

Abstract
ABSTRACT. Familial fatal insomnia (FFI) is fatal disorder characterized by damage to select thalamic nuclei, together with progressive insomnia and dysautonomia. In subjects carrying the D178N prion protein (PRNP) mutation, distinct phenotypes can be observed, depending on the methionine (Met) /valine (Val) codon 129 polymorphism. We report here a Chinese case of FFI with a D178N/Met129 genotype of the PRNP gene, who exhibited rapidly progressive dementia combined with behavioral disturbances and paroxysmal limb myoclonus. Our patient did not show refractory insomnia early in the disease course, nor demonstrate typical MRI and EEG alterations. There was remarkable family history of similar symptoms.
KEYWORDS: D178N, Familial fatal insomnia, Met129, prion protein
Abbreviations
- D178N
Asp178Asn
- FFI
familial fatal insomnia
- Met 129
Methionine 129
- PRNP
prion protein
Introduction
Familial fatal insomnia (FFI) is an autosomal dominant disorder mainly characterized by neuronal degeneration limited to select thalamic nuclei, together with progressive insomnia and dysautonomia.1 According to surveillance data from 2006, FFI accounts for about half of all genetic prion disease cases in China.2 FFI is caused by the D178N mutation of the prion protein (PRNP) gene,3 which is characterized by an increased propensity from conversion form PRNPC into PRNPSC and can perturb the stability and conformational dynamics of the protein.4 In subjects carrying the D178N PRNP mutation, distinct phenotypes are determined by the methionine(Met) /valine(Val) polymorphism at codon 129 on the mutated allele. D178N/Met129 haplotype causes FFI, while D178N/Val129 haplotype results on Creutzfeldt-Jakob disease (CJD).5,6 At present, descriptions of patients with the D178N/Met129 PRNP phenotype are few.7 We report a patient with the D178N/Met129 PRNP mutation, affected by FFI without typical refractory insomnia early in the disease course or MRI changes in the thalamus.
Case Presentation
A 51-year-old, right-handed male with 6 y of education was first seen in our geriatric psychiatry department because of memory difficulties. He presented with a 9-month history of progressive memory decline combined with gradual appearance of behavioral disturbances and paroxysmal limb twitching. He had been a merchant and underwent lumbar spine surgery 2 months prior to the onset of his symptoms.
Nine months before his initial presentation to our department, his relatives had started noticing neurological symptoms, such as memory loss and personality changes (quietness and apathy). Approximately 7 months before his admission to the local hospital, a rapid deterioration of his cognitive function, paroxysmal twitching of hands, neck, and shoulder, visual hallucinations, insomnia, and gluttony were observed. Subsequently, therapy with carbamazepine, olanzapine, and donepezil were started in order to control symptoms. Due to the appearance of a carbamazepine-induced rash, he was switched to sodium valproate. The paroxysmal twitching, visual hallucinations, and insomnia were partially alleviated, but cognitive decline and personality changes remained. On admission to the local hospital (December 4, 2014), his general physical examination was unremarkable. The cranial nerve examination was normal. Mild bradykinesia and clumsiness in his lower limbs were noted. On the Mini-Mental Status Exam (MMSE), the patient scored 13 out of 30 and exhibited severe impairments in orientation (3/10), attention and calculation (1/5), memory and recall (3/6), and no apparent impairments in language (9/9). (MMSE=13, at 7 months post-onset). Routine hematological and biochemical laboratory testing including electrolytes, chemistry, lipid profile, liver function, thyroid function, tumor markers, and viral antibodies were unremarkable. Autoimmune indexes of peripheral venous blood were all negative or within normal limits. Electrocardiography, chest radiography, and abdominal computed tomography were within normal limits. CSF examination (protein, glucose and cell count) was normal. Electroencephalogram (EEG) on the 6th day of admission showed intermittent appearance of diffuse 50 μV θ and 40 μV δ waves (Fig. 1). Video EEG on the 7th day of admission showed the intermittent appearance of 20–60 μ V θ and δ waves, predominantly in the right frontal-temporal lobe. His brain MRI, including T1 and T2 weighted images, performed on hospital day 5, revealed no obvious abnormality, but diffused weighted imaging showed a mild high signal intensity in right frontal cortex (Fig. 2). Autoimmune encephalitis could not be ruled out; however, treatment with high dose corticosteroid was ineffective. On hospital day 27, he was discharged without confirmed diagnosis or clinical improvement, but with apparent degeneration. Four days after discharge (January 4, 2015), he presented as an outpatient in our department and his relatives complained of aggravated symptoms, including poor memory, sluggishness, speech difficulty, unsteady gait, and worsening insomnia. The patient again received routine laboratory testing, which revealed no evidence of comorbid disease. The clinical neurological examination revealed increased muscle tone in bilateral upper extremities, bilateral hyperreflexia of the patellar reflex, intermittent positive Babinski sign on the right, cerebellum ataxia including deficits in tandem gait and dysmetria with finger-to-nose test bilaterally, and slowed gait. The patient scored 8 out of 30 on the MMSE, and exhibited severe impairments in orientation (0/10), attention and calculation (1/5), memory and recall (3/6), and language (4/9). Summary of MMSE changes including 7 and 8-month post onset of clinical disease was seen in Table 1. On the Montreal Cognitive Assessment examination (MoCA), the patient scored 5 out of 30, with deficits in visual-construction (0/4), alternate line (0/1), recall (0/5), attention and calculation (2/6), language (3/6), abstraction (0/2), and orientation (0/6) (MMSE=8, MoCA=5, at 8 months post-onset). Because of the patient's difficulty with cooperation, the brain MRI and polysomnography were not conducted. The comparison of signs, symptoms and studies between typical FFI case and this case was seen in Table 2.8
Figure 1.
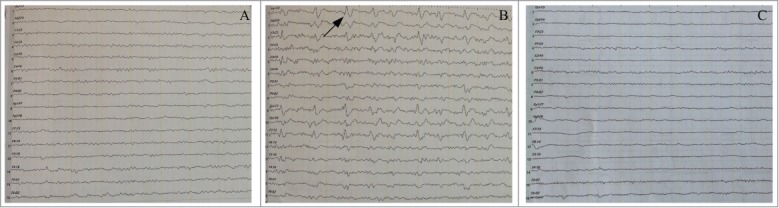
Electroencephalogram (EEG) of this case, typical case, and normal control. (A) The atypical EEG of this case at 7 months post-symptom onset showed intermittent appearance of 50 μV θ waves and 40 μV δ waves in a diffuse distribution. (B) The typical EEG of FFI patient showed many triphasic waves composed of 20–40 μV θ waves and 100 μV δ waves dominated in frontaltemporal regions, as indicated by arrow. (C) The normal EEG showed 10 μVα waves as the main background.
Figure 2.
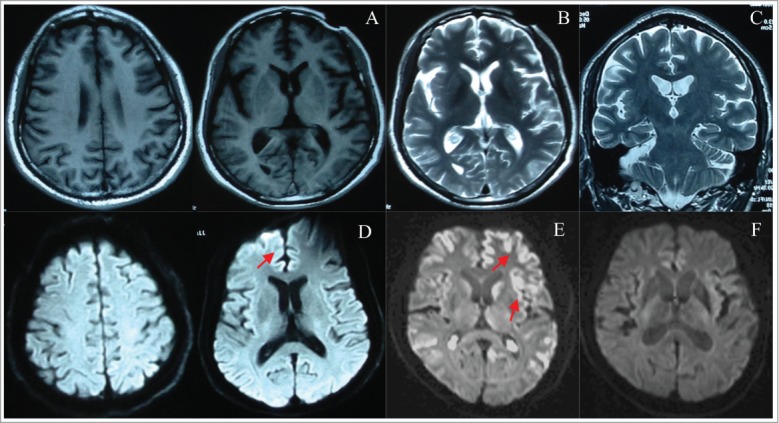
The brain MRI presentations of this case, typical case, and normal control. T1 weighted horizontal axial (A), T2 weighted horizontal axial (B), and T2 weighted coronal axial (C) brain MRI of this case showed no apparent abnormality, but diffused weighted horizontal axial (D) MRI of this case showed mild high signal intensity in frontal cortex with dominant involvement of right frontal lobe, as indicated by arrow. The typical diffused weighted MRI of FFI patient (E) showed apparent high signal intensity along cerebral cortex named as “ribbon sign” dominated in frontaltemporal lobes, as shown by arrow. The normal diffused weighted horizontal axial (F) MRI showed no apparent abnormality.
TABLE 1.
Summary of Mini Mental Status Examination (MMSE) changes
| Category (total score) | Month 7 post onset | Month 8 post onset |
|---|---|---|
| Orientation (10 points) | 3 | 0 |
| Attention and calculation (5 points) | 1 | 1 |
| Memory and recall (6 points ) | 3 | 3 |
| Language (9 points) | 9 | 4 |
TABLE 2.
The comparison of signs, symptoms and studies between typical FFI case and this case
| Typical case* | This case | |
|---|---|---|
| Organic sleep disturbances | √ | √ |
| Cognitive/amnestic deficits | √ | √ |
| Psychiatric changes | √ | √ |
| Ataxia | √ | √ |
| Myoclonus | √ | |
| Visual changes (double vision) | √ | |
| Dysarthria | √ | |
| Weight Loss | √ | |
| Vegetative signs | √ | |
| EEG changes (slowing) | √ | √ |
| MRI changes (supratentorial atrophy) | √ | |
| Polysomnography changes (REM, efficiency, and deep sleep reduction) | √ | n/a |
As reported in >50% of FFI patients.8
The family history was very remarkable, but this collateral information was not backed up by medical records or direct clinical examination. Several relatives had previously presented with cognitive and behavioral troubles. His father (II-1) and paternal uncle (II-5) both died at the age of 51, in the context of a rapidly progressive dementia accompanied by behavior changes and movement disorders. Relatives indicated that our patient had very similar clinical manifestations as his father and paternal uncle, who both died within 1 y after onset of symptoms. His paternal uncle's daughter (III-15) died at age 46. She exhibited psychotic and mood symptoms, and died within a few years after initial onset of these changes. His paternal uncle's son (III-16) died at age 22. He had a similar manifestation as our patient, and died within 1 y after onset of symptoms. The other relatives were clinically asymptomatic (Fig. 3).
Figure 3.

Pedigree of the family. The proband is indicated by an arrow (III-3). Three male subjects (II-1, II-5, III-16) had shown similar clinical manifestations as the proband, and died within one year in the context of a rapidly progressive dementia. One female subject (III-15) had been affected with psychotic and mood symptoms, and died within a few years. The other relatives were clinically asymptomatic.
The patient's rapidly progressive dementia, multiple neurological deficits, and informative family history are suggestive of a diagnosis of prion disease. Genomic DNA was extracted from peripheral blood leukocytes and direct sequencing of the PCR product showed a missense mutation at codon 178 of the PRNP gene (G to A), leading to a substitution of aspartate (Asp) by asparagine (Asn), and a common polymorphism of methionine (Met) at codon 129 in the PRNP gene (Fig. 4). No additional nucleotide exchanges were found in other regions of the PRNP sequence. Because of the patient's relatives refusing a brain biopsy, brain histology was not included to aid in the demonstration of this atypical case of FFI.
Figure 4.
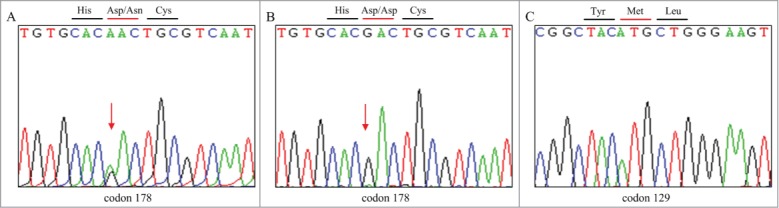
DNA sequence at codon 178 of PRNP gene from the patient (A) and a control (B). DNA sequence at codon 129 of PRNP gene from the patient (C).The red arrow indicates a point mutation causing a substitution of GAC (Asp) by AAC (Asn) at codon 178. Sequence in a control is homozygous for GAC (Asp) at codon 178. Direct sequencing product of our patient showed Met at codon 129 of the PRNP gene.
Discussion
The patient exhibited progressive memory decline combined with the gradual appearance of behavioral disturbances and paroxysmal limb twitching. Previously normal cognitive functioning deteriorated into a severe dementia rapidly within 8 months, which was entirely indicative of a rapidly progressive dementia. The patient's family history was very informative, lacked direct medical examinations and concrete diagnoses. Three male subjects (II-1, II-5, III-16) had displayed similar clinical manifestations to the proband, and died within one year in the context of rapidly progressive dementias. Although the MRI and EEG at 7 months post-onset did not reveal typical findings related to FFI, the clinical findings are still suggestive of a diagnosis of family hereditary prion disease. Geschwind MD had conducted comprehensive evaluations of 178 cases of rapidly progressive dementia from 2001 to 2007. He concluded that 62% of all patients had prion disease, and in 38% of the non prion RPD patients, neurodegenerative diseases accounted for 39% of cases, autoimmune for 22%, and unknown cause for 12%.9 Rapidly progressive neurodegenerative diseases such as Lewy Body Dementia (LBD), Frontal Temporal disease (FTD), and Cortical Basal-ganglia degeneration (CBD), were included our patient's differential diagnoses, due to patient's overall cognitive decline, visual hallucinations, and extra-pyramidal symptoms. Autoimmune encephalitis was also considered, given his acute onset and progression of symptoms, but this was ruled out due to lack of autoimmune antibody index and an ineffective trial of high dose corticosteroid therapy. Finally, the patient's missense mutation at codon 178 of the PRNP gene helped us to confirm the diagnosis of prion disease. Our MRI and EEG findings did not demonstrate typical changes. However, these tests tend to show non-specific changes that are not necessarily helpful in making the diagnosis of FFI. Sleep studies are the most sensitive and specific,8 but unfortunately were unavailable in this case.
Fatal familial insomnia (FFI) and familial hereditary Creutzfeldt-Jakob disease (CJD), 2 clinically and pathologically distinct diseases, are linked to the same mutation of D178N in the PRNP gene. The involvement of D178N either results in FFI or CJD, depending upon the presence of Met or Val at codon 129, respectively.10,11,12 The D178N, Met 129 allele segregated with FFI in all 15 affected members of 5 kindreds, whereas the D178N, Val129 allele segregated with familial CJD in all 15 affected members of 6 kindreds.11 The PRNP mutation D178N/Met129, combined with increased insomnia in the middle stages of the disease in the context of a rapidly progressive dementia, leads to a final diagnosis of FFI. Feng B found that the D178N variant was more susceptible to oxidation than the wild-type prion protein, and Met oxidation accelerates the aggregation and enhances the neurotoxicity of the D178N variant.13 As we know, FFI is characterized by severe insomnia early in the disease course, and the prominent damage occurs in the thalamus and inferior olives.14 There were hereditary pedigrees reported with the D178N/Met129 genotype and a clinical presentation of cerebellar ataxia without overt insomnia.1,14 From the clinical standpoint, our patient exhibited remediable insomnia in the early stage of illness, increased insomnia in the middle stages, and no observable MRI changes in thalamus or inferior olives. However, rapidly progressive dementia and other psychiatric symptoms dominated the course of illness. Clinically, FFI patients sometimes present with memory impairment as the initial symptom and the disease progression is similar to CJD, with a high frequency of cerebellar, myoclonus, extrapyramidal and pyramidal signs.15 Zarranz JJ claimed that FFI and CJD may be the extremes of a spectrum rather than 2 discrete and separate entities.16 Genetic prion diseases may not be easy to diagnosis and may be erroneously clinically classified as other dementias.1 In line with Guerrero RJ's opinion, PRNP screening is warranted in dementia cases with a rapid progression in order to arrive at a correct clinical assessment, counsel family members, and eventually, to apply efficient specific treatments.1
Materials and Methods
Autoimmune Indexes
Peripheral venous blood was collected from the patient. Autoimmune indexes were as follows: Anti-nuclear antibody through Ormond indirect fluorescence method, anti-double-stranded-DNA antibody through Ormond ELISA method, anti-ribonucleoprotein antibody, anti-Smith antibody, anti-SS-A antibody, anti-Ro-52 antibody, anti-SS-B antibody, anti-Scl−70 antibody, anti-PM-Scl antibody, anti-Jo-1 antibody, anti-centromere antibody, anti-PCNA antibody, anti-nucleosome antibody, anti-histone antibody, anti-ribosomal-p-protein antibody, and anti-mitochondrial antibody through Ormond immunoblot method, anti-cardiolipin antibody through ORG ELISA method, anti-β2-glycoprotein-1 immunoglobin G/M through AESKU ELISA method, immunoglobin G/A/M, and complement C3/C4 through Tyco transmission turbidimetric method, anti-streptolysin and rheumatoid factor through DESAY transmission turbidimetric method, and κ/λ light chain through Siemens scattering turbidimetry method.
Genetic Procedures
Blood samples were collected after written consent for PRNP mutation analysis. Genomic DNA was prepared and amplified from peripheral blood according to standard procedures. One hundred nanograms of the extracted DNA were amplified by Polymerase Chain Reaction (PCR) using specific PRNP primers (forward primer A: 5′- CCTTGGAGCAGGAGAAA -3′ and reverse primer A: 5′- GGTTGTGGTGACCGTGT -3′, forward primer B: 5′- TCAGTGGAACAAGCCGAGTA -3′ and reverse primer B: 5′- CCTAACAAACCTGGCAGAAA -3′).
EEG Recordings
The EEG activity was recorded, continuously by using electrodes set in an elastic cap (Electro-Cap International, Inc.). The patients were instructed to stay sit with closed eyes and relaxed. The ground electrode was placed in front of Fz. The left and right mastoids served as reference for all electrodes. The recordings were used off-line to re-reference the scalp recordings to the common average.
MRI Scans
The participant was instructed to lie down and remain motionless, keep eyes closed, and relax. Structural images were acquired by Siemens Verio 1.5T. A routine MRI sequences including high-resolution T1 weighted image, T2 weighted image, and diffused weighted images were conducted.
Ethics and Patient Consent
We received approval from the regional ethical standards committee on human experimentation for our experiment using human materials. We also received written informed consent for research from the patient participating in the study.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by a grant of Natural Science Foundation of China (81301139), Natural Science Foundation of Shanghai (13ZR1435900), and Flight Project of Shanghai Mental Health Center (2014-FX-04), National Key Clinical Disciplines at Shanghai Mental Health Center (Office of Medical Affairs, Ministry of Health, 2011–873; OMA-MH, 2011–873).
REFERENCES
- 1.Guerreiro RJ, Vaskov T, Crews C, Singleton A, Hardy J. A case of dementia with PRNP D178Ncis-129M and no insomnia. Alzheimer Dis Assoc Disord 2009; 23:415–7; PMID:19571725; http://dx.doi.org/ 10.1097/WAD.0b013e3181ae3a76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao C, Shi Q, Tian C, Chen C, Han J, Zhou W, Zhang BY, Jiang HY, Zhang J, Dong XP. The epidemiological, clinical, and laboratory features of sporadic Creutzfeldt–Jakob disease patients in China: surveillance data from 2006 to 2010. PLoS One 2011; 6:e24231; PMID:21904617; http://dx.doi.org/ 10.1371/journal.pone.0024231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tian C, Liu D, Sun QL, Chen C, Xu Y, Wang H, Xiang W, Kretzschmar HA, Li W, Chen C, el al. Comparative analysis of gene expression profiles between cortex and thalamus in Chinese fatal familial insomnia patients. Mol Neurobiol 2013; 48:36–48; PMID:23430483; http://dx.doi.org/ 10.1007/s12035-013-8426-6 [DOI] [PubMed] [Google Scholar]
- 4.Jetha NN, Semenchenko V, Wishart DS, Cashman NR, Marziali A. Nanopore analysis of wild-type and mutant prion protein (PrP(C)): single molecule discrimination and PrP(C) kinetics. PLoS One 2013; 8:e54982; PMID:23393562; http://dx.doi.org/ 10.1371/journal.pone.0054982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lugaresi E, Medori R, Montagna P, Baruzzi A, Cortelli P, Lugaresi A. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med 1986; 315:997–1003; PMID:3762620; http://dx.doi.org/ 10.1056/NEJM198610163151605 [DOI] [PubMed] [Google Scholar]
- 6.Marcon G, Indaco A, Di Fede G, Suardi S, Finato N, Moretti V, Fociani P, Zerbi P, Pincherle A, Redaelli V, et al.. Panencephalopathic Creutzfeldt-Jakob disease with distinct pattern of prion protein deposition in a patient with D178N mutation and homozygosity for valine at codon 129 of the prion protein Gene. Brain Pathol 2014; 24:148–51; PMID:24118545; http://dx.doi.org/ 10.1111/bpa.12095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Capellari S, Strammiello R, Saverioni D, Kretzschmar H, Parchi P. Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol 2011; 121:21–37; PMID:20978903; http://dx.doi.org/ 10.1007/s00401-010-0760-4 [DOI] [PubMed] [Google Scholar]
- 8.Krasnianski A, Bart M, Sanchez Juan PJ, Heinemann U, Meissner B, Varges D, Schulze-Sturm U, Kretzschmar HA, Schulz-Schaeffer WJ, Zerr I. Fatal familial insomnia: Clinical features and early identification. Ann Neurol 2008; 63:658–61; PMID:18360821; http://dx.doi.org/ 10.1002/ana.21358 [DOI] [PubMed] [Google Scholar]
- 9.Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol 2008; 64:97–108; PMID:18668637; http://dx.doi.org/ 10.1002/ana.21430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prusiner SB: Prions. Proc Natl Acad Sci USA: 1998; 95:13363–83; PMID:9811807; http://dx.doi.org/ 10.1073/pnas.95.23.13363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, Cortelli P, Julien J, Vital C, Pendelbury WW, et al.. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science 1992; 258:806–8; PMID:1439789; http://dx.doi.org/ 10.1126/science.1439789 [DOI] [PubMed] [Google Scholar]
- 12.Apetri AC, Vanik DL, Surewicz WK. Polymorphism at residue 129 modulates the conformational conversion of the D178N variant of human prion protein. Biochemistry 2005; 44:15880–8; PMID:16313190; http://dx.doi.org/ 10.1021/bi051455++ [DOI] [PubMed] [Google Scholar]
- 13.Feng B, Wang Z, Liu T, Jin R, Wang S, Wang W, Xiao G, Zhou Z. Methionine oxidation accelerates the aggregation and enhances the neurotoxicity of the D178N variant of the human prion protein. Biochim Biophys Acta 2014; 1842:2345–56; PMID:25281825; http://dx.doi.org/ 10.1016/j.bbadis.2014.09.012 [DOI] [PubMed] [Google Scholar]
- 14.Taniwaki Y, Hara H, Doh-Ura K, Murakami I, Tashiro H, Yamasaki T, Shigeto H, Arakawa K, Araki E, Yamada T, et al.. Familial Creutzfeldt-Jakob disease with D178N-129M mutation of PRNP presenting as cerebellar ataxia without insomnia. J Neurol Neurosurg Psychiatry 2000; 68:388; PMID:10787305; http://dx.doi.org/ 10.1136/jnnp.68.3.388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown P, Goldfarb LG, Kovanen J, Haltia M, Cathala F, Sulima M, Gibbs CJ Jr, Gajdusek DC. Phenotypic characteristics of familial Creutzfeldt-Jakob disease associated with the codon 178Asn PRNP mutation. Ann Neurol 1992; 31:282–5; PMID:1353342 [DOI] [PubMed] [Google Scholar]
- 16.Zarranz JJ, Digon A, Atares B, Rodriguez-Martinez AB, Arce A, Carrera N, Fernández-Manchola I, Fernández-Martínez M, Fernández-Maiztegui C, Forcadas I, et al.. Phenotypic variability in familial prion diseases due to the D178N mutation. J Neurol Neurosurg Psychiatry 2005; 76:1491–6; PMID:16227536; http://dx.doi.org/ 10.1136/jnnp.2004.056606 [DOI] [PMC free article] [PubMed] [Google Scholar]