

ABSTRACT
Despite major efforts devoted to understanding the phenomenon of prion transmissibility, it is still poorly understood how this property is encoded in the amino acid sequence. In recent years, experimental data on yeast prion domains allow to start at least partially decrypting the sequence requirements of prion formation. These experiments illustrate the need for intrinsically disordered sequence regions enriched with a particularly high proportion of glutamine and asparagine. Bioinformatic analysis suggests that these regions strike a balance between sufficient amyloid nucleation propensity on the one hand and disorder on the other, which ensures availability of the amyloid prone regions but entropically prevents unwanted nucleation and facilitates brittleness required for propagation.
KEYWORDS: amyloids, neurodegenerative diseases, prions, protein intrinsic disorder, Q/N-rich domains, yeast
Abbreviations
- AD
Alzheimer's disease
- TSE
transmissible spongiform encephalopathy
- PD
Parkinson's disease
- CJD
Creutzfeldt-Jakob disease
- fALS
familial amyotrophic lateral sclerosis
- PFD
prion forming domain
In the cell, proteins attain the native structure through a delicate and balanced network of interactions, where protein folding and aggregation exert as competing pathways.1,2 In a protein energy landscape, amyloid-like aggregates represent an energy minimum, being usually thermodynamically more stable than the native conformation. This has lead to the hypothesis that the amyloid structures reflects a universal mode of assembly of polypeptide chains and that native protein structures are evolutionary selected metastable states.2 Amyloids are aggregates displaying fibrillar structure, which is constituted by repetitions of a specific protein in a regular β-sheet conformation that runs perpendicular to the fibril axis.3 In humans, amyloids are linked to diseases ranging from neurodegenerative conditions such as Alzheimer's disease (AD), Parkinson's disease (PD) and Creutzfeldt-Jakob disease (CJD), to non-neuronal systemic and localized disorders.3 On the other hand functional amyloids, i.e. proteins that exploit the amyloid fold for evolutionary selected biological functions, have been discovered in diverse species, including human.4 The roles fulfilled by these functional amyloids range from obligate amyloid structures required for scaffolding and/or movement to conditional amyloids such as the yeast prions that can be triggered by environmental factors.5 Whether obligate or conditional, the natural selection of amyloid structure as a functional motif indicates that these properties are likely sequence specific. Whereas the attainment and sustainment of the native structure relies on cooperative interactions involving most, if not all, of the sequence of a protein domain,6,7 the now widely accepted ‘short stretch hypothesis’ states that amyloid formation in contrast is nucleated by short regions of the amino acid sequence named aggregation hot-spots (HS), Aggregation Prone Regions (APR) or Aggregation Prone Sequences (APS).8,9 The short stretch model led to the development of over 20 algorithms that more or less successfully predict protein aggregation and amyloid formation based on the identification of specific β-aggregation and amyloid-pro-ne regions in the polypeptide sequences.10–12 In disease-associated amyloids these regions are generally between 5 and 10 residues in length. 13
Prions are considered a subclass of amyloids in which protein aggregation becomes self-perpetuating and infectious. The phenomenon is known mostly as a neuronal pathology in mammals but in fungi prions play a crucial role in epigenetic inheritance.14–16 Importantly, despite the overlapping conformational properties of amyloids and prions, only a handful of amyloids are currently considered to display at least partial prion capacity under natural conditions.16 As a result, β-aggregation and amyloid predictors are still a long way from correctly detecting prion sequences in proteomes.17 In fact, the sequence characteristics that make a protein sequence a prion have been elusive for years. Moreover, at first glance, the sequence features conferring prion capacity to prion protein in mammals (PrP) appear to differ remarkably from those determining prion behavior in fungi.
Yeast prions are the best characterized transmissible amyloids, thus being excellent model systems to address the determinants of concomitant amyloid formation and propagation.18 In these proteins, prion formation from an initially soluble state involves a structural amyloid conversion driven by specific, relatively large, unstructured domains enriched in glutamine/asparagine (Q/N) residues.18 Interestingly, protein domains displaying this sequence signature are over-represented in eukaryotic proteomes relative to prokaryotes, suggesting that prion-like conformational transition might have evolved as a mechanism for regulating gene function at the protein level in eukaryotes.19 It should be mentioned, however, that PrP, the archetypical mammalian prion, lacks these sequential features.
In order to explore the repertoire of prion proteins in Saccharomyces cerevisiae the Lindquist group conducted a genome-wide bioinformatics survey using a hidden Markov sequence model to identify putative candidates on the basis of their compositional similarity to known prion forming domains (PFDs)20 and used experimental validation to identify the bone-fide PFDs in their predictions. These results are at the core of several algorithms for prion domain prediction, all relying on the analysis of amino acid sequences.17,21–24 These programs are constructed on 2 alternative models for amyloid formation by prion-like domains (Figure 1): (1) The compositional model relying on the establishment of a large number of weak interactions 17 and (2) our model, which proposes ‘classical’ nucleation by short amyloidogenic stretches, whose amyloid propensity is modulated by the structural context.24 Despite the mechanistic difference between algorithms, the advent of accurate computational tools to detect yeast prion domains opens new and exciting possibilities, allowing the exploration of proteomes for the discovery of novel and hitherto unexpected Q/N-enriched domains that may drive conformational conversion in novel prion proteins. Indeed, recent studies have revealed that over 250 human proteins display prion-like stretches in regions with high presence of uncharged polar residues and glycine, including several heterogeneous nuclear ribonucleoproteins (hnRNPs) related to neurodegenerative diseases such as familial amyotrophic lateral sclerosis (fALS).25
Figure 1.
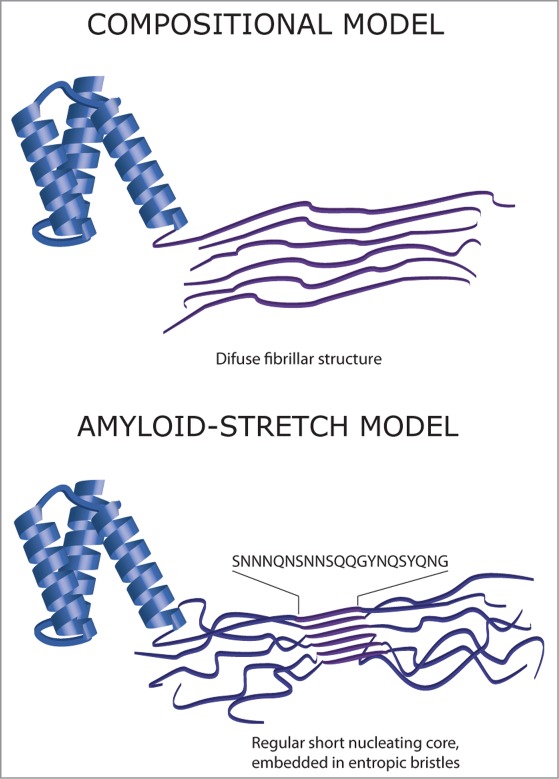
Two models for amyloid structure formation in Q/N-rich prion-like domains. The compositional model relies on the establishment a large number of weak interactions whereas the amyloid-stretch model proposes the existence of a preferential short nucleating sequence whose amyloid propensity is modulated by its structural context.
In the light of these advances, the requirements for a polypeptide sequence to act as a prion begin to be defined. In our view, for a protein sequence to become a Q/N enriched prion, 3 essential conditions appear to be required:
Requirement 1: A short amyloid-prone region able to trigger amyloid formation in a sequence specific manner. These amyloid cores should, however, possess distinctive features, since both a high aggregation rate and an elevated fragmentation capacity are necessary in prions in order to attain the number of propagons or seeds required for spreading and propagation.26,27 Thus, while a certain amyloid nucleation capacity favoring a sufficiently high aggregation rate is absolutely necessary, the final amyloid aggregate should at the same time display brittleness, a property that facilitates an increase in the number of nucleation events per cell. Accordingly, in contrast to most amyloids, the aggregation reaction should not be nucleated in PFDs by an extremely strong and highly hydrophobic amyloid core.
Requirement 2: The amyloid-prone region has to be located in a structurally disordered region, that permits its self-assembly without the necessity of conformational unfolding. The PFDs of all known Q/N enriched yeast prions display this property.18,20 The location of the amyloid core in large unstructured regions favors the acquisition of the β-cross motif without large conformational rearrangements and may at the same time promote the brittleness mentioned in requirement 1. Moreover, the disordered region may act as a so-called ‘entropic bristle’,27,28 which would reduce the overall aggregation propensity and could allow for a better biological control of the nucleation event, which is discussed more in detail in requirement 3.
Requirement 3: PFDs have to posses an amino acid composition allowing the protein to remain in a soluble state under physiological conditions while keeping intact a cryptic amyloid capacity. Stress situations promoting increased local protein concentration, as well as the presence of preformed amyloid seeds, might alter the delicate equilibrium between native/soluble and amyloid/insoluble states, providing means to control the nucleation of amyloid aggregation and hence the onset of the prion phenotype. If we recapitulate requirements (1) and (2), clustering in the same sequence region amino acid residues with a significant amyloid propensity together with residues promoting structural disorder would favor prion capacity. Only five amino acid residues seem to unite these 2 essential properties, i.e., amyloid propensity and structural disorder, according to FoldIndex29 and Waltz30 algorithms: N, Q, Y, S and W. (Figure 2). Interestingly enough N, Q, Y, S are, in this order, the most over-represented residues in bona-fide prion domains, relative to their frequency in the protein universe,22 with odds ratios of 5.70, 4.13, 1.72 and 1.66, respectively. In this context, N and Q residues show medium amyloid propensity, allowing the formation of amyloids with moderate strength, while at the same time are the amyloidogenic residues that more benefice disorder. This provides a rational basis for the strong over-representation of these particular residues in the PFDs of yeast. In good agreement with its higher frequency, N is the residue that best balances amyloid and disorder propensity and thus the preferred residue to support prion behavior. It is important to point out, however, that poly-N or poly-Q, these later being involved in a number of ataxias,31 are not expected to display a prion-like behavior since they lack requirement 1, that is, a specific region able to selectively nucleate ordered amyloid formation.
Figure 2.
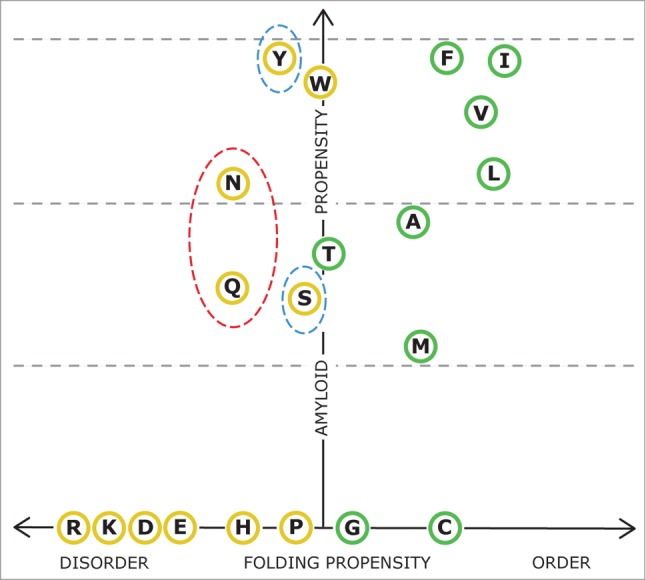
Balance between amyloid and structural propensities in natural amino acids. Residues rendering ordered and disordered 21-residues long homo-polymers according to FoldIndex14 are shown in green and yellow circles, respectively. The amyloid propensity of these stretches was calculated with Waltz.30 The four more over-represented residues in yeast PFDs are circled by discontinuous lines, red indicates odd ratios > 4.0 and blue odd ratios > 1.5, relative to the composition of the protein universe.22
Hydrophobic amino acids are under-represented in Q/N-rich yeast PFDs, likely because a high proportion of these residues would render the protein excessively aggregation-prone and/or result in too strong amyloid assemblies. Despite the presence of a reduced number of hydrophobic residues in PFDs has been shown to bust prion formation and amyloid formation,32 Y is the only hydrophobic residue over-represented in these domains. It has been proposed that this might respond to the fact that aromatic residues might facilitate both prion formation and chaperone dependent prion propagation.33 However, F is indeed under-represented in PFDs with an odds ratio of 0.72 and the Y/F relationship between odds ratios in PFDs is 2.4, suggesting that the additional hydroxyl group in Y should provide a certain advantage, which in our opinion is allowing a better balance of amyloid propensity and intrinsic disorder. Despite its aromatic character, W is one of the most under-represented residues in prion domains with and odd ratio of 0.091, only C, which is able to crosslink covalently polypeptide chains, being less frequent.22 The absence of W in PFDs is best explained by its particular structure, wherein the indole group may not be easily placed in β-cross structures due to of steric impediments, being indeed depleted relative to F and Y in functional and pathogenic amyloids.34
The two alternative models used to identify prion domains (Figure 1) coincide in the requirement of a relatively large disordered region in yeast PFDs. However, one prion model support the view of amyloid formation in PFDs resulting from a bias in sequence composition favoring a large number of weak interactions over a wide sequence stretch17 whereas the alternative model supports prion behavior to emerge from the preferential nucleation by specific and localized short amyloid-prone stretches embedded in the wider disordered region.24 Despite the apparent contradiction between these 2 views, indeed the second model just pursues to delimitate the aggregation driving force of the amyloid cores embedded in the prions domains defined by the compositional model. In this way, the first composition based methods to predict potential yeast prions proposed a minimal core of 60 residues.20 This further evolved into a method employing a 41 amino acid sliding window for compositional analysis, denoting that the initial 60 residues window was larger than actually required.17 We proposed to reduce this size even further to account for a 21 amino acid core, based on the length of the core of HET-s PFD, the unique protein for which an atomic-resolution structure of the infectious fibrillar state is available to date,24,35 which displays a β-arcade conformation.36 The excellent performance of our method, based on a preferential nucleation by short amyloid-prone stretches, lead us to believe not only that a 21 residues core is indeed sufficient for prediction, but also that the ‘classical’ short stretch nucleation model applies to prions in a similar manner as it does for ‘classic’ amyloids, the main differences being that, in prions, amyloid nucleating stretches might fold into β-strand-turn-β-strand elements and that their potency is strongly modulated by the entropy of the sequence context in which they are embedded, i.e. the degree of structural disorder will determine both the sensitivity for amyloid nucleation as well as the ability of formed fibrils to break up and provide additional propagons.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Pol Ventura for his comments on Figure 2.
Funding
This work was supported in part by Ministerio de Economia y Competividad, Spain [BFU2013-44763 to S.V.]; by SUDOE, INTERREG IV B, FEDER [SOE4/P1/E831to S.V.]; by ICREA [ICREA Academia 2009 to S.V.]; by the Ramón y Cajal Programme from Ministerio de Ciencia e Innovación [RYC-2011-07987 to R.S.]. The Switch Laboratory was supported by grants from VIB, University of Leuven, the Funds for Scientific Research Flanders (FWO), the Flanders Institute for Science and Technology (IWT) and the Federal Office for Scientific Affairs of Belgium (Belspo, IAP network P7/16).
REFERENCES
- 1.Jahn TR, Radford SE. The Yin and Yang of protein folding. Febs J 2005; 272:5962–70; PMID:16302961; http://dx.doi.org/ 10.1111/j.1742-4658.2005.05021.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jahn TR, Radford SE. Folding versus aggregation: polypeptide conformations on competing pathways. Arch Biochem Biophys 2008; 469:100–17; PMID:17588526; http://dx.doi.org/ 10.1016/j.abb.2007.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 2006; 75:333–66; PMID:16756495; http://dx.doi.org/ 10.1146/annurev.biochem.75.101304.123901 [DOI] [PubMed] [Google Scholar]
- 4.Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid–from bacteria to humans. Trends Biochem Sci 2007; 32:217–24; PMID:17412596; http://dx.doi.org/ 10.1016/j.tibs.2007.03.003 [DOI] [PubMed] [Google Scholar]
- 5.Falsone A, Falsone SF. Legal but lethal: functional protein aggregation at the verge of toxicity. Fron Cell Neurosci 2015; 9:45; PMID:25741240; http://dx.doi.org/ 10.3389/fncel.2015.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levinthal C. Are there pathways for protein folding?. J Chim Phys 1968; 65:44–5 [Google Scholar]
- 7.Zwanzig R, Szabo A, Bagchi B. Levinthal's paradox. Proc Natl Acad Sci U S A 1992; 89:20–2; PMID:1729690; http://dx.doi.org/ 10.1073/pnas.89.1.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopez de la Paz M, Serrano L. Sequence determinants of amyloid fibril formation. Proc Natl Acad Sci U S A 2004; 101:87–92; PMID:14691246; http://dx.doi.org/ 10.1073/pnas.2634884100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanchez de Groot N, Pallares I, Aviles FX, Vendrell J, Ventura S. Prediction of “hot spots” of aggregation in disease-linked polypeptides. BMC Struct Biol 2005; 5:18; PMID:16197548; http://dx.doi.org/ 10.1186/1472-6807-5-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belli M, Ramazzotti M, Chiti F. Prediction of amyloid aggregation in vivo. EMBO Rep 2011; 12:657–63; PMID:21681200; http://dx.doi.org/ 10.1038/embor.2011.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castillo V, Grana-Montes R, Sabate R, Ventura S. Prediction of the aggregation propensity of proteins from the primary sequence: aggregation properties of proteomes. Biotechnol J 2011; 6:674–85; PMID:21538897; http://dx.doi.org/ 10.1002/biot.201000331 [DOI] [PubMed] [Google Scholar]
- 12.Espargaro A, Busquets MA, Estelrich J, Sabate R. Predicting the aggregation propensity of prion sequences. Virus Res 2015; S0168-1702(15)00119-7; PMID:25747492 [DOI] [PubMed] [Google Scholar]
- 13.Rousseau F, Serrano L, Schymkowitz JW. How evolutionary pressure against protein aggregation shaped chaperone specificity. J Mol Biol 2006; 355:1037–47; PMID:16359707; http://dx.doi.org/ 10.1016/j.jmb.2005.11.035 [DOI] [PubMed] [Google Scholar]
- 14.Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev 2009; 89:1105–52; PMID:19789378; http://dx.doi.org/ 10.1152/physrev.00006.2009 [DOI] [PubMed] [Google Scholar]
- 15.Chien P, Weissman JS, DePace AH. Emerging principles of conformation-based prion inheritance. Annu Rev Biochem 2004; 73:617–56; PMID:15189155; http://dx.doi.org/ 10.1146/annurev.biochem.72.121801.161837 [DOI] [PubMed] [Google Scholar]
- 16.Sabate R. When amyloids become prions. Prion 2014; 8; PMID:24831240; http://dx.doi.org/ 10.4161/pri.29238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toombs JA, Petri M, Paul KR, Kan GY, Ben-Hur A, Ross ED. De novo design of synthetic prion domains. Proc Natl Acad Sci U S A 2012; 109:6519–24; PMID:22474356; http://dx.doi.org/ 10.1073/pnas.1119366109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uptain SM, Lindquist S. Prions as protein-based genetic elements. Annu Rev Microbiol 2002; 56:703–41; PMID:12142498; http://dx.doi.org/ 10.1146/annurev.micro.56.013002.100603 [DOI] [PubMed] [Google Scholar]
- 19.Michelitsch MD, Weissman JS. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc Natl Acad Sci U S A 2000; 97:11910–5; PMID:11050225; http://dx.doi.org/ 10.1073/pnas.97.22.11910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009; 137:146–58; PMID:19345193; http://dx.doi.org/ 10.1016/j.cell.2009.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Espinosa Angarica V, Angulo A, Giner A, Losilla G, Ventura S, Sancho J. PrionScan: an online database of predicted prion domains in complete proteomes. BMC Genomics 2014; 15:102; PMID:24498877; http://dx.doi.org/ 10.1186/1471-2164-15-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Espinosa Angarica V, Ventura S, Sancho J. Discovering putative prion sequences in complete proteomes using probabilistic representations of Q/N-rich domains. BMC Genomics 2013; 14:316; PMID:23663289; http://dx.doi.org/ 10.1186/1471-2164-14-316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lancaster AK, Nutter-Upham A, Lindquist S, King OD. PLAAC: a web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 2014; 30(17):2501–2; PMID:24825614; http://dx.doi.org/ 10.1093/bioinformatics/btu310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabate R, Rousseau F, Schymkowitz J, Ventura S. What makes a protein sequence a prion? PLoS Comput Biol 2015; 11:e1004013; PMID:25569335; http://dx.doi.org/ 10.1371/journal.pcbi.1004013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al.. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013; 495:467–73; PMID:23455423; http://dx.doi.org/ 10.1038/nature11922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature 2006; 442:585–9; PMID:16810177; http://dx.doi.org/ 10.1038/nature04922 [DOI] [PubMed] [Google Scholar]
- 27.Derdowski A, Sindi SS, Klaips CL, DiSalvo S, Serio TR. A size threshold limits prion transmission and establishes phenotypic diversity. Science 2010; 330:680–3; PMID:21030659; http://dx.doi.org/ 10.1126/science.1197785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grana-Montes R, Marinelli P, Reverter D, Ventura S. N-terminal protein tails act as aggregation protective entropic bristles: the SUMO case. Biomacromolecules 2014; 15:1194–203; PMID:24564702; http://dx.doi.org/ 10.1021/bm401776z [DOI] [PubMed] [Google Scholar]
- 29.Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL, et al.. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005; 21:3435–8; PMID:15955783; http://dx.doi.org/ 10.1093/bioinformatics/bti537 [DOI] [PubMed] [Google Scholar]
- 30.Maurer-Stroh S, Debulpaep M, Kuemmerer N, Lopez de la Paz M, Martins IC, Reumers J, Morris KL, Copland A, Serpell L, Serrano L, et al.. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat Meth 2010; 7:237–42; PMID:NOT_FOUND; http://dx.doi.org/ 10.1038/nmeth.1432 [DOI] [PubMed] [Google Scholar]
- 31.Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 1997; 19:333–44; PMID:9292723; http://dx.doi.org/ 10.1016/S0896-6273(00)80943-5 [DOI] [PubMed] [Google Scholar]
- 32.Ross ED, Toombs JA. The effects of amino acid composition on yeast prion formation and prion domain interactions. Prion 2010; 4:60–5; PMID:20495349; http://dx.doi.org/ 10.4161/pri.4.2.12190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez Nelson AC, Paul KR, Petri M, Flores N, Rogge RA, Cascarina SM, Ross ED. Increasing prion propensity by hydrophobic insertion. PLoS One 2014; 9:e89286; PMID:24586661; http://dx.doi.org/ 10.1371/journal.pone.0089286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gazit E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J 2002; 16:77–83; PMID:11772939; http://dx.doi.org/ 10.1096/fj.01-0442hyp [DOI] [PubMed] [Google Scholar]
- 35.Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Amyloid fibrils of the HET-s(218-289) prion form a beta solenoid with a triangular hydrophobic core. Science 2008; 319:1523–6; PMID:18339938; http://dx.doi.org/ 10.1126/science.1151839 [DOI] [PubMed] [Google Scholar]
- 36.Kajava AV, Baxa U, Steven AC. Beta arcades: recurring motifs in naturally occurring and disease-related amyloid fibrils. FASEB J 2010; 24:1311–9; PMID:20032312; http://dx.doi.org/ 10.1096/fj.09-145979 [DOI] [PMC free article] [PubMed] [Google Scholar]