

Abstract
The conformational diseases, linked to protein aggregation into amyloid conformations, range from non-infectious neurodegenerative disorders, such as Alzheimer's disease (AD), to highly infectious ones, such as human transmissible spongiform encephalopathies (TSEs). They are commonly known as prion diseases. However, since all amyloids could be considered prions (from those involved in cell-to-cell transmission to those responsible for real neuronal invasion), it is necessary to find an underlying cause of the different capacity to infect that each of the proteins prone to form amyloids has. As proposed here, both the intrinsic cytotoxicity and the number of nuclei of aggregation per cell could be key factors in this transmission capacity of each amyloid.
Keywords: Alzheimer's disease, amyloid, amyloid cytotoxicity, amyloid transmission, Creutzfeldt-Jakob disease, prion, transmissible spongiform encephalopathy
Abbreviations
- AD
Alzheimer's disease
- TSE
transmissible spongiform encephalopathy
- PD
Parkinson's disease
- HD
Huntington's disease
- FD
frontotemporal dementia
- ALS
amyotrophic lateral sclerosis
- AL
amyloid light-chain amyloidosis
- CJD
Creutzfeldt-Jakob disease
The conformational diseases are linked to the process of protein aggregation into amyloid conformations. This group of diseases ranges from neurodegenerative disorders, such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), and human transmissible spongiform encephalopathies (TSEs), which are commonly known as prion diseases; to non-neurodegenerative systemic and localized amyloidosis, which include amyloid light-chain amyloidosis (AL) and type II diabetes.1 Currently, some 36 million people worldwide suffer some form of dementia; the number is predicted to exceed 65 million by 2030 and to have tripled by 2050. Of all the different types of dementia, AD represents ∼70% of cases. In contrast, the incidence of sporadic Creutzfeldt-Jakob disease (CJD), the most common human TSE, is extremely low; it causes a death rate worldwide of about 1 case per million people each year.2-4
It is known that a key step in all conformational diseases is protein self-aggregation into amyloid-like fibers with a core formed of repetitive arrays of β-sheets oriented parallel to the fibril axis.5,6 However, although amyloids are considered to be universal and omnipresent structures, all of which share internal structural characteristics,1,7-10 prions represent only a tiny drop in the amyloid ocean. Specifically, they are those amyloids in which the aggregation process has become self-perpetuating and infectious; pathological in mammals and leading to protein-based genetic elements in fungi.11,12 Therefore, an essential question, which has remained unanswered for a long time, is: “Could AD and other conformational diseases become infectious?” Although it has long been accepted that AD and other dementias are the result of a widespread defect in neuron biochemistry that gradually leads to the accumulation of amyloid-like fibers, recent research suggests that amyloid-like proteins involved in these diseases could spread from one cell to another in the brain: they could be involved in cell-to-cell infection.13-21 However, these processes are far from the neuronal invasions produced in prion diseases, wherein prion infectivity is transferred from the spleen to the central nervous system (CNS) following a 2-phase model of transfer. The first phase is characterized by widespread colonization of lymphoreticular organs. The second involves the CNS (and also probably peripheral nerves), which in the presence of vesicle-associated infectivity, leads to the emergence of free-floating cell-free oligomeric or infectious protofibril particles.11,17 The infectious conformation of prion proteins is notoriously resistant to most physical, chemical, and enzymatic treatments, including heat, detergent dissolution, and protease. This high resistance coupled with low clearance rates seems to explain the spread of these proteins in infected individuals as well as the continued presence of prions.11,22-24 However, it is widely accepted that high resistance to denaturation is a generic property of amyloids. Thus, the essential question: “Why does an amyloid become infectious?” remains unanswered.
Fungal and yeast prions, which provide an excellent model from which to gain an understanding of amyloid formation and propagation,25,26 could shed some light on the key factors that are involved when an amyloid becomes a prion. Recent findings in the field have shown that the number of nuclei of aggregation could be a factor that affects the infection capacity of amyloid-prone proteins, just as their intrinsic cytotoxicity does. In both fungal and yeast prions, the number of nuclei of aggregation per cell27 determines, following Poisson's law, the probability of prion infectivity.28 Thus, high numbers of nuclei of aggregation per cell result in an increase in infectivity.
Since cytotoxicity is an intrinsic characteristic shared by all proteins involved in conformational diseases,1 the intrinsic cytotoxicity of each amyloid (irrespective of the number of nuclei of aggregation per cell) could be a key factor in the differentiation between infectious and non-infectious amyloids in humans. Recent findings related to the HET-s fungal protein—a protein composed of a globular domain (HeLo) appended to a prion-forming domain (PFD)—might shed some light on the effect of cytotoxicity on the prevalence of prion phenotypes. In the HET-s/HET-S heterokaryon incompatibility system (a self/non-self recognition phenomenon that occurs in the filamentous fungus Podospora anserina29), the incompatibility reaction between 2 genetically distinct strains is triggered when a strain expressing soluble HET-S is seeded by contact with another strain expressing the HET-s prion.30 Importantly, whereas in vitro HET-S (which differs from the prion HET-s sequence by only 13 residues out of 28931) forms amyloid aggregates that are extremely similar to those of its partner (see Fig. S1), the in vivo amyloid fibrils of HET-s and HET-S are prion and non-prion, respectively.32 Contrary to previously published findings related to HET-S that was purified under native conditions,33 when the protein is purified under denaturing conditions and then refolded (following the same protocol as that used to purify HET-s), aggregation mimicking to the in vitro case of HET-s can be observed. As suggested by constructed het-s/het-S chimeric alleles where the regions coding for the C- and N-terminal domains are been exchanged, the HeLo domain determines the phenotype of the protein.34 It has been observed that when the HeLo domain does not impede protein aggregation, the formation of amyloid-like fibrils is the more likely way for HET PFDs to aggregate. However, it is not clear what could explain the vast difference in their capacity to infect. Recent research into the HET-s self/non-self mechanism has helped to elucidate this intriguing question. This heterokaryon reaction, which can only be localized in dead heterokaryon cells and thus observed at the contact region between 2 genetically distinct strains, is the result of the extremely high cytotoxicity of the globular HeLo domain of HET-S when the PFD is aggregated in an amyloid-like conformation.32,35 The interaction between aggregated HET-s and soluble HET-S triggers the aggregation of the HET-S PFD (in a cross-seeding reaction), which entails the destabilization and misfolding of the HET-S HeLo domain.32,35 As a consequence of this destabilization, the HET-S HeLo domain exposes the 34 previously enclosed N-terminal residues, which are transformed into a trans-membrane domain that is quickly inserted into the membrane and triggers pore formation, membrane disruption and finally cell death.35 Importantly, in contrast to what occurs with HET-S, the amyloid aggregation of the HET-s PFD does not entail HET-s HeLo misfolding, thus cell viability is maintained.32,35 This extreme case illustrates how cytotoxicity can switch between a non-transmissible amyloid (HET-S fibrils) and a transmissible prion (HET-s fibrils). However, while this concept is certainly intriguing and can be beautifully applied to the HET-s/S system, it is unclear whether it applies to the majority of amyloidogenic proteins, including functional amyloids and those causing neurodegenerative diseases in humans.
Functional amyloids have been identified in bacteria (viz. Curlin,36 Chaplins,37 adhesin P138 and phenol soluble modulins39), fungi (viz. HET-s,34 hydrophobins,40 and the yeast prions Sup35p, Rnq1p or Ure2p41), animals (viz. Spidroin,42 eggshell chorion proteins43 and the neuron-specific isoform of CPEB44) and humans (viz. the intralumenal domain of Pmel17,45 cystatin-related epididymal spermatogenic protein46 and proteins involved in hormone storage in the pituitary glands).47 Amyloid-like aggregates (especially oligomeric β-sheet species) are cytotoxic and this toxicity seems to be inherent to the cross β-sheet structure.48 Given the toxic nature of amyloid-like aggregates in both intracellular and extracellular matrices, we have to expect that the cells producing functional amyloids overcome these harmful properties and avoid cellular damage. Thus, Pmel17 is synthesized in early melanosomes as a trans-membrane protein with no self-assembly capacity and its amyloid fragment is only released by proteolytic cleavage in the final stage and in specialized compartments, wherein amyloid aggregates are rapidly sequestered to its membrane. This highly controlled process drastically reduces the contact between toxic amyloid aggregates and susceptible structures, thus favoring cell survival. In the same way, it has been shown that other functional amyloids such as curli,49 spidroin42 and probably also CPEB prions44 could be under the influence of specific regulatory systems that minimize the intrinsic toxicity of the amyloid structures produced. It has also been shown that bacterial functional amyloids involved in surface adhesion, the processes of cell aggregation and biofilm formation, display high cytotoxicity for the cells they invade.36,38,39,50 However, the bacterial cell uses productive pathways that require specific assembly machinery to overcome the intrinsic toxicity of the amyloid aggregates.49 Importantly, all these systems are designed to isolate the aggregates, thereby counteracting the prion propensity of these functional amyloids.
At this point, special consideration should be given to the particular case of yeast prions. In recent years, the possible inherent toxicity of yeast prions has aroused considerable controversy in this field.26,51 Yeast prions could either be considered as harmful to the yeast,52,53 or conversely, they could be associated with beneficial phenotypes.54 Sup35 protein (Sup35p), probably the most common studied yeast prion, serves to illustrate the controversy. The prion Sup35p in the yeast Saccharomyces cerevisiae mediates the activity of the cytoplasmic nonsense suppressor known as [PSI+].55 It was initially reported that although higher tolerance to heat and chemical stress was shown by [PSI+], no difference in growth rates was displayed between [psi-] and [PSI+] under mild conditions. Moreover, [PSI+] was able to reversibly increase the translation termination efficiency (reducing [PSI+] persistence) in response to an environmental stress (in the presence of ethanol).56 Although it has been suggested that inheritance of [PSI+] may allow yeast cells to exploit pre-existing genetic variation and thereby to thrive in fluctuating environments,57,58 it has proved difficult to reproduce the beneficial phenotypes that have been reported.59 Thus, while it seems likely that [PSI+] would be at least mildly deleterious in most environments, this may be countered on rare occasions (i.e., under stress conditions) by its evolutionary properties.60 In contrast, the lack of any reproducible benefit provided by yeast prions suggests that the prions may not be beneficial for the yeast at all. In view of these findings, it could be assumed that, although slightly harmful to the cell, the possible toxicity of yeast prions that exist spontaneously in nature would be very low and thus should not drastically compromise cell survival. The existence of extremely lethal [PSI+] phenotypes is also possible; however, lethal [PSI+] are only compatible with cell growth when they also express a minimal Sup35 C-terminal, corresponding to RFM domains without the N-terminal prion domain. In the same way, although the Ure2p prion is important for Ure2p to function in stabilizing the protein against degradation in vivo,61 [URE3] is also a rare molecular degenerative disease.52 Thus, in yeast, as observed in HET-S, highly cytotoxic amyloid-like aggregates are incompatible with prion self-perpetuation as a consequence of their intrinsic toxicity, which provokes cell death.
In view of these facts, it seems likely that, as observed in the HET-s/HET-S system, toxicity is a key factor for the existence of prions among other functional amyloids. However, at this point, a key question remains unanswered: “Could this scenario be the case for the amyloid proteins that cause neurodegenerative diseases in humans?” Importantly, crucial differences have been reported between extracellular amyloid-like aggregates of PrPSc and Aβ peptide. Aβ, considered to date as a non-prion protein, is certainly the most studied amyloid. For years, the extracellular accumulation of Aβ fibrils in brain was considered the main source of the toxicity linked to AD. However, accumulating evidence suggests that prefibrillar soluble oligomers, generated at early stages of the fibrillation process, could be the primary cytotoxic species.62-64 Consistent with this hypothesis, the presence of these highly toxic oligomers greatly hinders the capacity of Aβ to act as a prion.
In contrast to the case of Aβ, it has been suggested that PrPSc might not be directly responsible of PrP cytotoxicity; it may act only on the deregulation of membrane-anchored PrPC (PrPC-GPI). The alteration of PrPC, proposed as a signaling molecule, would trigger a chain of reactions that would involve processes ranging from membrane alteration to cellular dysfunction and cell death.11,65,66 In recent years, the involvement of oligomers in toxicity and infection processes has been widely debated. On the one hand, in agreement with the increasingly accepted hypothesis that oligomeric species, and not mature fibers , are the primary causative agent of cytotoxicity in conformational diseases, it has been shown that PrP oligomers are the most cytotoxic PrP species.67,68 On the other hand, it has also been claimed that oligomers composed of about 20 PrP monomers could be the most infectious PrP particles.69 Since high toxicity prevents the appearance of the prion, the existence of highly toxic and infective aggregates would be extremely unlikely. How then can the existence of these aggregates be explained? One possible answer to this question would further confirm the proposed hypothesis. While the infectivity assays were carried out using oligomers from infected brain (in vivo material), the toxicity assays were performed using oligomers produced in vitro. Importantly, it is known that PrP aggregates produced in vitro lack high levels of infectivity;70 thus, toxicity and infectiousness assays have been carried out using 2 completely different types of oligomers. In agreement with the present hypothesis, highly toxic oligomers (formed in vitro) are non-infectious. In recent years, the possibility of there being several pathways in the PrP aggregation process has provoked considerable controversy in the field. In order to explain PrP infection, associated with a prolonged incubation period followed by rapid cytotoxic evolution, 2 differentiated and uncoupled pathways have been proposed for in vivo prion propagation (first step) and toxicity (second step).71 The second neurotoxic step could be linked to the increment of lethal PrP oligomers (PrP),L formed as an intermediate or the final product of secondary reactions during prion propagation.72 Since, in vitro, α-helix PrP monomers have been identified as highly toxic species,73 the conformation of these PrPL could be crucial to gain an understanding of the neurotoxic process associated with the spread of PrPSc.
In the last few years, increased evidence has led to the belief that prion capacity could be a more generic property of amyloid-prone proteins, extending the idea that some of the most prevalent human conformational diseases could be related to amyloid-prone proteins that display a certain prion capacity.13 Thus, Aβ, huntingtin and tau proteins have been proposed as putative prions that display a certain infectivity in experimental transmission assays (from cell-to-cell and host-to-graft transmission in animal models to transmission by intracerebral inoculation).13 More interesting is the case of α-synuclein, amyloid-A and apolipoprotein-A; which show prion capacity in natural transmission assays.13 However, although in vivo, these assays were performed under non-physiological conditions and the proteins tested should only be considered prion-like under each of the specific conditions. In more natural conditions, limiting factors that impede infectivity may include protein concentrations and secretion and penetration mechanisms, as well as cellular localization. Importantly, based on these recent data, inter-individual infectivity should only be taken into account for extracellular proteins such as amyloid-A74 and apolipoprotein-A.75 Despite the tentative nature of the evidence, the possibility that proteins associated with the most prevalent conformational human illnesses such as α-synuclein, tau or huntingtin act as prions is emerging as an interesting topic in the field. The case of α-synuclein is particularly interesting; here transmission from neuron to neuron or oligodendrocytes has been reported.76-78 Nevertheless, these transmissions are far from neuronal invasion, probably due to limitations on the spread of α-synuclein aggregates. On the one hand, the fact that both mature fibers and oligomers (the most transmissible material) display high neuronal toxicity79–82 and on the other, the added difficulty of having to be secreted to reach distant neurons, could be key factors that impede the prion capacity of α-synuclein aggregates. Very importantly at this point, it has been shown that cells normally secrete α-synuclein into their surrounding media; this opens up the possibility that oligomers of α-synuclein could be spread and act as prions.83 In the same way, similar scenarios may be considered for tau or huntingtin aggregates. Although certain evidence shows that neural death could occur in the overexpression of these proteins without the detection of aggregated material,84 tau and huntingtin aggregates display considerable neurotoxicity, right through from oligomers to mature fibers.85-90 As shown in α-synuclein, recent evidence suggests a slight extracellular tau presence, which could undergo protein spreading.91
As denoted by the arrows in Figure 1, if the HET-s/HET-S extreme case was a generic feature across the field of amyloids, it would suggest that amyloid aggregates might switch between non-infectious (or negligibly infectious) and infectious material, depending on the number of nuclei of aggregation and their cytotoxicity. In this way, while under natural conditions Aβ can be considered a non-infectious protein, when Aβ aggregates (both soluble and insoluble ones) are directly injected into brain, the peptide becomes a prion.92,93 This switching behavior could be explained by the concentration of Aβ aggregates in the extracellular matrix. Thus, whereas in natural conditions cellular death appears in accordance with cytotoxic species generation, in this particular case the abrupt incorporation of Aβ aggregates favors spreading that is faster than the onset of toxicity, thereby leading to widespread distribution.
Figure 1.
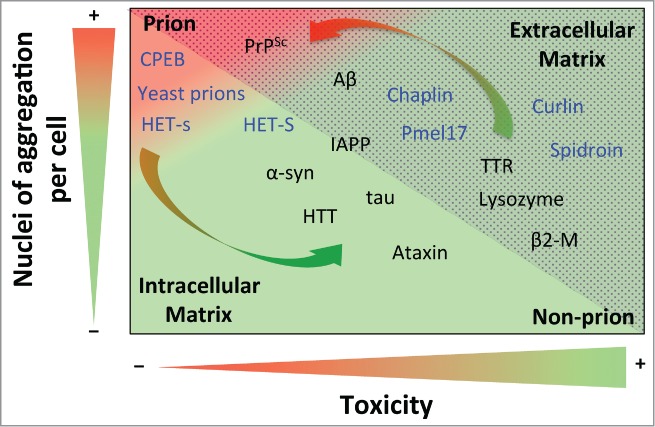
Prion propensity of amyloid-prone proteins. Effect of the number of aggregation nuclei per cell and cytotoxicity on the capacity of amyloids to infect; from low transmission (green) to high infective capacity (red). The proteins included in the graph represent the different subclasses of amyloids from extracellular (dotted area) and intracellular (shaded area) matrices. The functional amyloids are in blue. The arrows show switching possibilities between prion and non-prion. Note that the location of the amyloid-prone proteins in the graph is only to illustrate their prion or non-prion tendency. Protein abbreviations: amyloid β-peptide (Aβ), prion protein (PrPSc), cytoplasmic polyadenylation element-binding protein (CPEB), α-synuclein (α-syn), huntingtin (HTT), islet amyloid polypeptide (IAPP), (Pmel17), transthyretin (TTR) and β2-microglobulin (β2-M).
The number of nuclei of aggregation per cell is a parameter that is closely linked to replication efficiency; although it is also highly dependent on other parameters, such as resistance to biological clearance, bioavailability, transport and spreading.13 Consequently, the concentration of nuclei will determine the severity of toxicity or infection, depending on the properties of each kind of nuclei. Although not generic, it has been shown that the secondary structure and order of oligomeric aggregates is directly correlated with nucleation.94 It has been suggested that oligomers, and to a lesser extent mature amyloid fibers , could mediate the main cytotoxic effect and infection activity. Since the action of the oligomer depends on its intrinsic toxicity and the concentration of nuclei formed, 2 different scenarios can be envisaged. On one hand, when highly toxic, the oligomers mediate the main cytotoxic effect in conformational diseases.1,95 On the other hand, since oligomers are more highly dispersible,96 they should be considered as a potential source of prion infectivity,11 being directly related to the degree of infectivity when they display low toxicity. Thus, while slightly toxic oligomers (e.g., in PrPSc) would tend to drastically increase the appearance of prions, highly toxic ones (e.g., in Aβ) favor a continued non-prion condition. Increments in the amount of oligomeric species (favoring prion propensity) could be compensated by high intrinsic toxicity (reducing prion propensity). The fact that only a few proteins are considered prions suggests that the intrinsic amyloid toxicity of almost all amyloid-like species is sufficient to avert the apparition of prions.
In dementia, neuronal death is mostly associated with the intrinsic cytotoxicity of each amyloid, which is more pronounced in certain oligomeric species. However, the severity of this damage could be related to the number of toxic nuclei per cell. Since the presence of highly toxic aggregates leads to rapid cell death, it would dramatically reduce infectivity, thereby limiting the infection capacity to cell-to-cell transmission. In this case, the number of toxic nuclei would determine both the rate at which the cell itself dies and the capacity to kill nearby cells by membrane–membrane contact. In addition, since one proposed mechanism for amyloid neurotoxicity is membrane disruption resulting in cell death,97 it could be envisaged that contact between these toxic aggregates and the wall of neighboring cells would be sufficient to trigger cell death without requiring aggregation processes in nearby cells. In contrast, aggregates with a low toxicity would lead to greater rates of cell survival, thereby increasing the probability that the cells release nuclei in a veritable neuronal invasion. In this latter scenario, the infectivity of the cell would depend on the number of nuclei per cell as well as on its capacity to release them. On this basis, since all proteins involved in dementia can be considered as neurotoxic elements (at least certain of their amyloid aggregates can) which finally lead to neuronal death, it should be expected that, when the neurotoxicity process is started, the most transmissible amyloids, such as PrPSc, are involved in the most rapidly progressive and fatal diseases; as indeed happens. In contrast, the Aβ peptide could be an example of the opposite case. Mature Aβ fibers , considered as material with a low dispersibility, and oligomeric Aβ species, which are highly dispersible, display low and high cytotoxicity, respectively. This tips the balance toward the formation of a non-prion amyloid and reduces the possibility of Aβ becoming a prion. Thus, although severe AD symptomatology can be observed as a consequence of neurotoxic effects of oligomers, the intrinsic toxicity of these dispersible species greatly impedes their spread, thereby limiting Aβ diffusion to cell-to-cell process.
In the cell, protein folding and aggregation are competing pathways controlled by a delicate multi-step equilibrium that is highly dependent on both intrinsic and extrinsic factors. In the same way, amyloid aggregation is a consequence of a balance between a multitude of conformational states and inter-conversion between them in a complex network of equilibriums.1 Alterations in the cellular environment (e.g., under conditions of stress or modifications in the levels of transcription and protein expression) could be crucial in the selection of a particular self-assembly pathway, determining the final amyloid structure or structures (as a consequence of polymorphisms); and thus the capacity to infect. In the same way, both changes in the cellular environmental and the intrinsic cell properties of each patient may directly or indirectly induce different self-aggregation pathways, resulting in different amyloid structures. The different cell resistances to amyloid toxicity and the resultant diverse transmission propensities and cytotoxicity levels could have serious repercussions for medicine (see arrows in Fig. 1). On the one hand, this could explain why severe symptomatology (e.g., in AD) can be associated with less neuronal damage than expected and vice versa. On the other hand, it could also help elucidate why the severity and progression of each incidence of dementia depends on the individual patient and disease. Furthermore, as an important repercussion for dementia therapies, it could explain why treatments involving a reduction in amyloid toxicity, cell protection or the disruption of amyloid plaques can produce effects that are the opposite of those expected.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
I am the beneficiary of a contract under the Ramón y Cajal program financed by the Spanish Ministerio de Ciencia e Innovación.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 2006; 75:333-66; PMID: 16756495; http://dx.doi.org/ 10.1146/annurev.biochem.75.101304.123901 [DOI] [PubMed] [Google Scholar]
- 2. Byers AL, Yaffe K. Depression and risk of developing dementia. Nat Rev Neurol 2011; 7:323-31; PMID: 21537355; http://dx.doi.org/ 10.1038/nrneurol.2011.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Porzoor A, Macreadie IG. Application of Yeast to Study the Tau and Amyloid-beta Abnormalities of Alzheimer's Disease. J Alzheimers Dis 2013; 35:217-25; PMID: 23396350 [DOI] [PubMed] [Google Scholar]
- 4. Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol 2011; 7:137-52; PMID: 21304480; http://dx.doi.org/ 10.1038/nrneurol.2011.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carulla N, Caddy GL, Hall DR, Zurdo J, Gairi M, Feliz M, Giralt E, Robinson CV, Dobson CM. Molecular recycling within amyloid fibrils. Nature 2005; 436:554-8; PMID: 16049488; http://dx.doi.org/ 10.1038/nature03986 [DOI] [PubMed] [Google Scholar]
- 6. Kodali R, Wetzel R. Polymorphism in the intermediates and products of amyloid assembly. Curr Opin Struct Biol 2007; 17:48-57; PMID: 17251001; http://dx.doi.org/ 10.1016/j.sbi.2007.01.007 [DOI] [PubMed] [Google Scholar]
- 7. de Groot NS, Sabate R, Ventura S. Amyloids in bacterial inclusion bodies. Trends Biochem Sci 2009; 34:408-16; PMID: 19647433; http://dx.doi.org/ 10.1016/j.tibs.2009.03.009 [DOI] [PubMed] [Google Scholar]
- 8. Wang L, Maji SK, Sawaya MR, Eisenberg D, Riek R. Bacterial inclusion bodies contain amyloid-like structure. PLoS Biol 2008; 6:e195; PMID: 18684013; http://dx.doi.org/ 10.1371/journal.pbio.0060195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Villar-Pique A, Sabate R, Lopera O, Gibert J, Torne JM, Santos M, Ventura S. Amyloid-like protein inclusions in tobacco transgenic plants. PLoS One 2010; 5:e13625; PMID: 21049018; http://dx.doi.org/ 10.1371/journal.pone.0013625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rousseau F, Schymkowitz J, Serrano L. Protein aggregation and amyloidosis: confusion of the kinds? Curr Opin Struct Biol 2006; 16:118-26; PMID: 16434184; http://dx.doi.org/ 10.1016/j.sbi.2006.01.011 [DOI] [PubMed] [Google Scholar]
- 11. Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev 2009; 89:1105-52; PMID: 19789378; http://dx.doi.org/ 10.1152/physrev.00006.2009 [DOI] [PubMed] [Google Scholar]
- 12. Chien P, Weissman JS, DePace AH. Emerging principles of conformation-based prion inheritance. Annu Rev Biochem 2004; 73:617-56; PMID: 15189155; http://dx.doi.org/ 10.1146/annurev.biochem.72.121801.161837 [DOI] [PubMed] [Google Scholar]
- 13. Soto C. Transmissible proteins: expanding the prion heresy. Cell 2012; 149:968-77; PMID: 22632966; http://dx.doi.org/ 10.1016/j.cell.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Colby DW, Prusiner SB. De novo generation of prion strains. Nat Rev Microbiol 2011; 9:771-7; PMID:21947062; http://dx.doi.org/ 10.1038/nrmicro2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One 2012; 7:e31302; PMID: 22312444; http://dx.doi.org/ 10.1371/journal.pone.0031302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 2012; 73:685-97; PMID: 22365544; http://dx.doi.org/ 10.1016/j.neuron.2011.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 2009; 78:177-204; PMID: 19231987; http://dx.doi.org/ 10.1146/annurev.biochem.78.082907.145410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol 2010; 6:702-6; PMID: 21045796; http://dx.doi.org/ 10.1038/nrneurol.2010.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 2010; 11:301-7; PMID: 20308987; http://dx.doi.org/ 10.1038/nrm2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol 2011; 70:532-40; PMID: 22028219; http://dx.doi.org/ 10.1002/ana.22615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eisele YS, Obermuller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 2010; 330:980-2; PMID: 20966215; http://dx.doi.org/ 10.1126/science.1194516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brown P, Rau EH, Johnson BK, Bacote AE, Gibbs CJ, Jr., Gajdusek DC. New studies on the heat resistance of hamster-adapted scrapie agent: threshold survival after ashing at 600 degrees C suggests an inorganic template of replication. Proc Natl Acad Sci U S A 2000; 97:3418-21; PMID: 10716712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McKinley MP, Bolton DC, Prusiner SB. A protease-resistant protein is a structural component of the scrapie prion. Cell 1983; 35:57-62; PMID: 6414721; http://dx.doi.org/ 10.1016/0092-8674(83)90207-6 [DOI] [PubMed] [Google Scholar]
- 24. Soto C. Prion hypothesis: the end of the controversy? Trends Biochem Sci 2011; 36:151-8; PMID: 21130657; http://dx.doi.org/ 10.1016/j.tibs.2010.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Uptain SM, Lindquist S. Prions as protein-based genetic elements. Annu Rev Microbiol 2002; 56:703-41; PMID: 12142498; http://dx.doi.org/ 10.1146/annurev.micro.56.013002.100603 [DOI] [PubMed] [Google Scholar]
- 26. Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T. Prions of fungi: inherited structures and biological roles. Nat Rev Microbiol 2007; 5:611-8; PMID: 17632572; http://dx.doi.org/ 10.1038/nrmicro1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature 2006; 442:585-9; PMID: 16810177; http://dx.doi.org/ 10.1038/nature04922 [DOI] [PubMed] [Google Scholar]
- 28. Malato L, Dos Reis S, Benkemoun L, Sabate R, Saupe SJ. Role of Hsp104 in the propagation and inheritance of the; Het-s prion. Mol Biol Cell 2007; 18:4803-12; PMID: 17881723; http://dx.doi.org/ 10.1091/mbc.E07-07-0657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci U S A 1997; 94:9773-8; PMID: 9275200; http://dx.doi.org/ 10.1073/pnas.94.18.9773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. Amyloid aggregates of the HET-s prion protein are infectious. Proc Natl Acad Sci U S A 2002; 99:7402-7; PMID: 12032295; http://dx.doi.org/ 10.1073/pnas.072199199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nazabal A, Dos Reis S, Bonneu M, Saupe SJ, Schmitter JM. Conformational transition occurring upon amyloid aggregation of the HET-s prion protein of Podospora anserina analyzed by hydrogen/deuterium exchange and mass spectrometry. Biochemistry 2003; 42:8852-61; PMID: 12873146; http://dx.doi.org/ 10.1021/bi0344275 [DOI] [PubMed] [Google Scholar]
- 32. Mathur V, Seuring C, Riek R, Saupe SJ, Liebman SW. Localization of HET-S to the cell periphery, not to; het-s aggregates, is associated with; het-s-HET-S toxicity. Mol Cell Biol 2012; 32:139-53; PMID: 22037764; http://dx.doi.org/ 10.1128/MCB.06125-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Greenwald J, Buhtz C, Ritter C, Kwiatkowski W, Choe S, Maddelein ML, Ness F, Cescau S, Soragni A, Leitz D, et al. The mechanism of prion inhibition by HET-S. Mol Cell 2010; 38:889-99; PMID: 20620958; http://dx.doi.org/ 10.1016/j.molcel.2010.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Balguerie A, Dos Reis S, Ritter C, Chaignepain S, Coulary-Salin B, Forge V, Bathany K, Lascu I, Schmitter JM, Riek R, et al. Domain organization and structure-function relationship of the HET-s prion protein of Podospora anserina. EMBO J 2003; 22:2071-81; PMID: 12727874; http://dx.doi.org/ 10.1093/emboj/cdg213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seuring C, Greenwald J, Wasmer C, Wepf R, Saupe SJ, Meier BH, Riek R. The mechanism of toxicity in HET-S/HET-s prion incompatibility. PLoS Biol 2012; 10:e1001451; PMID: 23300377; http://dx.doi.org/ 10.1371/journal.pbio.1001451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dueholm MS, Albertsen M, Otzen D, Nielsen PH. Curli functional amyloid systems are phylogenetically widespread and display large diversity in operon and protein structure. PLoS One 2012; 7:e51274; PMID: 23251478; http://dx.doi.org/ 10.1371/journal.pone.0051274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sawyer EB, Claessen D, Haas M, Hurgobin B, Gras SL. The assembly of individual chaplin peptides from Streptomyces coelicolor into functional amyloid fibrils. PLoS One 2011; 6:e18839; PMID: 21526199; http://dx.doi.org/ 10.1371/journal.pone.0018839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oli MW, Otoo HN, Crowley PJ, Heim KP, Nascimento MM, Ramsook CB, Lipke PN, Brady LJ. Functional amyloid formation by Streptococcus mutans. Microbiology 2012; 158:2903-16; PMID: 23082034; http://dx.doi.org/ 10.1099/mic.0.060855-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schwartz K, Syed AK, Stephenson RE, Rickard AH, Boles BR. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog 2012; 8:e1002744; PMID: 22685403; http://dx.doi.org/ 10.1371/journal.ppat.1002744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kwan AH, Winefield RD, Sunde M, Matthews JM, Haverkamp RG, Templeton MD, Mackay JP. Structural basis for rodlet assembly in fungal hydrophobins. Proc Natl Acad Sci U S A 2006; 103:3621-6; PMID: 16537446; http://dx.doi.org/ 10.1073/pnas.0505704103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009; 137:146-58; PMID: 19345193; http://dx.doi.org/ 10.1016/j.cell.2009.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kenney JM, Knight D, Wise MJ, Vollrath F. Amyloidogenic nature of spider silk. Eur J Biochem 2002; 269:4159-63; PMID: 12180993; http://dx.doi.org/ 10.1046/j.1432-1033.2002.03112.x [DOI] [PubMed] [Google Scholar]
- 43. Iconomidou VA, Vriend G, Hamodrakas SJ. Amyloids protect the silkmoth oocyte and embryo. FEBS Lett 2000; 479:141-5; PMID: 10981723; http://dx.doi.org/ 10.1016/S0014-5793(00)01888-3 [DOI] [PubMed] [Google Scholar]
- 44. Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 2003; 115:879-91; PMID: 14697205; http://dx.doi.org/ 10.1016/S0092-8674(03)01020-1 [DOI] [PubMed] [Google Scholar]
- 45. Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol 2006; 4:e6; PMID: 16300414; http://dx.doi.org/ 10.1371/journal.pbio.0040006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Whelly S, Johnson S, Powell J, Borchardt C, Hastert MC, Cornwall GA. Nonpathological extracellular amyloid is present during normal epididymal sperm maturation. PLoS One 2012; 7:e36394; PMID: 22570708; http://dx.doi.org/ 10.1371/journal.pone.0036394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, Singru PS, Nilsson KP, Simon R, Schubert D, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009; 325:328-32; PMID: 19541956; http://dx.doi.org/ 10.1126/science.1173155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lashuel HA, Lansbury PT, Jr. Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q Rev Biophys 2006; 39:167-201; PMID: 16978447; http://dx.doi.org/ 10.1017/S0033583506004422 [DOI] [PubMed] [Google Scholar]
- 49. Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002; 295:851-5; PMID: 11823641; http://dx.doi.org/ 10.1126/science.1067484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barnhart MM, Chapman MR. Curli biogenesis and function. Annu Rev Microbiol 2006; 60:131-47; PMID: 16704339; http://dx.doi.org/ 10.1146/annurev.micro.60.080805.142106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Villar-Pique A, Espargaro A, Sabate R, de Groot NS, Ventura S. Using bacterial inclusion bodies to screen for amyloid aggregation inhibitors. Microb Cell Fact 2012; 11:55; PMID: 22553999; http://dx.doi.org/ 10.1186/1475-2859-11-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McGlinchey RP, Kryndushkin D, Wickner RB. Suicidal; PSI+ is a lethal yeast prion. Proc Natl Acad Sci U S A 2011; 108:5337-41; PMID: 21402947; http://dx.doi.org/ 10.1073/pnas.1102762108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wickner RB, Edskes HK, Bateman D, Kelly AC, Gorkovskiy A. The yeast prions; PSI+ and; URE3 are molecular degenerative diseases. Prion 2011; 5:258-62; PMID: 22052353; http://dx.doi.org/ 10.4161/pri.17748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Halfmann R, Jarosz DF, Jones SK, Chang A, Lancaster AK, Lindquist S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 2012; 482:363-8; PMID: 22337056; http://dx.doi.org/ 10.1038/nature10875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jensen MA, True HL, Chernoff YO, Lindquist S. Molecular population genetics and evolution of a prion-like protein in Saccharomyces cerevisiae. Genetics 2001; 159:527-35; PMID: 11606530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eaglestone SS, Cox BS, Tuite MF. Translation termination efficiency can be regulated in Saccharomyces cerevisiae by environmental stress through a prion-mediated mechanism. EMBO J 1999; 18:1974-81; PMID: 10202160; http://dx.doi.org/ 10.1093/emboj/18.7.1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. True HL, Berlin I, Lindquist SL. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature 2004; 431:184-7; PMID: 15311209; http://dx.doi.org/ 10.1038/nature02885 [DOI] [PubMed] [Google Scholar]
- 58. True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 2000; 407:477-83; PMID: 11028992; http://dx.doi.org/ 10.1038/35035005 [DOI] [PubMed] [Google Scholar]
- 59. Namy O, Galopier A, Martini C, Matsufuji S, Fabret C, Rousset JP. Epigenetic control of polyamines by the prion; PSI+. Nat Cell Biol 2008; 10:1069-75; PMID: 19160487; http://dx.doi.org/ 10.1038/ncb1766 [DOI] [PubMed] [Google Scholar]
- 60. Lancaster AK, Bardill JP, True HL, Masel J. The spontaneous appearance rate of the yeast prion; PSI+ and its implications for the evolution of the evolvability properties of the; PSI+ system. Genetics 2010; 184:393-400; PMID: 19917766; http://dx.doi.org/ 10.1534/genetics.109.110213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shewmaker F, Mull L, Nakayashiki T, Masison DC, Wickner RB. Ure2p function is enhanced by its prion domain in Saccharomyces cerevisiae. Genetics 2007; 176:1557-65; PMID: 17507672; http://dx.doi.org/ 10.1534/genetics.107.074153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cheon M, Chang I, Mohanty S, Luheshi LM, Dobson CM, Vendruscolo M, Favrin G. Structural reorganisation and potential toxicity of oligomeric species formed during the assembly of amyloid fibrils. PLoS Comput Biol 2007; 3:1727-38; PMID: 17941703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Necula M, Kayed R, Milton S, Glabe CG. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J Biol Chem 2007; 282:10311-24; PMID: 17284452; http://dx.doi.org/ 10.1074/jbc.M608207200 [DOI] [PubMed] [Google Scholar]
- 64. Sakono M, Zako T. Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J 2010; 277:1348-58; PMID: 20148964; http://dx.doi.org/ 10.1111/j.1742-4658.2010.07568.x [DOI] [PubMed] [Google Scholar]
- 65. Brandner S, Isenmann S, Kuhne G, Aguzzi A. Identification of the end stage of scrapie using infected neural grafts. Brain Pathol 1998; 8:19-27; PMID: 9458163; http://dx.doi.org/ 10.1111/j.1750-3639.1998.tb00130.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996; 379:339-43; PMID: 8552188; http://dx.doi.org/ 10.1038/379339a0 [DOI] [PubMed] [Google Scholar]
- 67. Novitskaya V, Bocharova OV, Bronstein I, Baskakov IV. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J Biol Chem 2006; 281:13828-36; PMID: 16554307; http://dx.doi.org/ 10.1074/jbc.M511174200 [DOI] [PubMed] [Google Scholar]
- 68. Simoneau S, Rezaei H, Sales N, Kaiser-Schulz G, Lefebvre-Roque M, Vidal C, Fournier JG, Comte J, Wopfner F, Grosclaude J, et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 2007; 3:e125; PMID: 17784787; http://dx.doi.org/ 10.1371/journal.ppat.0030125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature 2005; 437:257-61; PMID: 16148934; http://dx.doi.org/ 10.1038/nature03989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hill AF, Antoniou M, Collinge J. Protease-resistant prion protein produced in vitro lacks detectable infectivity. J Gen Virol 1999; 80 (Pt 1):11-4; PMID: 9934677 [DOI] [PubMed] [Google Scholar]
- 71. Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011; 470:540-2; PMID: 21350487; http://dx.doi.org/ 10.1038/nature09768 [DOI] [PubMed] [Google Scholar]
- 72. Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007; 318:930-6; PMID: 17991853; http://dx.doi.org/ 10.1126/science.1138718 [DOI] [PubMed] [Google Scholar]
- 73. Zhou M, Ottenberg G, Sferrazza GF, Lasmezas CI. Highly neurotoxic monomeric alpha-helical prion protein. Proc Natl Acad Sci U S A 2012; 109:3113-8; PMID: 22323583; http://dx.doi.org/ 10.1073/pnas.1118090109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang B, Une Y, Fu X, Yan J, Ge F, Yao J, Sawashita J, Mori M, Tomozawa H, Kametani F, et al. Fecal transmission of AA amyloidosis in the cheetah contributes to high incidence of disease. Proc Natl Acad Sci U S A 2008; 105:7263-8; PMID: 18474855; http://dx.doi.org/ 10.1073/pnas.0800367105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Korenaga T, Yan J, Sawashita J, Matsushita T, Naiki H, Hosokawa M, Mori M, Higuchi K, Fu X. Transmission of amyloidosis in offspring of mice with AApoAII amyloidosis. Am J Pathol 2006; 168:898-906; PMID: 16507905; http://dx.doi.org/ 10.2353/ajpath.2006.050350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 2009; 106:13010-5; PMID: 19651612; http://dx.doi.org/ 10.1073/pnas.0903691106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, Covert M, Melki R, Kirkegaard K, Brahic M. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann Neurol 2012; 72:517-24; PMID: 23109146; http://dx.doi.org/ 10.1002/ana.23747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 2014; 62:387-98; PMID: 24382629; http://dx.doi.org/ 10.1002/glia.22611 [DOI] [PubMed] [Google Scholar]
- 79. Karpinar DP, Balija MB, Kugler S, Opazo F, Rezaei-Ghaleh N, Wender N, Kim HY, Taschenberger G, Falkenburger BH, Heise H, et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson's disease models. EMBO J 2009; 28:3256-68; PMID: 19745811; http://dx.doi.org/ 10.1038/emboj.2009.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci 2007; 27:3338-46; PMID: 17376994; http://dx.doi.org/ 10.1523/JNEUROSCI.0285-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Taschenberger G, Garrido M, Tereshchenko Y, Bahr M, Zweckstetter M, Kugler S. Aggregation of alphaSynuclein promotes progressive in vivo neurotoxicity in adult rat dopaminergic neurons. Acta Neuropathol 2012; 123:671-83; PMID: 22167382; http://dx.doi.org/ 10.1007/s00401-011-0926-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Roostaee A, Beaudoin S, Staskevicius A, Roucou X. Aggregation and neurotoxicity of recombinant alpha-synuclein aggregates initiated by dimerization. Mol Neurodegener 2013; 8:5; PMID: 23339399; http://dx.doi.org/ 10.1186/1750-1326-8-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DM, Ikeda S, et al. Alpha-synuclein implicated in Parkinson's disease is present in extracellular biological fluids, including human plasma. FASEB J 2003; 17:1945-7; PMID: 14519670 [DOI] [PubMed] [Google Scholar]
- 84. de Silva R, Farrer M. Tau neurotoxicity without the lesions: a fly challenges a tangled web. Trends Neurosci 2002; 25:327-9; PMID: 12079751; http://dx.doi.org/ 10.1016/S0166-2236(02)02170-7 [DOI] [PubMed] [Google Scholar]
- 85. Bates G. Huntingtin aggregation and toxicity in Huntington's disease. Lancet 2003; 361:1642-4; PMID: 12747895; http://dx.doi.org/ 10.1016/S0140-6736(03)13304-1 [DOI] [PubMed] [Google Scholar]
- 86. Farias G, Cornejo A, Jimenez J, Guzman L, Maccioni RB. Mechanisms of tau self-aggregation and neurotoxicity. Curr Alzheimer Res 2011; 8:608-14; PMID: 21605046; http://dx.doi.org/ 10.2174/156720511796717258 [DOI] [PubMed] [Google Scholar]
- 87. Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron 2010; 66:631-45; PMID: 20547123; http://dx.doi.org/ 10.1016/j.neuron.2010.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener 2011; 6:39; PMID: 21645391; http://dx.doi.org/ 10.1186/1750-1326-6-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nucifora LG, Burke KA, Feng X, Arbez N, Zhu S, Miller J, Yang G, Ratovitski T, Delannoy M, Muchowski PJ, et al. Identification of novel potentially toxic oligomers formed in vitro from mammalian-derived expanded huntingtin exon-1 protein. J Biol Chem 2012; 287:16017-28; PMID: 22433867; http://dx.doi.org/ 10.1074/jbc.M111.252577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nekooki-Machida Y, Kurosawa M, Nukina N, Ito K, Oda T, Tanaka M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc Natl Acad Sci U S A 2009; 106:9679-84; PMID: 19487684; http://dx.doi.org/ 10.1073/pnas.0812083106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Le MN, Kim W, Lee S, McKee AC, Hall GF. Multiple mechanisms of extracellular tau spreading in a non-transgenic tauopathy model. Am J Neurodegener Dis 2012; 1:316-33; PMID: 23383401 [PMC free article] [PubMed] [Google Scholar]
- 92. Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M. Soluble Abeta seeds are potent inducers of cerebral beta-amyloid deposition. J Neurosci 2011; 31:14488-95; PMID: 21994365; http://dx.doi.org/ 10.1523/JNEUROSCI.3088-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Stohr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ, Prusiner SB, Giles K. Purified and synthetic Alzheimer's amyloid beta (Abeta) prions. Proc Natl Acad Sci U S A 2012; 109:11025-30; PMID: 22711819; http://dx.doi.org/ 10.1073/pnas.1206555109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ono K, Condron MM, Teplow DB. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci U S A 2009; 106:14745-50; PMID: 19706468; http://dx.doi.org/ 10.1073/pnas.0905127106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Choi YJ, Chae S, Kim JH, Barald KF, Park JY, Lee SH. Neurotoxic amyloid beta oligomeric assemblies recreated in microfluidic platform with interstitial level of slow flow. Sci Rep 2013; 3:1921; PMID: 23719665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rijal Upadhaya A, Capetillo-Zarate E, Kosterin I, Abramowski D, Kumar S, Yamaguchi H, Walter J, Fändrich M, Staufenbiel M, Thal DR. Dispersible amyloid beta-protein oligomers, protofibrils, and fibrils represent diffusible but not soluble aggregates: their role in neurodegeneration in amyloid precursor protein (APP) transgenic mice. Neurobiol Aging 2012; 33:2641-60; PMID: 22305478; http://dx.doi.org/ 10.1016/j.neurobiolaging.2011.12.032 [DOI] [PubMed] [Google Scholar]
- 97. Sabate R, Espargaro A, Barbosa-Barros L, Ventura S, Estelrich J. Effect of the surface charge of artificial model membranes on the aggregation of amyloid beta-peptide. Biochimie 2012; 94:1730-8; PMID: 22542639; http://dx.doi.org/ 10.1016/j.biochi.2012.03.027 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.