

Abstract
Myeloperoxidase (MPO) is a key antimicrobial enzyme, playing a normal role in host defense, but also contributing to inflammatory conditions including neuroinflammatory diseases such as Parkinson’s and Alzheimer’s. We synthesized and characterized more than 50 quinazolin-4(1H)-one derivatives and showed that this class of compounds inhibits MPO with IC50 values as low as 100 nM. Representative compounds showed partially reversible inhibition that was competitive with respect to Amplex Red substrate and did not result in the accumulation of MPO Compound II. Members of this group show promise for therapeutic development for the treatment of diseases in which inflammation plays a pathogenic role.
Keywords: Myeloperoxidase, thioxo-dihydroquinazolin-one, inflammation, NADPH-oxidase, therapeutic
Myeloperoxidase (MPO) is localized in the azurophilic granules of granulocytes. During phagocytosis of a microorganism, MPO is secreted into the phagolysosome, where it catalyzes the reaction between hydrogen peroxide and halides to form hypohalite ions, especially hypochlorite, a powerful microbicidal agent. Hypochlorite, a highly potent oxidant, reacts with biomolecules, causing oxidation and/or chlorination, for example, to form 3-chloro-tyrosine in proteins. In addition, MPO directly catalyzes the H2O2-dependent oxidation of biomolecules including proteins and DNA. These reactions play a key role in host defense by neutrophils and macrophages, resulting (along with other reactions) in the killing of invading microbes.
MPO-dependent reactions are also damaging to host tissues, can be pro-inflammatory, and can play a role in the initiation and progression of inflammatory diseases. For example, MPO itself and its oxidation/chlorination products such as 3-chlorotyrosine are detected in atherosclerotic plaques where they may play a pathogenic role. MPO has also been implicated in other diseases including stroke, renal injury, lung inflammation, Alzheimer’s disease, Parkinson’s disease, and others.1 Disease occurrence and severity may also be influenced by MPO overexpression. For example, the MPO allele bearing a −463G/A polymorphism results in increased transcription and abnormally high MPO levels.2 This polymorphism is associated with increased risk of coronary artery disease, sarcoidosis, Alzheimer’s disease, and several cancers. Thus, there is ample rationale to develop small molecule inhibitors of MPO that may prove useful for drug development.3−6
As part of a screen to identify inhibitors of human neutrophil NADPH oxidase 2 (NOX2; see PubChem AID 1274 and AID 1275), we discovered a compound that inhibited NADPH-dependent L-012 chemiluminescence in neutrophil membranes. The L-012 assay was previously reported as a sensitive method to measure superoxide (O2–•).7 Later, we reported that the L-012 signal requires not only a source of reactive oxygen (e.g., a NOX) but also a peroxidase.8 We found that the compound failed to inhibit NOX2-generated O2–• but instead targeted MPO (vide infra). The present study characterizes the inhibition and explores structural features that correlate with bioactivity among members of this group of MPO inhibitors.
Results
Medicinal Chemistry
The hit compound 1c (see Table 1) inhibited L-012 oxidation (IC50 of 0.8 μM, Table 2, Supporting Information (SI)). Medicinal chemistry was undertaken to improve potency and to investigate structure–activity relationships (SARs). Modifications were made in the left-hand and middle rings as well as in the side chain. Based on synthetic strategy, the thioxodihydroquinazolinone compounds9−12 described here are subdivided into Series 1 and Series 2 (further subdivided into Series 2A and 2B, Figure 1). The modifications focused mainly on three regions (X, Y, and Z) of molecules (Figure 1).
Table 1. IC50 Values for Inhibition of MPO and Xanthine Oxidase (XO) Activitya.
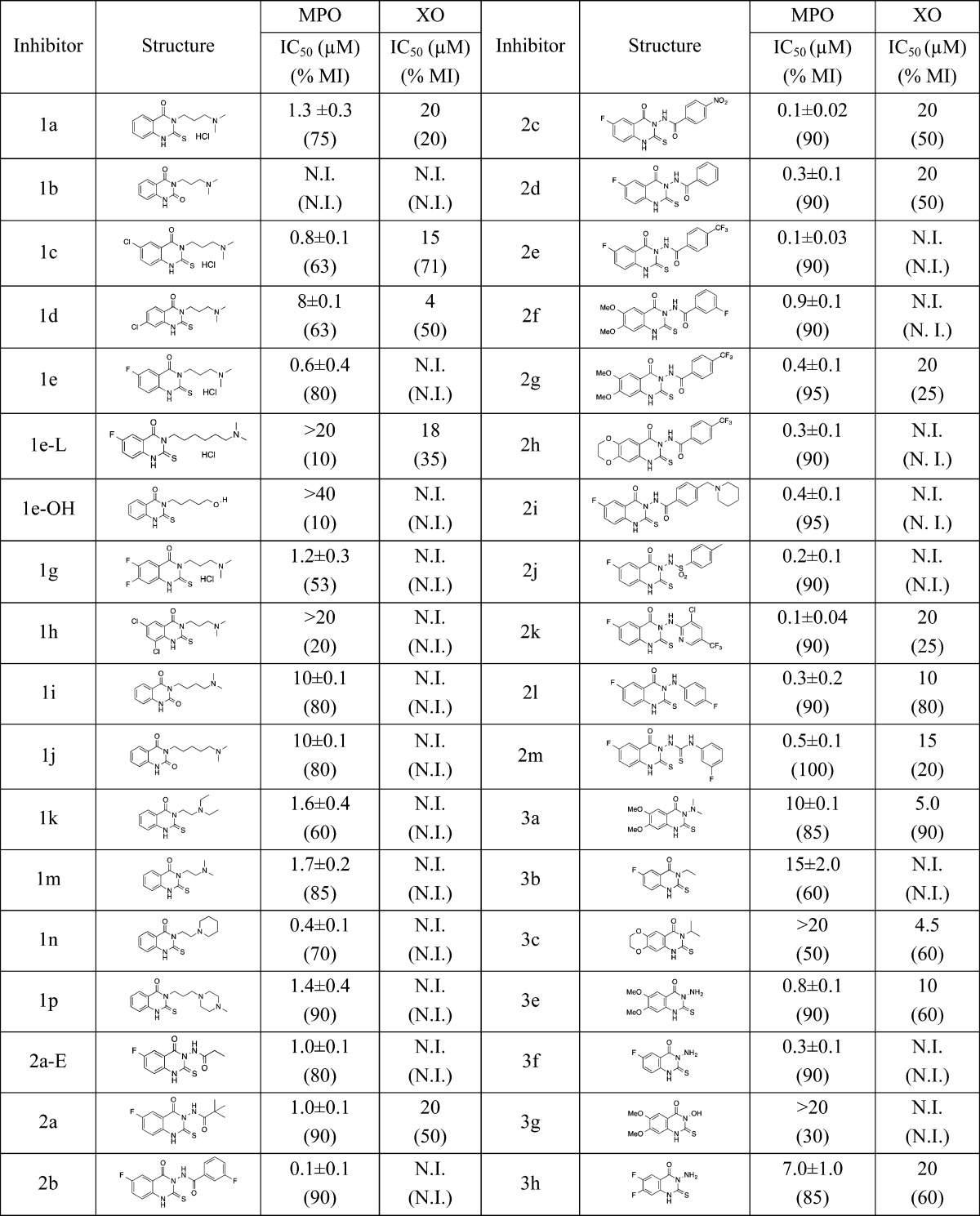
MPO (Amplex Red assay) and XO activities were measured under the same pH and buffer conditions (see Methods, SI), using a concentration range up to 100 μM for each inhibitor. IC50 values represent the average ± SEM of two to six determinations. N.I., no inhibition. % maximum inhibition values (% MI) are indicated in parentheses below the IC50 values.
Figure 1.
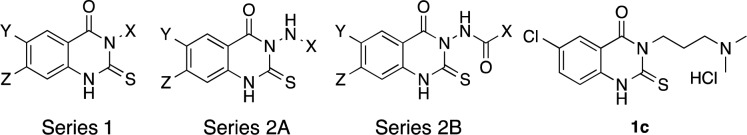
General structures of Series 1 and 2 compounds and compound 1c.
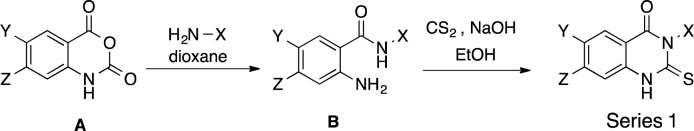
Series 1 compounds were synthesized as in Scheme 1 from commercially available isatoic anhydrides (A), which were treated with alkylamine containing varying alkyl chain lengths and varying terminal units (amines or other substitute groups) to generate 2-aminobenzamides (B). B was then treated in situ with NaOH and CS2 to generate the final Series 1 compounds. Compounds were purified using silica column chromatography (SI) and/or recrystallization.
Scheme 1. General Synthesis of Series 1 Compounds.
Series 2 compounds were synthesized according to Scheme 2, beginning with a benzamine (C), which was treated with thiophosgene in the presence of triethylamine to provide isothiocyanate (D). D was refluxed with an alkyl hydrazine and triethylamine in THF to generate Series 2A compounds. Treatment of D with hydrazine and triethylamine followed by a substituted chlorocarbonyl reagent generated series 2B compounds (Scheme 2).
Scheme 2. General Synthesis of Series 2 Compounds.

Approximately 38 Series 1 thioxodihydroquinazolinone analogues were first synthesized (some of them shown in Table 2, SI). Some compounds containing nitrogen at position X (Table 2, SI) showed good activity in the screening assay and formed the basis for designing Series 2A and 2B compounds (Figure 1).
In preliminary studies (Table 2, SI), compounds were tested by measuring L-012 oxidation, in a cell-free system from human neutrophils (METHODS).13 The xanthine/xanthine oxidase assay system14 was used to eliminate false positives from assay interference. Representative compounds in both series were then tested for inhibition of O2–• generation by NOX2 using NADPH-dependent, SOD-inhibitable cytochrome c reduction, an assay that is specific for O2–•.15 Because no inhibition was seen (not shown), some of the more promising compounds were then tested for inhibition of purified human MPO. All compounds tested showed inhibition of MPO, indicating that for these compounds, the initial assay using neutrophil membranes and L-012 was detecting inhibition of MPO and not NOX2. Examples of MPO inhibition curves for two compounds from series 1 (1c) and series 2 (2b) are shown in Figure 2. Recombinant MPO was inhibited by 1c with an IC50 of 0.8 μM and 60% maximum inhibition at high concentrations of the compound, and by 2b with an IC50 of 0.1 μM and 90% maximum inhibition. These were also tested for assay interference using horseradish peroxidase in place of MPO in the Amplex Red assay (Figure 2); no inhibition was seen even at high concentrations of compounds. Summary results for 36 representative compounds are shown in Table 1. Table 1 also provides data on possible assay interference, using xanthine/xanthine oxidase as a source for oxidants; in general, the compounds did not show assay interference in the concentration range that inhibited MPO.
Figure 2.

Concentration dependence for inhibition of human MPO and horseradish peroxidase (HRP). Activity was measured using the Amplex Red method at the indicated concentrations of 1c (squares) and 2b (circles). Assays were performed with 0.17 nM MPO (filled symbols) or 20 nM HRP (open symbols). Points show the mean ± SEM of three determinations.
Structure–Activity Correlations
Inspection of Table 1 allows some conclusions with regard to structural features that are important for optimal inhibition of MPO. First, sulfur was essential for bioactivity since replacement of sulfur with oxygen reduced or eliminated inhibition (compounds 1a vs 1b). Within limits, the side-chain (X in Scheme 1) or the presence of additional carbons on the terminal nitrogen had small effects on potency, suggesting that the binding site can accommodate modest variations in structure. However, increasing the aliphatic side-chain to five carbons (compound 1e-L, Table 1) completely eliminated activity, suggesting a size limitation to the binding cavity. A side-chain containing two to three carbons appeared to be optimal (e.g., compare 1a with 1e-L, 1i, 1j, 1k, and 1m). Analogues with a 3-carbon link and a piperazine tail (1p) or a piperidine tail (1n) showed modestly increased potency.
The effect of substituents on the left side phenyl ring was evaluated. A halide at position 6 of the left-hand phenyl ring frequently improved bioactivity (e.g., compare 1a with 1c and 1e in Table 1). Halides in the 7- or 8-positions on the same ring or the presence of two halides failed to improve activity compared with a halide at position 6 (e.g., see 1d, 1g, 1h). For example, a fluoro group at position 6 (1e, Table 1) improved activity, compared with 1a, whereas compound 1g with fluoro group at both 6- and 7-positions was less potent than the 6-monofluoro derivative. A chloro group at the 8-position (1h) eliminated inhibitory activity. Replacement of 6-fluoro substituent with a 6,7-dimethoxyl substitutes (2g) or dioxane group (2h) decreased activity compared with 2b suggesting somewhat stringent structural requirements at the binding site for the left side of the molecule.
A free amino group directly linked to one of the ring nitrogens in place of the N,N-dimethylaminopropyl side chain sometimes improved potency (e.g., compare 3f vs 1e), while a dimethylamino (3a) or a hydroxyl (3g) group weakened the potency compared with the free amino group. Ethyl (3b) and isopropyl (3c) groups linked to the ring nitrogen decreased potency (Table 1). Based on the improved activity of compounds with nitrogen directly linked to the ring nitrogen, Series 2A and 2B compounds were synthesized to maintain this structural feature while varying the side chain and left-side substituents (2a–2m, Table 1). Structural features of Series 2 inhibitors include an N–N linkage (Series 2A) or N–N–CO (Series 2B). An attached aromatic ring distal to the N–N linkage improved potency compared with an aliphatic side-chain (e.g., compare 2a and 2a-E with others in Series 2). As a group, compounds with this feature showed IC50 values for MPO inhibition that were consistently less than 400 nM. Some (2b, 2c, 2e, 2j, and 2k) showed IC50 values of 100–200 nM with 90% maximum inhibition. With regard to the importance of the benzamide linker, a sulfonamide linkage (2j) did not have a significant effect on potency. Analogues with a phenyl amine link (2k and 2l) instead of an amide showed similar potency compared with compounds containing an amide linkage. Inhibitor 2k with o-chloro and p-trifluoromethyl substituent groups, and 2l with a p-fluoro group showed excellent potency against purified MPO (Table 1). Compounds 1c, 2b, 2k, and 2i were also evaluated using the taurine chlorination assay (Methods, SI), and all were found to be active, with IC50 values about 2–3-fold higher than with the Amplex Red assay.
Inhibitor 1c was soluble in water to 90 μg/mL, but the solubility of 2b, 2c, and 2e were fairly modest, around 30 μg/mL (data not shown), which might prove problematic for future drug development. To increase solubility, a derivative with a methylene piperidinyl group (2i) was synthesized. This derivative maintained good potency (0.4 μM IC50), and its solubility was improved 50-fold to 1500 μg/mL, suggesting that this strategy may be useful for future development of these compounds.
Kinetic Mechanism of Inhibition
The kinetic mechanism of inhibition of MPO-dependent oxidation of Amplex Red was investigated for the inhibitors 1c and 2b. When H2O2 concentration was varied at several fixed inhibitor concentrations, the apparent Vmax changed but the Km for H2O2 did not, indicating noncompetitive inhibition. Figure 1, SI, shows a representative experiment using inhibitor 1c, and the same result was obtained for 2b. This shows that the inhibitors do not block the access of H2O2 to heme. In contrast, when Amplex Red concentration was varied at various inhibitor concentrations, the Vmax remained the same, but the Km for Amplex Red shifted to higher concentrations. Figure 2, SI, shows a representative kinetic analysis for 2b, and the same result was seen for 1c. These results support a model in which the inhibitor binds at the substrate-binding site, preventing access of Amplex Red.
Reversibility of Inhibition
Several known inhibitors of MPO (including the control inhibitor 4-aminobenzoic acid hydrazide ABAH) covalently modify MPO and therefore act as irreversible inhibitors.16,17 Reversibility was investigated using an ELISA assay in which plate-bound MPO is incubated with the inhibitor, and the ELISA plate is then washed to remove any inhibitor that is noncovalently bound, followed by assay of activity. As shown in Figure 3, ABAH nearly fully inhibits activity, and only a small amount of activity is regained upon washing the plate. In contrast, a reversible inhibitor of MPO,18 salicylhydroxamic acid (SHA), partially inhibited the enzyme, which then regained full activity after washing. Compound 1c and compound 2k also recovered significant activity upon washing. This indicates that in contrast to several other MPO inhibitors,16 representative members of this chemical series do not irreversibly inactivate MPO.
Figure 3.

Determination of reversibility of MPO inhibition; ABAH (6 μM), SHA (100 μM), 1c (50 μM), and 2k (50 μM), all in DMSO (1% final) or DMSO alone (Control), were incubated with MPO bound to wells of an ELISA plate for 1 h in the presence of 50 μM H2O2. Wells were either washed to remove inhibitor (gray bars) or remained unwashed (black bars) prior to measuring MPO activity as in Methods, SI. Results show mean MPO activity (ΔA652 nm/min values ± SEM, n = 3).
Effects of Representative Inhibitors on Absorption Spectra of Myeloperoxidase During Turnover
MPO (Fe3+) reacts with H2O2 to generate Compound I, which can react with chloride to generate HOCl, regenerating MPO (Fe3+), or it can react with a 1-electron-donor to produce Compound II. A second electron returns the enzyme to MPO (Fe3+). Some compounds inhibit MPO-dependent chlorination by acting as an efficient substrate for Compound I but not Compound II, trapping the enzyme as Compound II (removing it from the chlorination cycle).19 Under our conditions, H2O2 generated a small quantity of Compound II (solid line, Figure 4), which was markedly increased by 5-fluorotryptamine (5-FTA), known to inhibit MPO by trapping the enzyme as Compound II.19 Inhibitors 1c, 2b, or 3f caused a slight decrease rather than an increase in Compound II (Figure 4), indicating that their mechanism of inhibition does not involve trapping of MPO as Compound II.
Figure 4.

Effect of compounds 1c, 2b, 3f, and 5-fluorotryptamine on MPO intermediates. Difference spectra resulting from the addition of 250 μM H2O2 to purified MPO (1.2 μM) in the absence or presence of 10 μM inhibitor, 1% DMSO, and 150 mM NaCl in 50 mM potassium phosphate buffer, pH 7.4.
Summary
Thioxo-dihydroquinazolin-one derivatives were identified as novel inhibitors of MPO. These compounds are similar in some ways to the thioxanthine class of MPO inhibitors17,20−24 in that both have a thiourea moiety as part of a ring system, but the thioxanthines act as covalent inhibitors of MPO.17 Members of this class of compounds may prove useful for the future development of drugs targeting diseases in which inflammation plays a role, including neurodegenerative diseases, atherosclerosis, and others. These compounds, like the aromatic hydroxamates,18 differ from many other MPO inhibitors,16,17,19,24,25 in that they act reversibly rather than irreversibly and do so without the generation of MPO Compound II. Because inhibition by compounds that act by trapping the enzyme as Compound II might be reversed by endogenous reductants, e.g., ascorbate,26 they may lose effectiveness in vivo. The inhibitors described here do not trap the enzyme as Compound II and may provide advantages in some clinical settings.
Acknowledgments
We thank Dr. Minoru Tamura for the p67Np47N chimera plasmid. We thank Ben Lambeth for writing the computer program for storage and retrieval of compounds and associated data.
Glossary
ABBREVIATIONS
- ABAH
aminobenzoic acid hydrazide
- DMSO
dimethyl sulfoxide
- DTPA
diethylenetriamine pentaacetic acid
- FAD
flavin adenine dinucleotide
- 5-FTA
5-fluorotryptamine
- HRP
horseradish peroxidase
- MPO
myeloperoxidase
- NADPH
nicotinamide adenine dinucleotide phosphate
- NOX2
NADPH oxidase 2
- p47phox
cytosolic 47 000 Da protein, subunit of the NOX2 complex
- p67Np47N
a chimeric protein of the N-terminal segment of the p67phox and p47phox subunits
- p67phox
cytosolic 67 000 Da protein, subunit of the NOX2 complex
- phox
phagocyte NOX
- PMA
phorbal 12-myristate 13-acetate
- PPB
potassium phosphate buffer, pH 7.4
- ROS
reactive oxygen species
- SDS
sodium dodecyl sulfate
- SHA
salicylhydroxamic acid
- SOD
superoxide dismutase
- TLC
thin layer chromatography
- TMB
tetramethylbenzidine
- XO
xanthine oxidase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00287.
Experimental details along with the chemical and physical characterization of the compounds (PDF)
Author Contributions
All authors contributed to the writing of the manuscript. Y.L., T.G., and A.S. synthesized, purified, and characterized the compounds. Y.L., T.G., A.S., and J.D.L. designed compounds. B.D., Y.Z., J.M., S.S., and J.D.L. carried out and/or designed enzymatic assays. J.D.L. was responsible for the oversight of the project. All authors have given approval to the final version of the manuscript.
Supported by NIH Grant R33 AI102197.
The authors declare no competing financial interest.
Supplementary Material
References
- Klebanoff S. J. Myeloperoxidase: friend and foe. J. Leukocyte Biol. 2005, 77 (5), 598–625. 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- Piedrafita F. J.; Molander R. B.; Vansant G.; Orlova E. A.; Pfahl M.; Reynolds W. F. An Alu element in the myeloperoxidase promoter contains a composite SP1-thyroid hormone-retinoic acid response element. J. Biol. Chem. 1996, 271 (24), 14412–20. 10.1074/jbc.271.24.14412. [DOI] [PubMed] [Google Scholar]
- Heinecke J. W.; Goldberg I. J. Myeloperoxidase: a therapeutic target for preventing insulin resistance and the metabolic sequelae of obesity?. Diabetes 2014, 63 (12), 4001–3. 10.2337/db14-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forghani R.; Kim H. J.; Wojtkiewicz G. R.; Bure L.; Wu Y.; Hayase M.; Wei Y.; Zheng Y.; Moskowitz M. A.; Chen J. W. Myeloperoxidase propagates damage and is a potential therapeutic target for subacute stroke. J. Cereb. Blood Flow Metab. 2015, 222, 1–9. 10.1038/jcbfm.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malle E.; Furtmuller P. G.; Sattler W.; Obinger C. Myeloperoxidase: a target for new drug development?. Br. J. Pharmacol. 2007, 152 (6), 838–54. 10.1038/sj.bjp.0707358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Antwerpen P.; Zouaoui Boudjeltia K. Rational drug design applied to myeloperoxidase inhibition. Free Radical Res. 2015, 49 (6), 711–20. 10.3109/10715762.2015.1027201. [DOI] [PubMed] [Google Scholar]
- Imada I.; Sato E. F.; Miyamoto M.; Ichimori Y.; Minamiyama Y.; Konaka R.; Inoue M. Analysis of reactive oxygen species generated by neutrophils using a chemiluminescence probe L-012. Anal. Biochem. 1999, 271 (1), 53–8. 10.1006/abio.1999.4107. [DOI] [PubMed] [Google Scholar]
- Zielonka J.; Lambeth J. D.; Kalyanaraman B. On the use of L-012, a luminol-based chemiluminescent probe, for detecting superoxide and identifying inhibitors of NADPH oxidase: a reevaluation. Free Radical Biol. Med. 2013, 65, 1310–4. 10.1016/j.freeradbiomed.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Rashood S. T.; Hassan G. S.; El-Messery S. M.; Nagi M. N.; Habib E.-S. E.; Al-Omary F. A. M.; El-Subbagh H. I. Synthesis, biological evaluation and molecular modeling study of 2-(1,3,4-thiadiazolyl-thio and 4-methyl-thiazolyl-thio)-quinazolin-4-ones as a new class of DHFR inhibitors. Bioorg. Med. Chem. Lett. 2014, 24 (18), 4557–4567. 10.1016/j.bmcl.2014.07.070. [DOI] [PubMed] [Google Scholar]
- Alafeefy A. M.; Ceruso M.; Al-Tamimi A.-M. S.; Del Prete S.; Capasso C.; Supuran C. T. Quinazoline–sulfonamides with potent inhibitory activity against the α-carbonic anhydrase from Vibrio cholerae. Bioorg. Med. Chem. 2014, 22 (19), 5133–5140. 10.1016/j.bmc.2014.08.015. [DOI] [PubMed] [Google Scholar]
- Behalo M. S.; Gad El-karim I. A.; Issac Y. A.; Farag M. A. Synthesis of novel pyridazine derivatives as potential antimicrobial agents. J. Sulfur Chem. 2014, 35 (6), 661–673. 10.1080/17415993.2014.950661. [DOI] [Google Scholar]
- Bello A. M.; Wei L.; Majchrzak-Kita B.; Salum N.; Purohit M. K.; Fish E. N.; Kotra L. P. Small molecule mimetics of an interferon-α receptor interacting domain. Bioorg. Med. Chem. 2014, 22 (3), 978–985. 10.1016/j.bmc.2013.12.049. [DOI] [PubMed] [Google Scholar]
- Smith S. M.; Min J.; Ganesh T.; Diebold B.; Kawahara T.; Zhu Y.; McCoy J.; Sun A.; Snyder J. P.; Fu H.; Du Y.; Lewis I.; Lambeth J. D. Ebselen and congeners inhibit NADPH oxidase 2-dependent superoxide generation by interrupting the binding of regulatory subunits. Chem. Biol. 2012, 19 (6), 752–63. 10.1016/j.chembiol.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiber A.; Oelze M.; August M.; Wendt M.; Sydow K.; Wieboldt H.; Kleschyov A. L.; Munzel T. Detection of superoxide and peroxynitrite in model systems and mitochondria by the luminol analogue L-012. Free Radical Res. 2004, 38 (3), 259–69. 10.1080/10715760410001659773. [DOI] [PubMed] [Google Scholar]
- Bromberg Y.; Pick E. Activation of NADPH-dependent superoxide production in a cell-free system by sodium dodecyl sulfate. J. Biol. Chem. 1985, 260 (25), 13539–45. [PubMed] [Google Scholar]
- Kettle A. J.; Gedye C. A.; Winterbourn C. C. Mechanism of inactivation of myeloperoxidase by 4-aminobenzoic acid hydrazide. Biochem. J. 1997, 321 (2), 503–8. 10.1042/bj3210503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiden A. K.; Sjogren T.; Svensson M.; Bernlind A.; Senthilmohan R.; Auchere F.; Norman H.; Markgren P. O.; Gustavsson S.; Schmidt S.; Lundquist S.; Forbes L. V.; Magon N. J.; Paton L. N.; Jameson G. N.; Eriksson H.; Kettle A. J. 2-thioxanthines are mechanism-based inactivators of myeloperoxidase that block oxidative stress during inflammation. J. Biol. Chem. 2011, 286 (43), 37578–89. 10.1074/jbc.M111.266981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes L. V.; Sjogren T.; Auchere F.; Jenkins D. W.; Thong B.; Laughton D.; Hemsley P.; Pairaudeau G.; Turner R.; Eriksson H.; Unitt J. F.; Kettle A. J. Potent reversible inhibition of myeloperoxidase by aromatic hydroxamates. J. Biol. Chem. 2013, 288 (51), 36636–47. 10.1074/jbc.M113.507756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantschko W.; Furtmuller P. G.; Zederbauer M.; Neugschwandtner K.; Lehner I.; Jakopitsch C.; Arnhold J.; Obinger C. Exploitation of the unusual thermodynamic properties of human myeloperoxidase in inhibitor design. Biochem. Pharmacol. 2005, 69 (8), 1149–57. 10.1016/j.bcp.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Pivonka D.; Tiden A.-K. W.; Anna-Karin; Viklund J.. From thioxanthine derivatives and their use as inhibitors of MPO. WO 2007120097 A1, 2007.
- Tiden A.-K. W.; Viklund J.. Preparation of thioxanthines as inhibitors of myeloperoxidase (MPO). WO 2007120098 A1, 2007.
- Tiden A.-K. W., Cecilia from preparation of 1-(2-hydroxyethyl)-2-thioxo-1,2,3,5-tetrahydropyrrolo[3,2-d]pyrimidin-4-one as a myeloperoxidase (MPO) inhibitor. WO 2007142577 A1, 2007.
- Tiden A.-K. W., Cecilia from preparation of 2-thioxanthines as myeloperoxidase (MPO) inhibitors. WO 2007142576 A1, 2007.
- Ward J.; Spath S. N.; Pabst B.; Carpino P. A.; Ruggeri R. B.; Xing G.; Speers A. E.; Cravatt B. F.; Ahn K. Mechanistic Characterization of a 2-Thioxanthine Myeloperoxidase Inhibitor and Selectivity Assessment Utilizing Click Chemistry–Activity-Based Protein Profiling. Biochemistry 2013, 52 (51), 9187–9201. 10.1021/bi401354d. [DOI] [PubMed] [Google Scholar]
- Soubhye J.; Aldib I.; Elfving B.; Gelbcke M.; Furtmuller P. G.; Podrecca M.; Conotte R.; Colet J. M.; Rousseau A.; Reye F.; Sarakbi A.; Vanhaeverbeek M.; Kauffmann J. M.; Obinger C.; Neve J.; Prevost M.; Zouaoui Boudjeltia K.; Dufrasne F.; Van Antwerpen P. Design, synthesis, and structure-activity relationship studies of novel 3-alkylindole derivatives as selective and highly potent myeloperoxidase inhibitors. J. Med. Chem. 2013, 56 (10), 3943–58. 10.1021/jm4001538. [DOI] [PubMed] [Google Scholar]
- Furtmuller P. G.; Jantschko W.; Regelsberger G.; Jakopitsch C.; Moguilevsky N.; Obinger C. A transient kinetic study on the reactivity of recombinant unprocessed monomeric myeloperoxidase. FEBS Lett. 2001, 503 (2–3), 147–50. 10.1016/S0014-5793(01)02725-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.