

ABSTRACT
In recent years, prion protein (PrPC) has been considered as a promising target molecule for cancer therapies, due its direct or indirect participation in tumor growth, metastasis, and resistance to cell death induced by chemotherapy. PrPC functions as a scaffold protein, forming multiprotein complexes on the plasma membrane, which elicits distinct signaling pathways involved in diverse biological phenomena and could be modulated depending on the cell type, complex composition, and organization. In addition, PrPC and its partners participate in self-renewal of embryonic, tissue-specific stem cells and cancer stem cells, which are suggested to be responsible for the origin, maintenance, relapse, and dissemination of tumors. Interference with protein–protein interaction has been recognized as an important therapeutic strategy in cancer; indeed, the possible interference in PrPC engagement with specific partners is a novel strategy. Recently, our group successfully used that approach to interfere with the interaction between PrPC and HSP-90/70 organizing protein (HOP, also known as stress-inducible protein 1 - STI1) to control the growth of human glioblastoma in animal models. Thus, PrPC-organized multicomplexes have emerged as feasible candidates for anti-tumor therapy, warranting further exploration.
Keywords: prion protein, ligand, cancer, therapy, peptide, cell signaling, cancer stem cell
Abbreviations
- HSP
heat shock proteins
- GBM
glioblastoma multiforme
- BBB
blood brain barrier
- CSC
cancer stem cell
Introduction
Prion protein (PrPC) has been studied thoroughly for decades, due its involvement in transmissible spongiform encephalopathies and conversion to infectious proteinaceous agents called prions.1 Despite intense discussion of the function of the normal protein, convincing data from different groups indicate that PrPC has important roles in the nervous and immune systems, regulating cellular processes such as cell death and survival, proliferation, and differentiation.2,3 PrPC is a glycosylphosphatidylinositol-anchored protein, and many of its described functions depend on specific interactions with partners on the plasma membrane (receptors or extracellular molecules), such as laminin, vitronectin, NCAM, caveolin, and HSP-90/70 heat shock organizing protein, also known as stress-inducible protein 1 (HOP/STI1)4 (a list of these ligands can be found at http://www.signaling-gateway.org/molecule/query?afcsid=A003935), which can modulate cellular signaling cascades. Due to these properties, we have proposed that PrPC plays a scaffolding role on the cell surface, recruiting diverse partners to organize signaling platforms.5 In this review we will discuss how these PrPC-organized complexes can be involved with the tumoral processes and the strategy to target the engagement of PrPC to specific ligands for therapeutic interventions.
PrPC in Tumor Biology: Processes and Mechanisms
The functions of PrPC in tumor cells have been addressed, and evidence suggests that this protein is an important player in tumor biology. Several studies have demonstrated the importance of PrPC in proliferation, apoptosis, invasion, metastasis, and drug resistance in different cancer types.6 In undifferentiated gastric tumors, a worse response to chemotherapy and lower patient survival rate are associated with higher PrPC expression levels.7 Positive correlations between PrPC expression and invasion, lymph node metastasis, and survival have also been confirmed in patients with gastric tumors, indicating that PrPC is an independent prognostic factor in these tumors.8 Proteomic evaluation of colorectal cancer cell lines identified PrPC as a putative biomarker for adenoma–carcinoma progression, discriminating low-risk adenomas and normal colon from high-risk adenomas and colorectal cancer.9 Accordingly, a previous study showed that PrPC expression could be used as a prognostic factor in patients with colorectal cancer.10 In breast cancer cell lines, resistance to TNF-induced cell death is associated with greater PrPC expression.11 Furthermore, lower sensitivity to neoadjuvant therapy has been observed in ER-negative breast tumors expressing higher (vs. lower) levels of PrPC.12
Despite this evidence of PrPC involvement in tumor biology, the mechanisms associated with these functions remain largely unexplored. In some cases, mechanistic approaches have shown that PrPC inhibits Bax-induced apoptosis through Bax conformational change prevention, impairing mitochondrial translocation and cytochrome c release in breast cancer cells.13 In cell lines derived from colon tumors, PrPC has been shown to regulate glucose transporter 1 expression through the activation of Fyn-HIF-2a, increasing glucose uptake, glycolysis, and cell survival/proliferation.14 PrPC activation of Fyn in breast cancer cells is also involved in epithelial–mesenchymal transition and results in a more aggressive phenotype.15 PrPC silencing in glioma cell lines causes increased autophagy due to induction of LC3-II, an increase in Beclin 1, and simultaneous decreases in p62, Bcl− 2, and the phosphorylation of 4E-BP1, a target of mTOR autophagy signaling.16 Interestingly, mTOR, a master player in cell signaling with pivotal role in tumorigenesis, is also involved in PrPC-dependent neuronal differentiation and neuroprotection through activation of PI3K/Akt pathways.17
PrPC Engagement to its Ligands: A Reliable Target for Therapeutic Intervention
The phenotypes and mechanisms described for PrPC in tumoral biology are in agreement with its roles in multicomplex protein formation and organization of diverse signaling platforms.5 PrPC priming can be induced after ligand binding, stimulating the acquisition of specific conformations and allowing additional interactions in the complex.5 The upregulation of PrPC or its primary ligands in different tumor types may favor the assembly of tissue-specific complexes with different protein compositions. These complexes could alter the pattern of cellular signaling and, consequently, processes such as proliferation, adhesion, migration, differentiation, and drug resistance. Indeed, we believe that the identification of PrPC ligands that coordinate these processes will help researchers focus on mechanisms that can be targeted for therapeutic interventions (as illustrated in Fig. 1).
Figure 1.
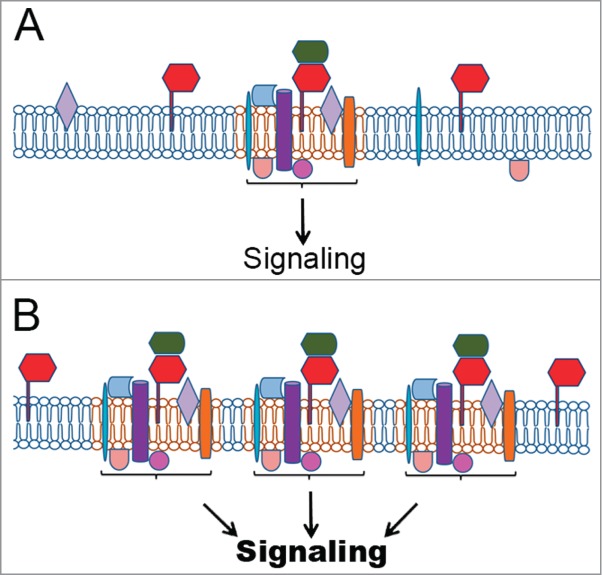
PrPC in signaling platforms and their regulation. (A) PrPC (red) interaction with a prime soluble ligand (green) modulates a group of multiple partners at the plasma membrane, organizing signaling platforms that regulate specific cellular functions. Other PrPC partners include integral transmembrane proteins (purple, blue or orange) or peripheral membrane proteins, including cytoplasmic (beige or rose) or proteins attached to outer leaflet of the plasma membrane (light blue or light purple). Importantly, the prime ligand can be a membrane protein in the same domain of PrPC localization (cis), in the membrane of another cell (trans), or on an extracellular vesicle surface. (B) The upregulation of PrPC allows the formation of additional tissue-specific complexes with the ability to regulate events associated to tumorigenesis. (C) Higher levels of the prime ligand can also improve the number of PrPC molecules able to organize signaling platforms, thereby inducing cancer processes. (D) Conversely, PrPC downregulation or silencing disorganizes these complexes, impairing PrPC-mediated signaling and, consequently, the tumoral process. (E) The inhibition of PrPC interaction with the prime ligand impairs the organization of these platforms and serves as a target for cancer therapy, potentially with fewer side effects.

In recent years, several independent studies have characterized novel PrPC complexes modulating specific tumoral cell behaviors. In breast cancer, PrPC engagement to multidrug resistance protein (P-gp) and caveolin is associated with drug resistance.18 In addition, the PrPC–P-gp complex may include CD44, a membrane receptor involved in cell adhesion, motility, and metastasis, promoting resistance to neoadjuvant therapy.19 However, the researchers proposing this association provided no clear explanation of the predominant nuclear localization of PrPC observed in tumor samples.19
In pancreatic tumors and melanomas, an unconventional transmembrane form of PrPC (pro-PrP) can interact with filamin A (a cytoplasmic protein involved in actin organization), perturbing cytoskeleton organization and conferring growth advantage.20 Pro-PrP also confers a worse prognosis for pancreatic ductal adenocarninomas.21 Thus, the identification of compounds that can interfere with this binding would be of great importance.
The PrPC complex formed with 37-kDa/67 kDa laminin receptor (LRP/LR, also known as MGr1-AG/37LRP) is well understood, and many of its functions in nervous system cells have been examined.22 LRP/LR is involved in various tumorigenic processes, and the upregulation of PrPC and LRP/LR in gastric tumors predicts poor prognosis.8 In this case, therapeutic interventions using antibodies to MGr1-AG/37LRP have been addresssed.23
Targeting the PrPC-HOP/STI1 Complex in Glioblastoma: Proof of Concept
The interaction of PrPC with HOP/STI1 is one of the best-characterized PrPC complexes and it has emerged as one of the most important in tumorigenesis. PrPC-HOP/STI1 interaction was first glimpsed in 1997, when we characterized a novel PrPC partner using complementary hydropathy theory;24 subsequently, we identified this ligand as the co-chaperone HOP/STI125 and demonstrated the involvement of the complex in neuroprotection, neurogenesis, and astrocyte proliferation, among others.4 HOP/STI1 was initially identified as a co-chaperone that cooperates with HSP70 and HSP90 to assist in the folding and stability of client proteins, with a key function in cellular homeostasis.26 Since that time, many functions in addition to its role as a co-chaperone and PrPC ligand have been attributed to this protein.26 HOP/STI1 expression is increased in tumor cell lines and tumoral tissues from the breast, colon, pancreas, liver, and ovary; in most of these tumors, greater HOP/STI1 expression is associated with more aggressive disease, poorer survival, and drug therapy resistance. Cytoplasmic HOP/STI1 can modulate migration and invasion due to its interaction with actin and tubulin at the cytoskeleton, as well as modulation of the expression of matrix metalloprotease 2 (see review by Baindur-Hudson et al.26). Extracellular HOP/STI1 was identified in conditioned media from different cell lines, and significantly higher levels of the protein are present in serum from patients with ovarian cancer.26 In this case, the secreted form of HOP/STI1 binds to ALK2 and activates the SMAD signaling pathway, promoting cell proliferation.26 More recently, our group demonstrated that HOP/STI1 is secreted in the membranes of exosome-like extracellular vesicles.27 Together, these data suggest the importance of HOP/STI1 as a prognostic biomarker in some tumors and as a target for therapeutic strategies. Furthermore, because the secreted form of HOP/STI1 may have different ligands at the cell surface, its use as a target for therapy must focus on specific interactions related to tumoral processes.
In 2014, our group demonstrated28 that HOP/STI1 and PrPC are upregulated in human glioblastoma (GBM), which was confirmed by sample analysis from the TGCA consortium. PrPC and HOP/STI1 expression levels were correlated with higher proliferation rates and poorer clinical outcome. Additionally, data demonstrated that the engagement of HOP/STI1 to PrPC promoted proliferation and tumor growth in GBM cell lines, and that total or partial PrPC ablation promoted tumor growth inhibition and improved survival of mice bearing GBM xenografts. To address the importance of PrPC-HOP/STI1 in GBM biology, we inhibited formation of the complex with a synthetic peptide corresponding to the HOP/STI1 binding site to PrPC (named HOP/STI1230–245). Alone, this peptide had no effect on proliferation; however, it competed with full-length HOP/STI1, displacing it from PrPC at the cell surface. The HOP/STI1230–245 peptide abolished proliferation induced by HOP/STI1-PrPC in GBM cell lines. The delivery of HOP/STI1230–245 into orthotopic xenografts hindered cell proliferation and induced apoptosis, leading to tumor growth inhibition and increased animal survival.28 Remarkably, in addition to its previously demonstrated neuroprotective function and positive effect on memory formation,29,30 the HOP/STI1230–245 peptide was able to prevent cognitive decline caused by tumor growth.28 Indeed, due its antitumoral and neuroprotective functions, the HOP/STI1230–245 peptide is a promising candidate for testing in brain tumor treatment alone or combined with other conventional therapies.
The use of peptides for the treatment of wide range of diseases has increased recently. Many features favor the use of peptides, rather than small-molecule (<500 Da) or large-molecule (>5000 Da) biological drugs (e.g., monoclonal antibodies or recombinant proteins). High specificity is perhaps the most important feature of peptides, as they can mimic structural domains responsible for protein–protein interactions, competing for their binding and activation. This property reduces the probability of adverse toxic effects, and interference in drug combinations is insignificant.31 The disadvantages of peptides as drug candidates are being investigated intensively, and points for both oral bioavailability and half-lives of these molecules in the organism. In the context of brain diseases, the blood–brain barrier (BBB) is an additional drug obstacle. Nevertheless, evidence of partial BBB disruption in GBM indicates that brain tumor cells could be left unprotected, increasing their vulnerability to drugs, including peptides.31 Modifications of the chemical structure of peptides,32 in particular the HOP/STI1230–245 peptide discussed here, such as replacement of L-amino acids by unnatural D-amino acids, cyclization, and peptidomimetics, could be performed to overcome these challenging conditions and increase peptide stability and half-life in the organism; the achievement of such goals would allow systemic administration and improve tumoral diffusion.
Considering large-molecule biological drugs, the use of antibodies could be a feasible approach to target the interaction of extracellular HOP/STI1 and PrPC in tumors. In some studies described here, antibodies against PrPC or HOP/STI1 successfully inhibited tumor growth in vivo in diverse organs other than the brain. A preliminary experiment using a HOP/STI1 antibody25 has been performed. As indicated in Figure 2, intra-tumor delivery of anti-HOP/STI1 into orthotopic xenografts of GBM cells slightly improved animal survival.
Figure 2.
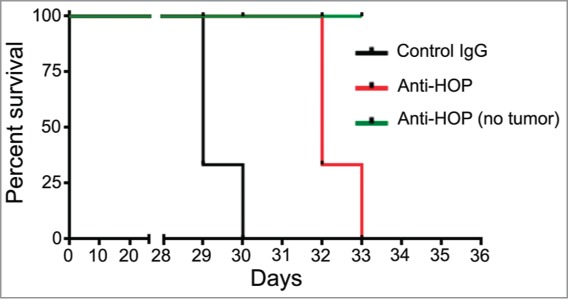
Disruption of PrPC-HOP/STI1 interaction using anti-HOP antibodies in xenografts increases the survival of animals with glioblastoma. Preliminary results indicate that disruption of the PrPC-HOP/STI1 complex using a specific antibody increases animal survival. Briefly, nude mice were injected orthotopically with a U87 cell line (5×105 cells) and treated with HOP/STI1-antibody25 for 28 d (240 ng/day) using osmotic micropumps. The complete methodology is described in Lopes et al.28 Kaplan–Meier survival curve of mice implanted with U87 cells. Log-rank p = 0.0023, n = 3 mice per group.
Importantly, however, the blockage of both PrPC and HOP/STI1 could be deleterious. Long-term,33 but not short-term,29 intracranial infusion of antibodies against PrPC, in particular those directed to the globular domain of PrPC, can be neurotoxic.33 The short-term use of polyclonal antibodies against full-length HOP/STI1 or the HOP/STI1230–245 peptide has not caused brain toxicity.29 However, we demonstrated that the constitutive deletion of HOP/STI1 is embryonically lethal, and heterozygous animals expressing half-levels of the protein presented higher sensitivity to brain injury,34 indicating the importance of this protein also in adults. Furthermore, maternally derived HOP/STI1 autoantibodies were detected in mothers of children with autism, suggesting that neurodevelopment is impaired by these autoantibodies.35 Indeed, interference with PrPC-HOP/STI1 interaction in tumors, particularly those in the central nervous system (CNS), using peptides that compete for their engagement should lead to better results than the use of antibodies against these molecules.
Targeting Cancer Stem Cells by Blocking PrPC Interactions
One of the most-studied recent themes in oncology is related to features that govern tumor origin, and cancer stem cells (CSCs) have emerged as a pivotal component able to initiate and maintain tumors.36 CSCs have been functionally defined as a small subpopulation of cells capable of self-renewal, differentiate into all cell types in a determined tumor, and tumor propagation when xenotransplanted into immunodeficient mice.36 An important characteristic of CSCs is their resistance to conventional therapies, which has been implicated in cancer recurrence and has made these cells a key target for therapy.36 Although the origin of CSCs remains unidentified, these cells share key properties with normal tissue-resident stem cells and are thought to arise through malignant transformation events in normal stem cells.36 Considering the emerging functions of PrPC in stemness, fundamental issues that must be addressed include its interaction with a prime ligand, the role of the complex in CSCs, and its possible use as a therapeutic target in cancer (Fig. 3).37,38 PrPC engages CD44, a stem cell marker, and their expression is correlated with resistance to chemotherapy in breast cancer cell lines.19 Moreover, the CD44+ PrPC+ subpopulation of colorectal tumor cells has CSCs properties, including tumorigenesis and metastasis capacities,39 indicating that PrPC contributes to tumor maintenance by modulating CSCs behaviors. The contribution of the PrPC-HOP/STI1 complex to CSCs self-renewal remains to be explored. Nevertheless, the HOP/STI1-PrPC complex is known to play an important role in self-renewal and proliferation of neural stem cells.40
Figure 3.
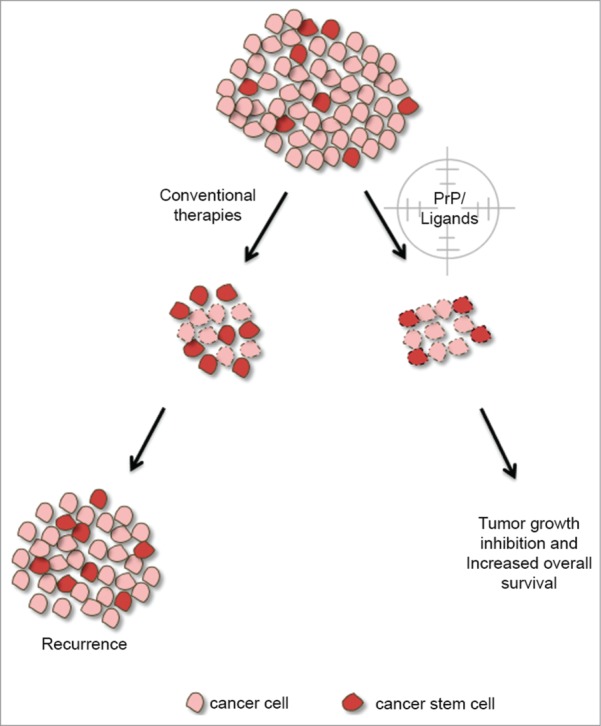
Targeting of PrPC and its partners in CSCs for cancer therapy. Conventional therapy targets tumor cells by destroying them or decreasing their proliferation. Tumor growth is governed by multiple cellular mechanisms in which PrPC plays a role. The progression of tumor development is related to the presence of cancer stem cells that have a pivotal role in cell resistance, culminating in tumor recurrence. Indeed, PrPC and its ligands in cancer stem cells could also be targeted, suggesting a promising approach for novel cancer therapies. Cells with dotted lines represent cells undergoing cell death.
Recently, Tomasetti and Vogelstein41 reported a positive correlation between cancer risk and the number of mitotic divisions of stem cells in different tissues, strengthening the importance of the participation of tissue-resident stem cells in tissue homeostasis or as a substrate that gives rise to tumors. In this scenario, tumors in which PrPC or HOP/STI1 has been described to play a significant role, such as colorectal, pancreatic, and hepatocellular tumors (discussed above), are related to tissues with more total stem cell divisions during their lifespan. These authors41 documented a much smaller number of CSCs divisions in GBMs than in the tumors discussed herein; however, PrPC expression is known to be more abundant in cells from brain tissue than in those from other tissues, which may contribute to the importance of PrPC-HOP/STI1 in brain stem cells.2
In conclusion, the mechanisms related to the roles of PrPC in cancer biology need to be better explored. Its predicted role as a scaffold protein participating in the organization of membrane platforms indicates that specific partners within tumor cells, extracellular matrix, and soluble factors secreted from tumor cells or the tumor microenvironment must be considered as good candidates for therapeutic interventions. Strikingly, our work exploring the engagement of PrPC with the secreted form of HOP/STI1 allows the development of strategies to target this complex specifically and control tumor growth. The success of this approach for other tumors and PrPC ligands should be evaluated to direct new discoveries in cancer biology.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
FUNDING
Funding was provided by São Paulo Research Foundation (FAPESP; 2011/13906-2 and 2009/14027-2) and National Council for Scientific and Technological Development (CNPq 2008/57904-0 and 2008/57887-9).
REFERENCES
- 1.Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol 2011; 3:a006833;PMID:21421910; http://dx.doi.org/ 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linden R, Martins VR, Prado MAM, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev 2008; 88:673-728;PMID:18391177; http://dx.doi.org/ 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- 3.Biasini E, Turnbaugh JA, Unterberger U, Harris DA. Prion protein at the crossroads of physiology and disease. Trends Neurosci 2012; 35:92-103;PMID:22137337; http://dx.doi.org/ 10.1016/j.tins.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martins VR, Beraldo FH, Hajj GN, Lopes MH, Lee KS, Prado MAM, Linden R. Prion protein: orchestrating neurotrophic activities. Curr Issues Mol Biol 2010; 12:63-86;PMID:19767651. [PubMed] [Google Scholar]
- 5.Linden R, Cordeiro Y, Lima LMTR. Allosteric function and dysfunction of the prion protein. Cell Mol Life Sci 2012; 69:1105-24;PMID:21984610; http://dx.doi.org/ 10.1007/s00018-011-0847-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mehrpour M, Codogno P. Prion protein: From physiology to cancer biology. Cancer Lett 2010; 290:1-23;PMID:19674833; http://dx.doi.org/ 10.1016/j.canlet.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Wang J-H, Du J-P, Zhang Y-H, Zhao X-J, Fan R-Y, Wang Z-H, Wu Z-T, Han Y. Dynamic changes and surveillance function of prion protein expression in gastric cancer drug resistance. World J Gastroenterol 2011; 17:3986-93;PMID:22046086; http://dx.doi.org/ 10.3748/wjg.v17.i35.3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou L, Shang Y, Liu C, Li J, Hu H, Liang C, Han Y, Zhang W, Liang J, Wu K. Overexpression of PrPc, combined with MGr1-Ag/37LRP, is predictive of poor prognosis in gastric cancer. Int J Cancer 2014; 135:2329-37;PMID:24706505; http://dx.doi.org/ 10.1002/ijc.28883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Wit M, Jimenez CR, Carvalho B, Belien JAM, Delis-van Diemen PM, Mongera S, Piersma SR, Vikas M, Navani S, Ponten F, et al.. Cell surface proteomics identifies glucose transporter type 1 and prion protein as candidate biomarkers for colorectal adenoma-to-carcinoma progression. Gut 2012; 61:855-64;PMID:21890811; http://dx.doi.org/ 10.1136/gutjnl-2011-300511. [DOI] [PubMed] [Google Scholar]
- 10.Antonacopoulou AG, Grivas PD, Skarlas L, Kalofonos M, Scopa CD, Kalofonos HP. POLR2F, ATP6V0A1 and PRNP Expression in Colorectal Cancer: New Molecules with Prognostic Significance? Anticancer Res 2008; 1228:1221-7. [PubMed] [Google Scholar]
- 11.Diarra-Mehrpour M, Arrabal S, Jalil A, Pinson X, Gaudin C, Piétu G, Pitaval A, Ripoche H, Eloit M, Dormont D, et al.. Prion protein prevents human breast carcinoma cell line from tumor necrosis factor α-induced cell death. Cancer Res 2004; 64:719-27;PMID:14744790; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-1735. [DOI] [PubMed] [Google Scholar]
- 12.Meslin F, Conforti R, Mazouni C, Morel N, Tomasic G, Drusch F, Yacoub M, Sabourin JC, Grassi J, Delaloge S, et al.. Efficacy of adjuvant chemotherapy according to Prion protein expression in patients with estrogen receptor-negative breast cancer. Ann Oncol 2007; 18:1793-8;PMID:17872899; http://dx.doi.org/ 10.1093/annonc/mdm406. [DOI] [PubMed] [Google Scholar]
- 13.Roucou X, Giannopoulos PN, Zhang Y, Jodoin J, Goodyer CG, LeBlanc A. Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ 2005; 12:783-95;PMID:15846375; http://dx.doi.org/ 10.1038/sj.cdd.4401629. [DOI] [PubMed] [Google Scholar]
- 14.Li Q-Q, Sun Y-P, Ruan C-P, Xu X-Y, Ge J-H, He J, Xu Z-D, Wang Q, Gao W-C. Cellular prion protein promotes glucose uptake through the Fyn-HIF-2α-Glut1 pathway to support colorectal cancer cell survival. Cancer Sci 2011; 102:400-6;PMID:21265952; http://dx.doi.org/ 10.1111/j.1349-7006.2010.01811.x. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Qian J, Wang F, Ma Z. Cellular prion protein accelerates colorectal cancer metastasis via the Fyn-SP1-SATB1 axis. Oncol Rep 2012; 28: 2029-34;PMID:22972305. [DOI] [PubMed] [Google Scholar]
- 16.Barbieri G, Palumbo S, Gabrusiewicz K, Azzalin A, Marchesi N, Spedito A, Biggiogera M, Sbalchiero E, Mazzini G, Miracco C, et al.. Silencing of cellular prion protein (PrPC) expression by DNA-antisense oligonucleotides induces autophagy-dependent cell death in glioma cells. Autophagy 2011; 7:840-53;PMID:21478678; http://dx.doi.org/ 10.4161/auto.7.8.15615. [DOI] [PubMed] [Google Scholar]
- 17.Roffe M, Beraldo FH, Bester R, Nunziante M, Bach C, Mancini G, Gilch S, Vorberg I, Castilho BA, Martins VR, et al.. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc Natl Acad Sci U S A 2010; 107:13147-52;PMID:20615969; http://dx.doi.org/ 10.1073/pnas.1000784107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li QQ, Cao XX, Xu JD, Chen Q, Wang WJ, Tang F, Chen ZQ, Liu XP, Xu ZD. The role of P-glycoprotein/cellular prion protein interaction in multidrug-resistant breast cancer cells treated with paclitaxel. Cell Mol Life Sci 2009; 66:504-15;PMID:19099191; http://dx.doi.org/ 10.1007/s00018-008-8548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng Y, Tao L, Xu J, Li Q, Yu J, Jin Y, Chen Q, Xu Z, Zou Q, Liu X. CD44/cellular prion protein interact in multidrug resistant breast cancer cells and correlate with responses to neoadjuvant chemotherapy in breast cancer patients. Mol. Carcinog 2014; 53:686-97;http://dx.doi.org/ 10.1002/mc.22021. [DOI] [PubMed] [Google Scholar]
- 20.Li C, Xin W, Sy M-S. Binding of pro-prion to filamin A: by design or an unfortunate blunder. Oncogene 2010; 29:5329-45;PMID:20697352; http://dx.doi.org/ 10.1038/onc.2010.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C, Yu S, Nakamura F, Yin S, Xu J, Petrolla AA, Singh N, Tartakoff A, Abbott DW, Xin W, et al.. Binding of pro-prion to filamin A disrupts cytoskeleton and correlates with poor prognosis in pancreatic cancer. J Clin Invest 2009; 119:2725-36;PMID:19690385; http://dx.doi.org/ 10.1172/JCI39542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mbazima V, Da Costa Dias B, Omar A, Jovanovic K, Weiss SFT. Interactions between PrP(c) and other ligands with the 37-kDa/67-kDa laminin receptor. Front Biosci 2010; 15:1150-63;http://dx.doi.org/ 10.2741/3667. [DOI] [PubMed] [Google Scholar]
- 23.Omar A, Jovanovic K, Da Costa Dias B, Gonsalves D, Moodley K, Caveney R, Mbazima V, Weiss SFT. Patented biological approaches for the therapeutic modulation of the 37 kDa/67 kDa laminin receptor. Expert Opin Ther Pat 2011; 21:35-53;PMID:21110766; http://dx.doi.org/ 10.1517/13543776.2011.539203. [DOI] [PubMed] [Google Scholar]
- 24.Martins VR, Graner E, Garcia-Abreu J, de Souza SJ, Mercadante AF, Veiga SS, Zanata SM, Neto VM, Brentani RR. Complementary hydropathy identifies a cellular prion protein receptor. Nat Med 1997; 3:1376-82;PMID:9396608; http://dx.doi.org/ 10.1038/nm1297-1376. [DOI] [PubMed] [Google Scholar]
- 25.Zanata SM, Lopes MH, Mercadante AF, Hajj GNM, Chiarini LB, Nomizo R, Freitas ARO, Cabral ALB, Lee KS, Juliano MA, et al.. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J 2002; 21:3307-16;PMID:12093732; http://dx.doi.org/ 10.1093/emboj/cdf325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baindur-Hudson S, Edkins A, Blatch G. Hsp70/Hsp90 organising protein (hop): beyond interactions with chaperones and prion proteins. Subcell Biochem 2015; 78:69-90;PMID:25487016; http://dx.doi.org/ 10.1007/978-3-319-11731-7_3. [DOI] [PubMed] [Google Scholar]
- 27.Hajj GNM, Arantes CP, Dias MVS, Roffé M, Costa-Silva B, Lopes MH, Porto-Carreiro I, Rabachini T, Lima FR, Beraldo FH, et al.. The unconventional secretion of stress-inducible protein 1 by a heterogeneous population of extracellular vesicles. Cell Mol Life Sci 2013; 70:3211-27;PMID:23543276; http://dx.doi.org/ 10.1007/s00018-013-1328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopes MH, Santos TG, Rodrigues BR, Queiroz-Hazarbassanov N, Cunha IW, Wasilewska-Sampaio AP, Costa-Silva B, Marchi FA, Bleggi-Torres LF, Sanematsu PI, et al.. Disruption of prion protein-HOP engagement impairs glioblastoma growth and cognitive decline and improves overall survival. Oncogene 2014; PMID:25151961; doi: 10.1038/onc.2014.261 [DOI] [PubMed] [Google Scholar]
- 29.Coitinho AS, Lopes MH, Hajj GNM, Rossato JI, Freitas AR, Castro CC, Cammarota M, Brentani RR, Izquierdo I, Martins VR. Short-term memory formation and long-term memory consolidation are enhanced by cellular prion association to stress-inducible protein 1. Neurobiol Dis 2007; 26:282-90;PMID:17329112; http://dx.doi.org/ 10.1016/j.nbd.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 30.Chiarini LB, Freitas ARO, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals. EMBO J 2002; 21:3317-26;PMID:12093733; http://dx.doi.org/ 10.1093/emboj/cdf324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serrano Lopez DR, Lalatsa A. Peptide pills for brain diseases? Reality and future perspectives. Ther Deliv 2013; 4:479-501;PMID:23557289; http://dx.doi.org/ 10.4155/tde.13.5. [DOI] [PubMed] [Google Scholar]
- 32.Craik DJ, Fairlie DP, Liras S, Price D. The Future of Peptide-based Drugs. Chem Biol Drug Des 2013; 81:136-47;PMID:23253135; http://dx.doi.org/ 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 33.Sonati T, Reimann RR, Falsig J, Baral PK, O'Connor T, Hornemann S, Yaganoglu S, Li B, Herrmann US, Wieland B, et al.. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013; 501:102-6;PMID:23903654; http://dx.doi.org/ 10.1038/nature12402. [DOI] [PubMed] [Google Scholar]
- 34.Beraldo FH, Soares IN, Goncalves DF, Fan J, Thomas AA, Santos TG, Mohammad AH, Roffé M, Calder MD, Nikolova S, et al.. Stress-inducible phosphoprotein 1 has unique cochaperone activity during development and regulates cellular response to ischemia via the prion protein. FASEB J 2013; 27:3594-607;PMID:23729591; http://dx.doi.org/ 10.1096/fj.13-232280. [DOI] [PubMed] [Google Scholar]
- 35.Braunschweig D, Krakowiak P, Duncanson P, Boyce R, Hansen RL, Ashwood P, Hertz-Picciotto I, Pessah IN, Van de Water J. Autism-specific maternal autoantibodies recognize critical proteins in developing brain. Transl Psychiatry 2013; 3:e277;PMID:23838888; http://dx.doi.org/ 10.1038/tp.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer 2013; 13:727-38;PMID:24060864; http://dx.doi.org/ 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 37.Martin-Lannerée S, Hirsch T, Hernandez-Rapp J, Halliez S, Vilotte J, Launay J, Mouillet-Richard S. PrP(C) from stem cells to cancerPrP(C) from stem cells to cancer. Front cell Dev Biol 2014; 2:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lopes MH, Santos TG. Prion potency in stem cells biology. Prion 2012; 6:142-6;PMID:22437733; http://dx.doi.org/ 10.4161/pri.19035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du L, Rao G, Wang H, Li B, Tian W, Cui J, He L, Laffin B, Tian X, Hao C, et al.. CD44-positive cancer stem cells expressing cellular prion protein contribute to metastatic capacity in colorectal cancer. Cancer Res 2013; 73:2682-94;PMID:23418321; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-3759. [DOI] [PubMed] [Google Scholar]
- 40.Santos TG, Silva IR, Costa-Silva B, Lepique AP, Martins VR, Lopes MH. Enhanced neural progenitor/stem cells self-renewal via the interaction of stress-inducible protein 1 with the prion protein. Stem Cells 2011; 29:1126-36;PMID:21608082; http://dx.doi.org/ 10.1002/stem.664. [DOI] [PubMed] [Google Scholar]
- 41.Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015; 347:78-81;PMID:25554788; http://dx.doi.org/ 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]