

Abstract
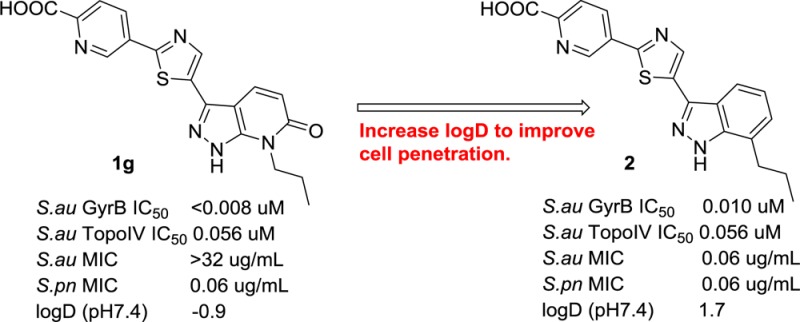
Antibacterials with a novel mechanism of action offer a great opportunity to combat widespread antimicrobial resistance. Bacterial DNA Gyrase is a clinically validated target. Through physiochemical property optimization of a pyrazolopyridone hit, a novel class of GyrB inhibitors were discovered. Guided by structure-based drug design, indazole derivatives with excellent enzymatic and antibacterial activity as well as great animal efficacy were discovered.
Keywords: Antibacterial, GyrB, topoisomerase, indazole, MRSA
Because of the increasing prevalence of bacterial antibiotic resistance, the current antibiotics continue to lose their efficacy. Therefore, the investigation and development of new antibacterials that are not subject to the existing mechanisms of resistance offer a solution to this growing unmet medical need. Bacterial DNA type II topoisomerases are well-established targets. Fluoroquinolones such as ciprofloxacin act on the GyrA subunit of the DNA gyrase complex. The GyrB subunit, the target of Novobiocin, offers an opportunity for overcoming the widespread cross-resistance to fluoroquinolones. Due to the clinical success of the fluoroquinolone class of antibacterials, GyrB has attracted a great deal of interest from both industrial and academic institutions.1−3 Representative GyrB inhibitors are depicted in Figure 1: the tricyclic pyrimidines 1a from Trius,4 aminobenzimidazoles 1b from Vertex,5,6 cyclothialidines 1c from Roche,7,8 pyrrolamides 1d from Astra-Zeneca,9−11 imidazopyridine 1e from Pfizer,12 and benzothiazole 1f from Prolysis.13
Figure 1.

Representative GyrB inhibitors.
Based on a fragment based screen against GyrB,14 carboxylic acid-containing pyrazolopyridone 1g was discovered and possesses potent Staphylococcus aureus (Sa) GyrB activity with an IC50 < 8 nM. This excellent enzymatic activity could be explained via the X-ray crystallographic structure of an analogous compound bound in the GyrB 24KDa ATPase pocket (compound 1g without the carboxylic acid, Figure 2, PDB code 5D7D). It engages a number of key interactions with GyrB ATP binding pocket: (1) a hydrogen bond donor–acceptor network with catalytic Asp81 and a stabilized water; (2) N-propyl group has favorable hydrophobic interaction within a small lipophilic pocket formed by Val79 and Ile175; (3) the distal pyridine moiety forms π–cation stacking with Arg84 and hydrogen bonding with Arg144 while placing the carboxylic acid in 1g in the solvent exposed region; (4) the central thiazole ring establishes hydrophobic interaction with Ile86, Ile102, and Leu10. Despite the excellent Sa GyrB binding potency, 1g does not show whole cell antibacterial activity. We reasoned that the lack of MIC could be due to poor cell membrane penetration resulted from polar functional groups and therefore a low logD. In an effort to improve the cell penetration for carboxylic acid-containing analogue 1g, we decided to replace the more polar pyrazolopyridone with indazole to increase the overall logD for better cell penetration (Figure 3). Thiazolylindazoles are a well-known class of GyrB inhibitors as demonstrated by the excellent work of Roche15 and Quorex/Pfizer.16 However, only 2-thiazolylindazoles were disclosed. In our pyrazolopyridone series,14 we found that 5-thiazolyl pyrazolopyridone analogues provided ∼64× better MICs than the corresponding 2-thiazolyl derivatives. Given the markedly observed MIC differences and the lack of detailed SAR disclosure from the previous works, we think that further SAR studies around the 5-thiazolylindazole scaffold are warranted.
Figure 2.
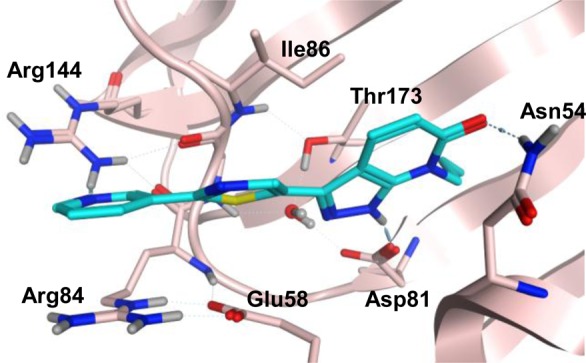
X-ray crystallographic structures of S.au GyrB ATP-binding pocket with pyrazolopyridone analogue. Protein carbons are in pink and ligand carbons are in blue.
Figure 3.
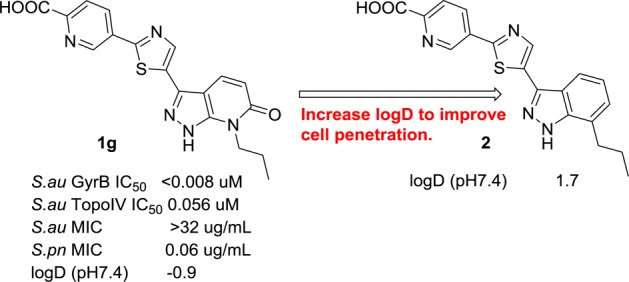
Indazole design strategy to improve cell penetration.
The synthesis of 2 is outlined in Scheme 1. All indazole analogues were prepared according to this general route. The synthesis starts from commercially available 3-bromo-6-cyanopyridine and 2-tributylstannylthiazole via a standard Stille coupling to give the biaryl 3 in 82% yield. The cyanide was then unmasked with HCl in methanol to give the methyl ester 4 in 87% yield. NBS mediated bromination provide bromothiazole 5 as one of the coupling fragments. The synthesis of the other building block starts from the commercially available 7-bromoindazole 6. THP protection followed by a standard Suzuki coupling provided intermediate 7 in 53% overall yield. We found that the yield for the Suzuki coupling was significantly better when the indazole NH was protected. Acid mediated THP deprotection gave 8 in 87% yield. Iodinaton of indazole 8 under strongly basic condition gave the 3-iodoindazole 9 in 75% yield. The indazole NH was reprotected in its THP form to give 10 in 65% yield. The conversion of iodoindazole to the trimethylstannane 11 provided the other building block. The key Stille coupling between 5 and 11 proceeded uneventfully to give 12 in 52% yield, which underwent THP deprotection under acidic conditions to provide the target compound 2 in 76% yield after purification.
Scheme 1. Synthesis of Indazoles.

Reagents and conditions: (a) Pd(PPh3)2Cl2, 1,4-dioxane, 110 °C,82%; (b) 4 M HCl in dioxane, MeOH, 75 °C, 87%; (c) NBS, acetonitrile, 90 °C, 78%; (d) 1. dihydropyran, TFA, 90 °C; 2. n-PrB(OH)2, Pd(dppf)Cl2, aq. Na2CO3, 1,4-dioxane, 110 °C, 53%; (e) TFA, DCM, RT, 87%; (f) KOH, I2, RT, 75%; (g) dihydropyran, TFA, 90 °C, 65%; (h) Pd(PPh3)2Cl2, Me6Sn2, 1,4-dioxane, 100 °C, 65%; (i) Pd(PPh3)2Cl2, P(furyl)3, 1,4-dioxane, 100 °C, 52%; (j) 6 M aq HCl, 100 °C, 76%.
Biological testing showed that 2 possessed an excellent Gram positive MIC profile in addition to retaining enzymatic activity (Table 1), therefore validating our hypothesis that increased logD over pyrazolopyridone analogue 1g would improve cell penetration. Based on the SAR knowledge gained from our previous pyrazolopyridone series, our efforts toward optimizing this class as potent Gram positive antibacterial agent were focused on physiochemical property exploration and further target binding affinity optimization. Specifically, our goals were to (1) optimize the interactions with Arg84 and Arg144 with different left-hand heterocycles; (2) tune the physiochemical properties via attaching solubilizing groups off the aforementioned heterocycles; (3) explore indazole phenyl ring SAR to enhance binding affinity.
Table 1. Optimization of Interactions with Arginine Residues: Left-Hand Aryl Group SARa.
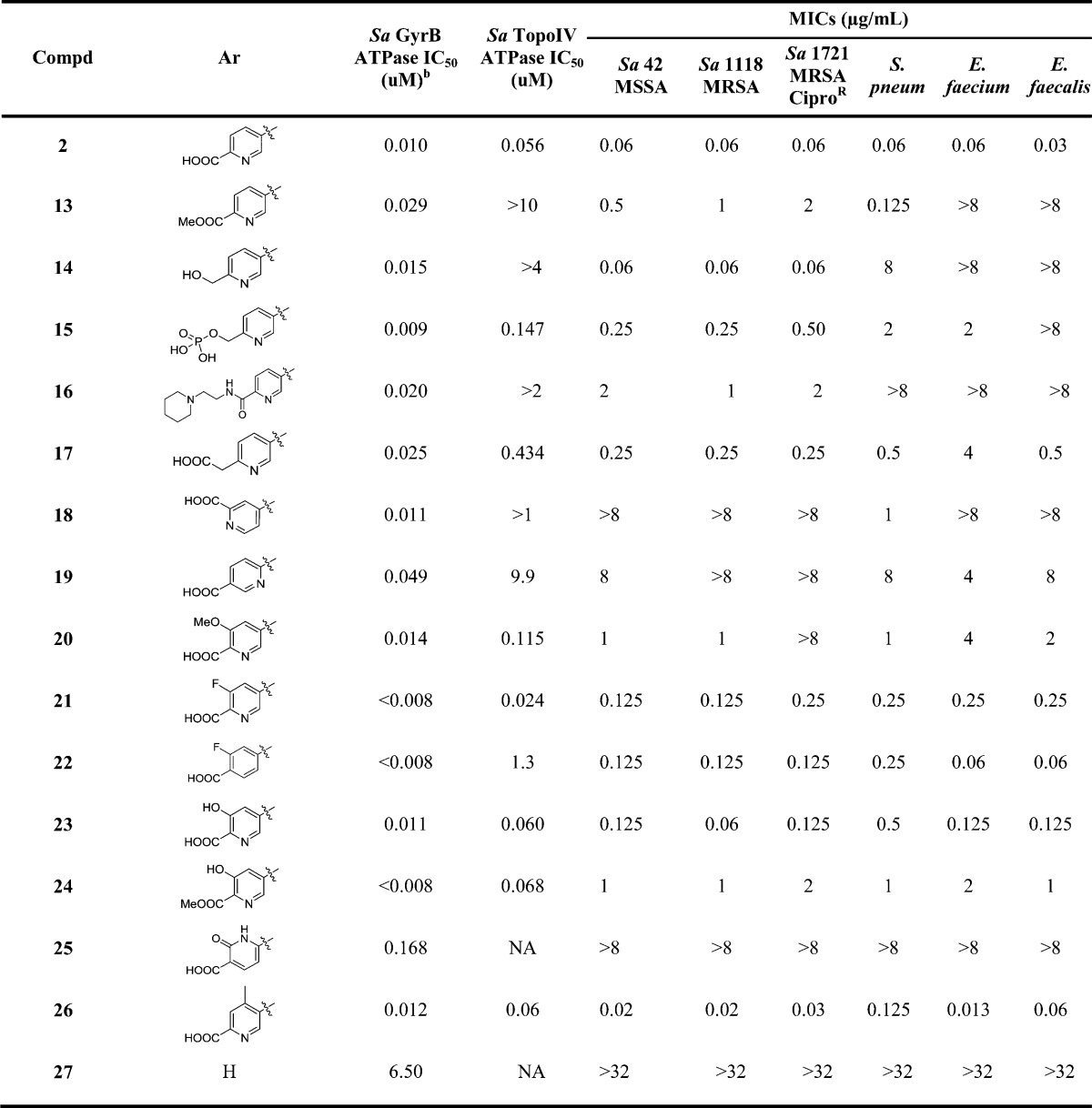
NA = not determined.
Our GyrB IC50 assay detection limit is ∼10 nM. A more accurate assay was developed later.17
The SAR around the left-hand heteroaryl is summarized in Table 1. Methyl ester 13 shows a much worse MIC profile than 2, due to weaker GyrB and Topo IV binding. Hydroxymethyl analogue 14 gives a similar IC50 for GyrB; however, it is much weaker against TopoIV. It maintains the excellent level of MICs for Sa while compromising those for Streptococcus pneumoniae (Spn) and Enterococcus faecium (Ef). Because of 14’s superb Sa MICs and poor aqueous solubility, its phosphate prodrug 15 was prepared. Interestingly, 15 shows a reasonable MIC profile with much improved MICs vs Spn and Ef.18 As a representative example of the amides we prepared, 16 shows weaker IC50s and MICs, demonstrating the important contribution of the salt bridge interaction between the carboxylic acid of 2 and Arg144 toward enzymatic activity. However, pyridine acetic acid analogue 17 has an 8× loss in TopoIV IC50 while displaying 4–8× weaker MICs. Moving the pyridine nitrogen atom or carboxylic acid around the ring leads to diminished GyrB inhibitions (18 and 19). These data suggest that both the rigidity and the bidentate hydrogen bond or salt bridge interaction with Arg144 are critical for GyrB inhibition and whole cell activity. Efforts to better engage Arg144 through an additional hydrogen bond acceptor such as m-F, OH, and OMe resulted in the syntheses of 20–24. All of these compounds give potent GyrB and TopoIV IC50s and slightly weaker MIC profile than 2. Fluorobenzoic acid analogue 22 is especially interesting due to its much improved aqueous solubility19 and excellent overall MIC profile. The carboxyl pyridone analogue 25 gives poor enzymatic activity and MICs. The o-Me analogue 26 was designed to increase the aqueous solubility by disrupting the coplanarity of the left-hand biaryl. Though it shows similar IC50s, its MICs are improved several folds in all the Sa strains and an Ef strain. Lastly, to gauge how much the left-hand carboxypyridine contributes to the overall enzyme affinity, the des-pyridine analogue 27 was prepared and only shows a 6.5 μM GyrB IC50 and no measurable MICs.
The limited antibacterial activity improvements through the left-hand heteroaryl modifications indicate that the carboxypyridine motif is already fairly optimal in terms of engaging Arg84 and Arg144, though several analogues show improvements in the overall physiochemical properties relative to 2. We next turned our attention to the indazole core region to improve the enzymatic and antibacterial activity. Based on our previous SAR in the pyrazolopyridone series,14 we chose to focus our efforts on C6 and C7 positions of the indazole core while leaving out C4 and C5. The C6 and C7 SAR is detailed in Table 2.
Table 2. Indazole Ring Substitution SAR.
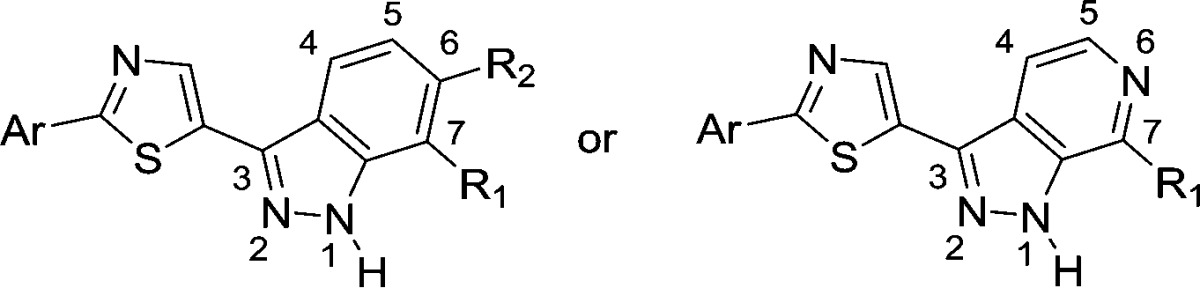
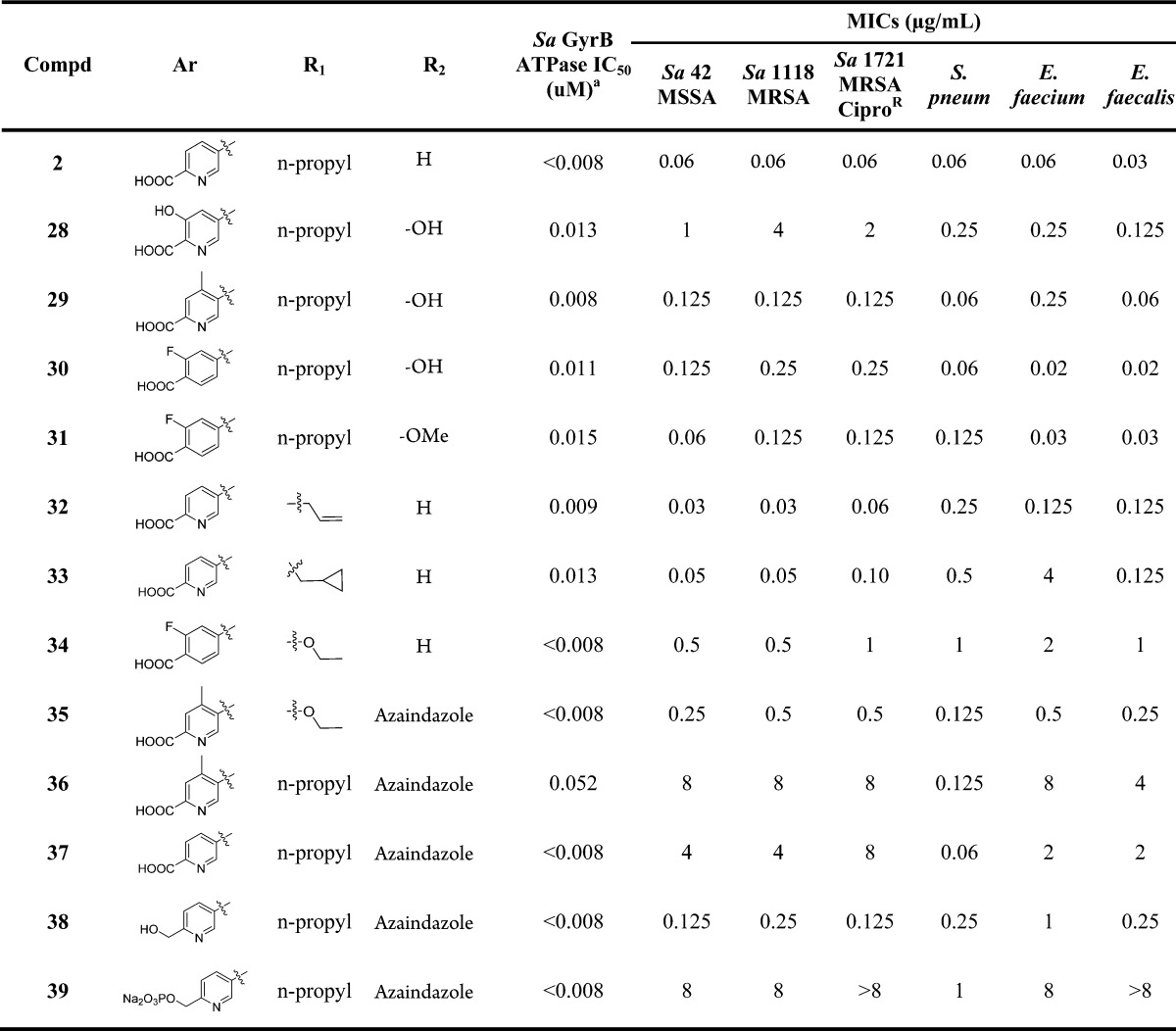
Our GyrB IC50 assay detection limit is ∼10 nM. A more accurate assay was developed later.17
At the corresponding C6 position of pyrazolopyridone 1g, there is a pyridone carbonyl that forms a hydrogen bond with Asn54 (Figure 2). Trius also reported that engaging this Asn54 was critical for the enhanced enzymatic and antibacterial activities of their tricyclic GyrB inhibitors.20 In an effort to re-engage Asn54 with the indazole nucleus, we designed 6-OH and 6-OMe 7-n-propylindazole, then combined them with a few best left-hand fragments and prepared analogues 28–31. We were able to resolve the crystal structure of 28 bound in the 24 kDa GyrB active site (Figure 4, PDB code 5D7R). In addition to the contacts observed with our previous pyrazolopyridone series, the crystal structure confirms that (1) the pyridine acid motif is engaged in a bidentate hydrogen bond interaction with Arg144 through both the carboxylic acid oxygen and pyridine nitrogen; (2) the hydroxyl group on the pyridine ring points to the solvent, and the π–cation interaction with Arg84 is also maintained; (3) the 6-hydroxyl group on the indazole ring indeed makes a hydrogen bond interaction with Asn54. It is hard to conclude if this newly formed hydrogen bond leads to improved binding affinity as they all have IC50s below our assay detection limits. Compounds 28 and 29 show slightly worse MICs than the C6 unsubstituted analogues 23 and 26, while fluorobenzoic acid analogues 30 and 31 display similar Sa and Spn MICs and superior Ef and Em MICs compared to 22.
Figure 4.
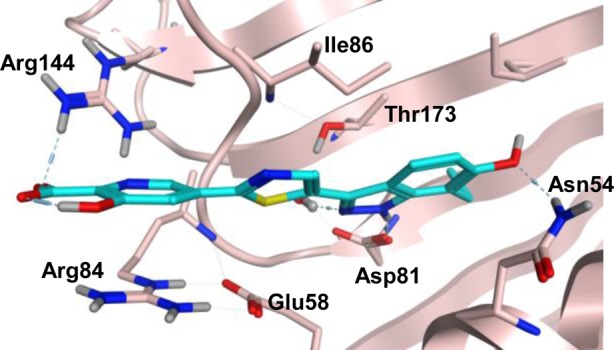
X-ray crystallographic structure of compound 28 bound in Sa GyrB ATPase binding pocket. Protein carbons are in brown, and ligand carbons are in blue.
As shown in Figure 4,21 the C7 n-propyl group directly points toward the small hydrophobic pocket formed by Val79 and Ile175. This lipophilic interaction is critical for GyrB binding affinity just like the pyridone N-propyl substituent. Among the analogues prepared, only C7 allyl analogue 32 gives a similar MIC profile to 2. Other analogues such as 33 and 34 have much weaker MICs than their corresponding n-propyl parents. A few 6-azaindazole derivatives 35–39 were also prepared in order to fine-tune the overall physiochemical properties with the expectation that the N6 of azaindazole is too far from Asn54 to make hydrogen bond contact. Though none of them gives better MIC profile than 2, 35 and 38 still possess impressive Gram positive MICs. To improve the solubility of 38 for IV delivery, the phosphate prodrug 39 was prepared, which had an aqueous solubility of >25 mg/mL.
A few selected compounds were progressed into mouse in vivo PK and efficacy studies, and the results are depicted in Table 3. Compound 2 shows impressive Sa septicemia efficacy with a mice protection PD50 of 1 mg/kg. Unfortunately, it does not show appreciable level of Spn lung efficacy due to the poor lung permeability and the solubility limitation, and possibly high clearance resulted from the indazole NH glucuronidation.22 Benzoic acid analogues 22 and 31 show similarly moderate levels of Spn lung efficacy. Though 31 displays slightly better Spn MIC and mouse exposure, it probably has poorer tissue penetration relative to 22, resulting in similar lung efficacy.
Table 3. Mice in Vivo Efficacy and IV PK.
| compd | Sa1118 MIC (μg/mL) | septicemia efficacy PD50 (mg/kg) | Spn MIC (μg/mL) | lung efficacy ED-2log/ED-3log (mg/kg) | DNAUCa (μg·h/mL)/(mg/kg) | Vdss (L/kg) | t1/2 (h) | Cl (mL/min/kg) |
|---|---|---|---|---|---|---|---|---|
| Novobiocin | 0.125 | 10 | 2 | >100/>100 | ND | ND | ND | ND |
| 2 | 0.06 | 1 | 0.06 | >25/>25c | 0.159 | 3.47 | 2.1 | 105 |
| 22 | 0.125 | NDb | 0.25 | 63.0/97.6 | 0.397 | 1.70 | 2.0 | 42 |
| 31 | 0.125 | ND | 0.125 | 60.6/103.7 | 0.893 | 0.32 | 0.31 | 19 |
| Moxifloxacin | ND | ND | 0.06 | 28.6/50.8 | ND | ND | ND | ND |
Mice were dosed IV at 2 mg/kg.
Not determined.
The highest achievable IV dose due to the solubility limitation.
In summary, the indazole class of GyrB inhibitors, derived from the pyrazolopyridone series and guided by structure-based drug design and physiochemical properties optimization, showed both excellent enzymatic and antibacterial activity against clinically important Gram-positive pathogens including MRSA, fluoroquinolone resistant MRSA, Streptococcus pneumonia, Enterococcus faecium, and Enterococcus faecalis. As a result of their excellent antibacterial activity and favorable PK properties, good animal efficacy was observed in various mouse infection models for selected compounds. Further preclinical evaluation of the indazole class of GyrB inhibitors is ongoing and will be reported in due course.
Acknowledgments
We would like to thank Dr. Ole Andersen and Dr. John Barker at Evotec AG for the crystallographic work, and Dr. Hongwu Gao’s team in Chempartner for analogue synthesis.
Glossary
ABBREVIATIONS
- ADME
absorption, distribution, metabolism, and excretion
- DCM
dichloromethane
- dppf
diphenylphosphinoferrocene
- Ef
Enterococcus faecalis
- Em
Enterococcus faecium
- GyrA
gyrase A
- GyrB
gyrase B
- MIC
minimum inhibitory concentration
- MRSA
methicillin-resistant Staphylococcus aureus
- MSSA
methicillin-susceptible Staphylococcus aureus
- NBS
N-bromosuccinimide
- PK
pharmacokinetics
- SAR
structure–activity relationship
- Sa
Staphylococcus aureus
- Spn
Streptococcus pneumonia
- TFA
trifluoroacetic acid
- THP
tetrahydropyran
- Topo IV
topoisomerase IV
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00266.
Experimental procedure for the synthesis of 2, the general procedure for GyrB and TopoIV IC50 determination, MIC protocol, X-ray crystal structure determination, an alternative view of 28/GyrB cocrystal structure, mouse septicemia and lung efficacy procedure, and a general PK protocol (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tomašić T.; Mašič L. P. Prospects for developing new antibacterials targeting bacterial type IIA topoisomerases. Curr. Top. Med. Chem. 2014, 14 (1), 130–51. 10.2174/1568026613666131113153251. [DOI] [PubMed] [Google Scholar]
- Oblak M.; Kotnik M.; Solmajer T. Discovery and development of ATPase inhibitors of DNA gyrase as antibacterial agents. Curr. Med. Chem. 2007, 14 (19), 2033–47. 10.2174/092986707781368414. [DOI] [PubMed] [Google Scholar]
- Collin F.; Karkare S.; Maxwell A. Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92 (3), 479–497. 10.1007/s00253-011-3557-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tari L. W.; Li X.; Trzoss M.; Bensen D. C.; Chen Z.; Lam T.; Zhang J.; Lee S. J.; Hough G.; Phillipson D.; Akers-Rodriguez S.; Cunningham M. L.; Kwan B. P.; Nelson K. J.; Castellano A.; Locke J. B.; Brown-Driver V.; Murphy T. M.; Ong V. S.; Pillar C. M.; Shinabarger D. L.; Nix J.; Lightstone F. C.; Wong S. E.; Nguyen T. B.; Shaw K. J.; Finn J. Tricyclic GyrB/ParE (TriBE) inhibitors: a new class of broad-spectrum dual-targeting antibacterial agents. PLoS One 2013, 8 (12), 1–13. 10.1371/journal.pone.0084409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronkin S. M.; Badia M.; Bellon S.; Grillot A. L.; Gross C. H.; Grossman T. H.; Mani N.; Parsons J. D.; Stamos D.; Trudeau M.; Wei Y.; Charifson P. S. Discovery of pyrazolthiazoles as novel and potent inhibitors of bacterial gyrase. Bioorg. Med. Chem. Lett. 2010, 20, 2828–2831. 10.1016/j.bmcl.2010.03.052. [DOI] [PubMed] [Google Scholar]
- Charifson P. S.; Grillot A.-L.; Grossman T. H.; Parsons J. D.; Badia M.; Bellon S.; Deininger D. D.; Drumm J. E.; Gross C. H.; LeTiran A.; Liao Y.; Mani N.; Nicolau D. P.; Perola E.; Ronkin S.; Shannon D.; Swenson L. L.; Tang Q.; Tessier P. R.; Tian S.-K.; Trudeau M.; Wang T.; Wei Y.; Zhang H.; Stamos D. Novel dual-targeting benzimidazole urea inhibitors of DNA gyrase and topoisomerase IV possessing potent antibacterial activity: intelligent design and evolution through the judicious use of structure-guided design and structure-activity relationships. J. Med. Chem. 2008, 51 (17), 5243–63. 10.1021/jm800318d. [DOI] [PubMed] [Google Scholar]
- Lewis R. J.; Singh O. M.; Smith C. V.; Skarzynski T.; Maxwell a; Wonacott a J.; Wigley D. B. The nature of inhibition of DNA gyrase by the coumarins and the cyclothialidines revealed by X-ray crystallography. EMBO J. 1996, 15, 1412–1420. [PMC free article] [PubMed] [Google Scholar]
- Angehrn P.; Goetschi E.; Gmuender H.; Hebeisen P.; Hennig M.; Kuhn B.; Luebbers T.; Reindl P.; Ricklin F.; Schmitt-Hoffmann A. A new DNA gyrase inhibitor subclass of the cyclothialidine family based on a bicyclic dilactam-lactone scaffold. Synthesis and antibacterial properties. J. Med. Chem. 2011, 54 (7), 2207–24. 10.1021/jm1014023. [DOI] [PubMed] [Google Scholar]
- Dumas J.; Sherer B.. (2-Pyridin-3-ylimidazo[1,2-b]pyridazin-6-yl) urea derivatives as antibacterial agents. Int. Pat. Appl. WO2009027733 A1.
- Basarab G.; Hill P.; Zhou F.. Piperidine compounds and uses thereof. Int. Pat. Appl. WO2008152418 A1.
- Illingworth R. N.; Uria-Nickelsen M.; Bryant J.; Eakin A. E. Presented at the 48th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), 2008, Poster F1-2028.
- Starr J. T.; Sciotti R. J.; Hanna D. L.; Huband M. D.; Mullins L. M.; Cai H.; Gage J. W.; Lockard M.; Rauckhorst M. R.; Owen R. M.; Lall M. S.; Tomilo M.; Chen H.; McCurdy S. P.; Barbachyn M. R. 5-(2-Pyrimidinyl)-imidazo[1,2-a]pyridines are antibacterial agents targeting the ATPase domains of DNA gyrase and topoisomerase IV. Bioorg. Med. Chem. Lett. 2009, 19 (18), 5302–06. 10.1016/j.bmcl.2009.07.141. [DOI] [PubMed] [Google Scholar]
- Haydon D. R.; Czaplewski L. G.; Palmer N. J.; Mitchell D. R.; Atherall J. F.; Steele C. R.; Ladduwahetty T.. Antibacterial compositions. Int. Pat. Appl. WO2007148093 A1.
- The fragment based GyrB inhibitors will be published in a separate communication.
- Boehm H.; Boehringer M.; Bur D.; Gmuender H.; Huber W.; Klaus W.; Kostrewa D.; Kuehne H.; Luebbers T.; Meunier-Keller N.; Mueller F. Novel Inhibitors of DNA Gyrase: 3D Structure Based Biased Needle Screening, Hit Validation by Biophysical Methods, and 3D Guided Optimization. A Promising Alternative to Random Screening. J. Med. Chem. 2000, 43, 2664–74. 10.1021/jm000017s. [DOI] [PubMed] [Google Scholar]
- Yager K.; Chu S.; Appelt K.; Li X.. Preparation of thiazolylindazoles as inhibitors of bacterial DNA gyrase B inhibitors. US20050054697 A1.
- Unpublished results.
- Prodrug was found to be stable in the MIC assay media.
- A complete disclosure of the physicochemical and ADME properties of the indazole derivatives will be reported in a future full paper.
- Li X.; Tari1 L. W.; Bensen D. C.; Trzoss M.; Lam T.; Zhang J.; Chen Z.; Lee S.-J.; Cunningham M.; Kwan B.; Nelson K.; Stidham M.; Brown-Driver V.; Hough G.; Phillipson D.; Nguyen T.; Lightstone F.; Wong S.; Shaw K. J.; Finn J. Presented at the 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy (ICCAC), 2012, Poster F-2017.
- An alternative view of the X-ray structure was included in the Supporting Information.
- Rose K.; Yang Y.; Sciotti R.; Cai H. Structure-activity relationship (SAR): effort towards blocking N-glucuronidation of indazoles (PF-03376056) by human UGT1A enzymes. Drug Metab. Lett. 2009, 3, 28–34. 10.2174/187231209787176371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.