 | 0.7 |
| 4i | 57.7 | 15.1 | 14.1 | 28.4 | 23.7 | 0.8 |
| 4j | 55.1 | 17.9 | 14.9 | 26.4 | 8.5 | 0.3 |
| 4k | 71.9 | 7.6 | 7.0 | 29.1 | 39.7 | 1.3 |
| 4l | 72.5 | 22.1 | 20.1 | 24.4 | 47.7 | 1.9 |
| 4m | 90.6 | 0.82 | 46.4 | 10.41 | 46.4 | 4.4 |
| 4n | 60.9 | 10.6 | 12.7 | ND | 12.7 | − |
| 4o | 78.3 | 11.8 | 10.8 | ND | 9.9 | − |
| 4p | 78.7 | 7.9 | 8.0 | 11.7 | 14.0 | 1.1 |
| 4q | 62.2 | 22.2 | 22.0 | 18.7 | 24.1 | 1.2 |
| 4r | 78.0 | 25.2 | 25.0 | 19.7 | 50.9 | 2.5 |
| 4s | 88.2 | 5.2 | 5.5 | 12.7 | 10.1 | 0.7 |
| 6a | 40.2 | ND | ND | 15.6 | 10.3 | 0.6 |
| 6b | 55.2 | ND | ND | 16.5 | 21.5 | 1.3 |
| 6c | 91.5 | 0.7 | 0.8 | 0.03 | 32.2 | 1073 |
| 6d | 71.5 | 3.1 | 3.0 | 1.15 | 26.0 | 22.6 |
| 6e | 55 | ND | ND | 10.5 | 29.4 | 2.8 |
ST, % inhibition of strand transfer activity of integrase enzyme.
ST, dose determination of molecules at which 50% of integrase activity was inhibited. RAL, raltegravir. EC, effective concentration. TI, therapeutic index (CC50/EC50). ND is not determined.
Figure 3.
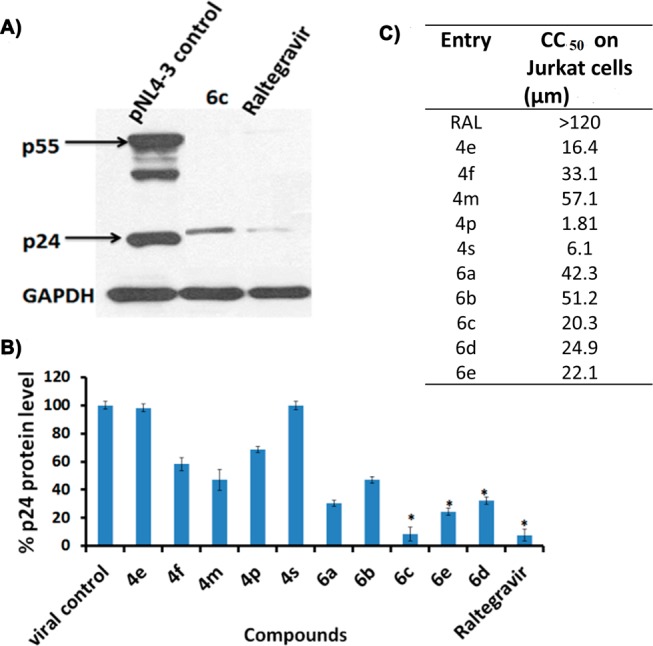
(A) One representative Western blot of jurkat cells treated with 6c or with no ligand (lane pNL4–3) and then infected at an MOI of 1 with VSV-G pseudotyped virus–pNL4–3 (WT–HIV-1). Cell supernatants were pelleted, solubilized in lysis buffer, detected, and analyzed using a p24 antibody. (B) Percent p24 viral protein level after treatment by compounds, calculated by densitometry analysis. *p < 0.05 compared to the control. (C) Cytotoxicity of the Jurkat cell line of the indicated compounds.
Molecular docking was carried out in silico with both R and S stereoisomers to understand the molecular recognition interactions. A two-metal model for HIV-1 IN CCD in complex with the small molecule, 1-(5-chloroindol-3-yl)-3- (tetrazoyl)-1,3-propandione-ene (5-CITEP), was used as a surrogate for an IN/viral DNA complex, as this model explains the molecular recognition interactions.28 The molecular docking poses along with interactive sites of the compounds with amino acids are provided (Supporting Table S1). The molecular docking study of 4m, 6c, and 6d demonstrated that nitro groups chelate with both the active site magnesium ions. Additionally these compounds show cation−π interaction with Mg2+ (Figure 4). Compound 4m has been found to be the best among the compounds 4a–s in in vitro as well as antiviral assays. The molecular docking scores of these compounds were in agreement with their activity. Additionally, the p-fluorophenyl group of compound 4m interacts with Lys159 and Lys156 to form hydrogen bonding networks (Figure 4b). Compound 6c perfectly fits in the active site, chelating both active site Mg2+ so that it blocks access of Mg2+ to the substrate. Additionally, it forms hydrogen bonds between the phenolic methoxy group and Ser119. Asn117, Asn120, and Lys159 were involved in interactions with the target DNA. Alves et al.29 reported the importance of Lys159 in anti-HIV IN activity by a quantum mechanics/molecular mechanics (QM/MM) study of the protein–ligand interaction for HIV-1 integrase inhibitors. Recently, Ser119 contribution to target site preferences was reported in the CCD of IN.30 Our results corroborate with the literature reports in terms of substrate–enzyme interactions.
Figure 4.
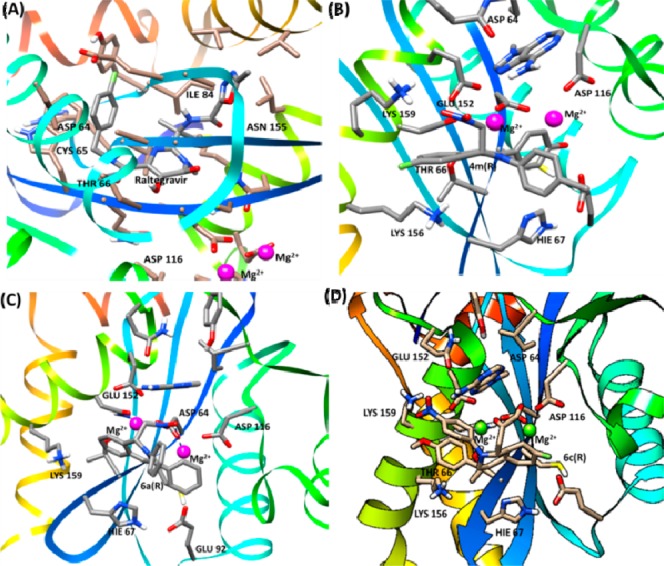
Docked poses of ligand-HIV IN CCD in complexes of (A) raltegravir, (B) 4m(R), (C) 6a(R), and (D) 6c(R). The chelation of two Mg2+ ions with the ligands is shown here. The most interactive residues in the docked complexes were found to be Asp116, Glu92, Lys159, Hie67, and Asp64.
Gratifyingly, 24 compounds were successfully synthesized using an operationally simple, direct approach. The cell based cytotoxicity assay in TZM-bl and Jurkat cells (Figure 3C) along with ST inhibition suggest that 4e, 4f, 4m, 4s, 4p, and 6a–e have good therapeutic efficacy in vitro. The probable reason for the diminished activity of 4e, 4f, 4m, 4s, 4p, 6a, 6b, 6d, and 6e in antiviral assays could be high protein binding. According to the molecular docking study, the addition of a nitro moiety instead of an acetone chain is an attractive approach to increase the inhibitory activity against HIV-1 IN, as the nitro group is a stronger nucleophile in which oxygen atoms can chelate with the active site Mg2+ ions in a more effective manner. In light of the above observations, undesired intramolecular hydrogen bonding could be a reason for the diminished activity of these acetone, hydroxyacetone, and hydroxybutanone analogues. Compounds 4a–s contain a number of oxygen atoms in the vicinity and had a maximum opportunity to chelate Mg2+ to inhibit enzymatic activity (due to keto–enol tautomerism) and form intramolecular hydrogen bonds, which drastically reduce the availability of oxygen atoms to chelate Mg2+. Hence, the introduction of nitro in the 1,2-dihydroisoquinoline derivatives is an attractive idea for the generation of new drug entities. Compound 6c showed a noteworthy 91% reduction of viral antigens as indicated in Figure 3B comparable to that of RAL. Interestingly, these compounds showed excellent inhibition of ST activity against RAL-resistant IN mutant G140S. The G140S mutation causes a conformational change that overcomes the ability of Q148H and other RAL-resistant mutants to replicate in cells. As these compounds have comparable activity against G140S, they are better substitutes to overcome the limitations developed against RAL. The docking posture of these compounds simplifies the presence of oxygen atoms in the vicinity of both the active site Mg2+ to form metal coordination. These compounds show cation−π interactions with the active site Mg2+ ions. Furthermore, the binding of these compounds at the active site of HIV-IN is stabilized by various electrostatic and van der Waals interactions with the active site amino acids (docking table). The CCD of IN is essential for 3′-processing of the viral DNA and ST reactions, and the substitution of any of the residues in the DDE motif dramatically inhibits the activity of IN. We propose that newly synthesized INTIs 6a–e primarily act through this mechanism.
In summary, a series of differently substituted 1,2-dihydroisoquinolines were synthesized in one-pot via a nitro-Mannich reaction from easily accessible o-alkynylal dehydes under mild reaction conditions and cobalt catalysis as an inexpensive methodology. Screening of the designed compounds on HIV-1 IN inhibition have confirmed 1,2-dihydroisoquinoline derivatives as potential HIV-1 INSTIs. The lead compounds disclosed in this study demonstrated (i) significant inhibition against the strand transfer processes of HIV-1 IN; (ii) significant antiviral activity, and (iii) comparatively reduced cytotoxicity in the TZM-bl cell line. Among 24 compounds screened, 4m and 6c stood out as potent HIV-1 inhibitors with significant ST inhibition. Dihydroisoquinoline 6c showed significant viral inhibition with better ST inhibition making it a potent HIV-1 inhibitor. The cross resistance of elvitegravir31 and dolutegravir32 with raltegravir cannot be ruled out. Hence, novel agents can prove to be an effective alternative to FDA approved INSTIs. The potential newer molecules presented in our study are indeed toxic compared to raltegravir but will provide a backbone to develop next-generation inhibitors against integrase.
Glossary
ABBREVIATIONS
- RAL
raltegravir
- IN
integrase
- INSTI
Integrase strand transfer inhibitor
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00230.
Experimental procedures, biological assays, characterization of compounds, and docking table (PDF)
Author Contributions
The manuscript was written via contributions of all authors. All authors have given approval of the final version of the manuscript.
The authors acknowledge the Department of Science and Technology for financial support and the Delhi University Instrumentation Centre for providing NMR facilities. P.Y. is thankful to DST-WOS B for financial support. S.S. and V.Ti. are thankful to CSIR, India for SRF.
The authors declare no competing financial interest.
Supplementary Material
References
- Pommier Y.; Johnson A. A.; Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug Discovery 2005, 4, 236–248. 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- Neamati N.; Sunder S.; Pommier Y. Design and discovery of HIV-1 integrase inhibitors. Drug Discovery Today 1997, 2, 487–498. 10.1016/S1359-6446(97)01105-7. [DOI] [Google Scholar]
- Rice P.; Craigie R.; Davies D. R. Retroviral integrases and their cousins. Curr. Opin. Struct. Biol. 1996, 6, 76–83. 10.1016/S0959-440X(96)80098-4. [DOI] [PubMed] [Google Scholar]
- Chiu T. K.; Davies D. R. Structure and function of HIV-1 integrase. Curr. Top. Med. Chem. 2004, 4, 965–977. 10.2174/1568026043388547. [DOI] [PubMed] [Google Scholar]
- Kulkosky J.; Jones K. S.; Katz R. A.; Mack J. P.; Skalka A. M. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retro transposon integrases and bacterial insertion sequence transposases. Mol. Cell. Biol. 1992, 12, 2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summa V.; Petrocchi A.; Bonelli F.; Crescenzi B.; Donghi M.; Ferrara M.; Fiore F.; Gardelli C.; Gonzalez P. O.; Hazuda D. J.; Jones P.; Kinzel O.; Laufer R.; Monteagudo E.; Muraglia E.; Nizi E.; Orvieto F.; Pace P.; Pescatore G.; Scarpelli R.; Stillmock K.; Witmer M. V.; Rowley M. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 2008, 51, 5843–5855. 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- Steigbigel R. T.; Cooper D. A.; Kumar P. N.; Eron J. E.; Schechter M.; Markowitz M.; Loutfy M. R.; Lennox J. L.; Gatell J. M.; Rockstroh J. K.; Katlama C.; Yeni P.; Lazzarin A.; Clotet B.; Zhao J.; Chen J.; Ryan D. M.; Rhodes R. R.; Killar J. A.; Gilde L. R.; Strohmaier K. M.; Meibohm A. R.; Miller M. D.; Hazuda D. J.; Nessly M. L.; DiNubile M. J.; Isaacs R. D.; Nguyen B. Y.; Teppler H. Raltegravir with optimized background therapy for resistant HIV-1 infection. N. Engl. J. Med. 2008, 359, 339–354. 10.1056/NEJMoa0708975. [DOI] [PubMed] [Google Scholar]
- Garrido C.; Villacian J.; Zahonero N.; Pattery T.; Garcia F.; Gutierrez F.; Caballero E.; Van H. M.; Soriano V.; de M. C. Broad phenotypic cross-resistance to elvitegravir in HIV-infected patients failing on raltegravir containing regimens. Antimicrob. Agents Chemother. 2012, 56, 2873–2878. 10.1128/AAC.06170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M.; Motomura T.; Aramaki H.; Matsuda T.; Yamashita M.; Ito Y.; Kawakami H.; Matsuzaki Y.; Watanabe W.; Yamataka K.; Ikeda S.; Kodama E.; Matsuoka M.; Shinkai H. Novel HIV-1 integrase inhibitors derived from quinolone antibiotics. J. Med. Chem. 2006, 49, 1506–1508. 10.1021/jm0600139. [DOI] [PubMed] [Google Scholar]
- Min S.; Song I.; Borland J.; Chen S.; Lou Y.; Fujiwara T.; Piscitelli S. C. Pharmacokinetics and safety of S/GSK1349572, a next-generation HIV integrase inhibitor, in healthy volunteers. Antimicrob. Agents Chemother. 2010, 54, 254–258. 10.1128/AAC.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura K.; Kodama E. N. Elvitegravir: a new HIV integrase inhibitor. Antivir. Chem. Chemother. 2009, 20, 79–85. 10.3851/IMP1397. [DOI] [PubMed] [Google Scholar]
- Ramkumar K.; Neamati N. Raltegravir: The evidence of its therapeutic value in HIV-1 infection. Core. Evid. 2009, 4, 131–147. 10.2147/CE.S6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mawsawi L. Q.; Al-Safi R. I.; Neamati N. Anti-infectives: clinical progress of HIV-1 integrase inhibitors. Expert Opin. Emerging Drugs 2008, 13, 213–225. 10.1517/14728214.13.2.213. [DOI] [PubMed] [Google Scholar]
- Abram M. E.; Hluhanich R. M.; Goodman D. D.; Andretta K. N.; Margot N. A.; Ye L.; Niedziela-Majka A.; Barnes T. L.; Novikov N.; Chen X.; Svarovskaia E. S.; McColl D. J.; White K. L.; Miller M. D. Impact of Primary Elvitegravir Resistance-Associated Mutations in HIV-1 Integrase on Drug Susceptibility and Viral Replication Fitness. Antimicrob. Agents Chemother. 2013, 57, 2654–2663. 10.1128/AAC.02568-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carganico A.; Dupke S.; Ehret R; Berg T.; Baumgarten A.; Obermeier M.; Walter H. New dolutegravir resistance pattern identified in a patient failing antiretroviral therapy. J. Int. AIDS Soc. 2014, 174, 19749. 10.7448/IAS.17.4.19749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp K.; Hang J. Q.; Rajendran S.; Yang Y.; Derosier A.; Wong K.; Overton H.; Parkes K. E.; Cammack N.; Martin J. A. Two-metal ion mechanism of RNA cleavage by HIV RNase H and mechanism-based design of selective HIV RNase H inhibitors. Nucleic Acids Res. 2003, 31, 6852–6859. 10.1093/nar/gkg881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchaud V.; Bailly F.; Lion C.; Calmels C.; Andreolá M.; Christ F.; Debyser Z.; Cotelle P. Investigation of a novel series of 2-hydroxyisoquinoline-1,3(2H,4H)-diones as human immunodeficiency virus type 1 integrase inhibitors. J. Med. Chem. 2014, 57, 4640–4660. 10.1021/jm500109z. [DOI] [PubMed] [Google Scholar]
- Billamboz M.; Bailly F.; Lion C.; Touati N.; Vezin H.; Calmels C.; Andreola M. L.; Christ F.; Debyser Z.; Cotelle P. Magnesium chelating 2-hydroxyisoquinoline-1,3(2H,4H)-diones, as inhibitors of HIV-1 integrase and/or the HIV-1 reverse transcriptase ribonuclease H domain: discovery of a novel selective inhibitor of the ribonuclease H function. J. Med. Chem. 2011, 54, 1812–1824. 10.1021/jm1014692. [DOI] [PubMed] [Google Scholar]
- Urvashi; Rastogi G. K.; Ginotra S. K.; Agarwal A.; Tandon V. An expedient approach to 1,2-dihydroisoquinoline derivatives via cobalt catalysed 6-endo dig cyclization followed by Mannich condensation of o-alkynylarylaldimines. Org. Biomol. Chem. 2015, 13, 1000–1007. 10.1039/C4OB02036G. [DOI] [PubMed] [Google Scholar]
- Ding Q.; Wu J. Lewis acid- and organocatalyst-cocatalyzed multicomponent reactions of 2-alkynylbenzaldehydes, amines, and ketones. Org. Lett. 2007, 9, 4959–4962. 10.1021/ol7020669. [DOI] [PubMed] [Google Scholar]
- Markina N. A.; Mancuso R.; Neuenswander B.; Lushington G. H.; Larock R. C. Solution-phase parallel synthesis of a diverse library of 1,2-dihydroisoquinolines. ACS Comb. Sci. 2011, 13, 265–271. 10.1021/co1000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David C. A.; Middleton T.; Montgomery D.; Lim H. B.; Kati W.; Molla A.; Xuei X.; Warrior U.; Kofron J. L.; Burns D. J. Microarray compound screening (microARCS) to identify inhibitors of HIV integrase. J. Biomol. Screening 2002, 7, 259–266. 10.1177/108705710200700309. [DOI] [PubMed] [Google Scholar]
- Delelis O.; Malet I.; Na L.; Tchertanov L.; Calvez V.; Marcelin A. G.; Subra F.; Deprez E.; Mouscadet J. F. The G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutation. Nucleic Acids Res. 2009, 37, 1193–1201. 10.1093/nar/gkn1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Gao F.; Mascola J. R.; Stamatatos L.; Polonis V. R.; Koutsoukos M.; Voss G.; Goefert P.; Gilbert P.; Greene K. M.; Bilska M.; Kothe D. L.; Salazar-Gozalez J. F.; Wei X.; Decker J. M.; Hahn B. H.; Montefiori Human immunodeficiency virusttype 1 env clones from acute and early subtype b infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 2005, 79, 10108–25. 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akari H.; Bour S.; Kao S.; Adachi A.; Strebel K. The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor κ-β-dependent expression of anti apoptotic factors. J. Exp. Med. 2001, 194, 1299–1311. 10.1084/jem.194.9.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata S.; Ali A.; Sood V.; Raja R.; Banerjea A. C. HIV-1 Rev downregulates Tat expression and viral replication via modulation of NAD(P)H:quinine oxidoreductase 1 (NQO1). Nat. Commun. 2015, 6, 7244–8244. 10.1038/ncomms8244. [DOI] [PubMed] [Google Scholar]
- Cornall A.; Sharma L.; Solomon A. A novel, rapid method to detect infectious HIV-1 from plasma of persons infected with HIV-1. J. Virol. Methods 2010, 165, 90–96. 10.1016/j.jviromet.2010.01.010. [DOI] [PubMed] [Google Scholar]
- Savarino A. In-Silico docking of HIV-1 integrase inhibitors reveals a novel drug type acting on an enzyme/DNA reaction intermediate. Retrovirology 2007, 4, 21. 10.1186/1742-4690-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves C. N.; Marti S.; Castillo R.; Andres J.; Moliner V.; Tunon I.; Silla E. A quantum mechanics/molecular mechanics study of the protein-ligand interaction for inhibitors of HIV-1 integrase. Chem. - Eur. J. 2007, 13, 7715–7724. 10.1002/chem.200700040. [DOI] [PubMed] [Google Scholar]
- Harper A. L.; Skinner L. M.; Sudol M.; Katzman M. Use of patient-derived human immunodeficiency virus type 1 integrases to identify a protein residue that affects target site selection. J. Virol. 2001, 75, 7756–7762. 10.1128/JVI.75.16.7756-7762.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez-Arias L. Molecular basis of human immunodeficiency virus type 1 drug resistance: Overview and recent developments. Antiviral Res. 2013, 98, 93–120. 10.1016/j.antiviral.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Canducci F.; Ceresola E. R.; Boeri E.; Spagnuolo V.; Cossarini F.; Castagna A.; Lazzarin A.; Clementi Massimo. Cross-resistance profile of the novel integrase inhibitor dolutegravir (S/GSK1349572) using clonal viral variants selected in patients failing raltegravir. J. Infect. Dis. 2011, 204, 1811–1815. 10.1093/infdis/jir636. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.