

The crystal structures of two constructs of RC1339/APRc from R. conorii, consisting of either residues 105–231 or 110–231 followed by a His tag, have been determined in three different crystal forms. The monomer of APRc assumes a retropepsin-like fold, but the dimer observed in all crystal forms is entirely different from the canonical dimer of retropepsins.
Keywords: aspartic proteases, enzyme evolution, bacterial proteases, dimeric proteins
Abstract
The crystal structures of two constructs of RC1339/APRc from Rickettsia conorii, consisting of either residues 105–231 or 110–231 followed by a His tag, have been determined in three different crystal forms. As predicted, the fold of a monomer of APRc resembles one-half of the mandatory homodimer of retroviral pepsin-like aspartic proteases (retropepsins), but the quaternary structure of the dimer of APRc differs from that of the canonical retropepsins. The observed dimer is most likely an artifact of the expression and/or crystallization conditions since it cannot support the previously reported enzymatic activity of this bacterial aspartic protease. However, the fold of the core of each monomer is very closely related to the fold of retropepsins from a variety of retroviruses and to a single domain of pepsin-like eukaryotic enzymes, and may represent a putative common ancestor of monomeric and dimeric aspartic proteases.
1. Introduction
Aspartic proteases of MEROPS (Rawlings & Barrett, 2000 ▸) families A1 (pepsin-like) and A2 (retropepsins) are by far the best characterized and most characteristic members of this class of enzymes, mainly owing to their critical roles in different physiological and pathophysiological conditions and to their involvement in the life cycle of various pathogenic microorganisms and viruses (Dunn et al., 2002 ▸; Dunn, 2002 ▸; Wlodawer & Gustchina, 2000 ▸). Several aspartic proteases have been explored as therapeutic targets, with retropepsins constituting the most successful example owing to the design of more than ten drugs that are used clinically for the treatment of HIV/AIDS (Dash et al., 2003 ▸; Dunn et al., 2002 ▸). Pepsins and retropepsins share the same catalytic apparatus dependent on the presence of two aspartates located in highly conserved sequence motifs that form the structural feature known as the ψ loop (Rawlings & Barrett, 2013 ▸; Wlodawer et al., 2013 ▸). The members of the two families exhibit similar protein folds, although their sequence similarity is low beyond the active-site region. Indeed, pepsin-like proteases are bilobal, with each domain sharing similar secondary-structure elements and contributing one catalytic aspartate to the active site located in the cleft formed between the two domains (Dunn, 2002 ▸). On the other hand, retropepsins are homodimers and the single active site is formed at the dimer interface by conserved residues originating from each monomer, which in turn share a similar topology with the N-terminal domain of pepsin-like enzymes (Dunn et al., 2002 ▸; Wlodawer & Gustchina, 2000 ▸). This common secondary-structure template among domains/monomers supports the view that pepsin-like aspartic proteases and retropepsins are evolutionarily related and that the former may have arisen by gene duplication and fusion of an ancestral form of the latter (Rao et al., 1991 ▸; Tang et al., 1978 ▸).
The nature of this primordial single-lobed aspartic protease has been a matter of debate over the years, mostly owing to the lack of compelling evidence for the presence of both family A1 and A2 members in prokaryotes (Cascella et al., 2005 ▸; Rao et al., 1991 ▸; Rawlings & Bateman, 2009 ▸). However, this argument was first challenged by the discovery of pepsin homologs in a restricted number of bacteria (Rawlings & Bateman, 2009 ▸) and the observation that at least one of these genes encodes an active enzyme (Simões et al., 2011 ▸). More recently, Cruz and coworkers reported the identification of a gene coding for a membrane-embedded, single-lobed aspartic protease that is highly conserved in the genomes of 55 species of Rickettsia (Cruz et al., 2014 ▸). Using R. conorii gene homolog rc1339 as a working model, the authors provided evidence that the encoded product, named APRc, indeed shares several enzymatic properties with viral retropepsins, and it was assigned to MEROPS family A32, which comprises retropepsin-like enzymes found in bacteria, whereas families A28 (type peptidase DNA-damage inducible protein 1 from Saccharomyces cerevisiae) and A33 (type peptidase skin aspartic protease from Mus musculus) contain those identified in eukaryotes (Rawlings & Barrett, 2013 ▸). The common properties include autolytic activity, optimum pH, specificity preferences and inhibition by HIV-1 protease inhibitors. Moreover, APRc was shown to be expressed in two pathogenic species of Rickettsia and to be integrated into the outer membrane of both species. In vitro processing of two conserved autotransporter adhesin/invasion proteins Sca5/OmpB and Sca0/OmpA by APRc suggests its participation in a proteolytic pathway relevant to the rickettsial life cycle and makes it a potential target for the design of novel antibiotics targeting rickettsia (Cruz et al., 2014 ▸).
Despite the rather low sequence similarity to viral retropepsins, the resemblance of enzymatic features and predicted conservation of secondary structure suggested that APRc might indeed represent a more primordial form of these proteases (Cruz et al., 2014 ▸). To further validate this hypothesis and to provide an understanding of the evolutionary relationship with viral and eukaryotic retropepsins, we report here the elucidation of the structure of the soluble domain of APRc. Although the topology of the dimer observed in the crystals appears to reflect a crystal-packing artifact and not the biologically relevant interface, our results unequivocally demonstrate that a monomer of APRc follows the canonical fold observed in all retropepsins, either of viral or eukaryotic origin, for which structural data are available (Dunn et al., 2002 ▸; Li, DiMaio et al., 2011 ▸; Sirkis et al., 2006 ▸).
2. Materials and methods
2.1. Protein expression and purification
Expression constructs bearing the activation product APRc105–231 (a construct coding amino acids 105–231) previously identified in Cruz et al. (2014 ▸) and a shorter version of this soluble domain (APRc110–231; a construct coding amino acids 110–231) were both generated using the construct pET-APRc1–231-His as a template (Cruz et al., 2014 ▸). Sequences were amplified with a forward primer containing an NdeI restriction site (5′-CATATGTATAAATGGAGTACCGAAGTT-3′ for pET-APRc105–231-His and 5′-CATATGGAAGTTGGCGAAATTATCATTGC-3′ for pET-APRc110–231-His) and the same reverse primer containing an NotI restriction site (5′-CTCGAGATAATTCAGAATCAGCAGATCTTT-3′), and amplification products were cloned into pGEM-T Easy (Promega). The inserts were subsequently digested with NdeI/NotI and subcloned into pET-23a expression vector (Invitrogen) in frame with a C-terminal His tag (constructs pET-APRc105–231-His and pET-APRc110–231-His). Both constructs were confirmed by DNA sequencing. Expression and purification of both forms of soluble APRc were performed essentially as described in Cruz et al. (2014 ▸) with minor modifications. Briefly, both expression vectors were transformed into Escherichia coli BL21(DE3) strain and gene expression was induced with 0.1 mM IPTG for 3 h at 30°C when the cultures reached an OD600 of 0.7. After expression, the cells were harvested by centrifugation (9000g, 20 min at 4°C) and the pellets were resuspended in 20 mM phosphate buffer pH 7.5, 0.5 M NaCl, 0.01 M imidazole (buffer A). For pET-APRc105–231-His, lysozyme was added (100 µg ml−1) and the cells were lysed by freezing (−20°C) and thawing and were subsequently incubated with DNase (1 µg ml−1) and MgCl2 (5 mM) for 1 h at 4°C. For pET-APRc110–231-His, the cell suspension was lysed by three passages through an EmulsiFlex (Avestin; 69 MPa). The total lysates were then centrifuged at 27 216g for 20 min at 4°C and the resulting supernatant was filtered (0.2 µm) before loading onto a HisTrap HP 5 ml column (GE Healthcare Life Sciences) pre-equilibrated in buffer A. Protein elution was performed with a stepwise gradient of imidazole concentration (0.05, 0.1 and 0.5 M) in the same buffer. Fractions containing the protein of interest (from the 0.1 M imidazole step) were pooled and the buffer was exchanged by an overnight dialysis step into either 20 mM phosphate buffer pH 7.5 (APRc105–231-His) or 20 mM HEPES buffer pH 7.4 (APRc110–231-His). Dialyzed protein fractions were further purified by ion-exchange chromatography on a Mono S 5/50 column (GE Healthcare Life Sciences) and protein elution was carried out with a linear gradient of NaCl (0–1 M) in 20 mM phosphate buffer pH 7.5 (APRc105–231-His) or 20 mM HEPES buffer pH 7.4 (APRc110–231-His). For both recombinant forms, the eluted protein was then loaded onto a Superdex 200 10/300 GL size-exclusion chromatography column (GE Healthcare Life Sciences) previously equilibrated with 20 mM HEPES pH 7.4 containing 0.1 M NaCl. Expression of selenomethionine-labeled (SeMet) APRc105–231-His was performed in M9 medium instead of LB. 1 l of M9 medium contains 6.8 g Na2HPO4, 3 g KH2PO4, 0.59 g NaCl and 1 g NH4Cl. Before use, 10 ml 20% glucose, 2 ml 1 M MgSO4, 0.1 ml 1 M CaCl2, 0.1 ml 0.5% thiamine (vitamin B1), 20 ml of a 19-amino-acid mixture (2 mg ml−1 of each except for methionine) and 0.2 ml 1 M zinc acetate were added to the M9 medium. After the cells had grown at 37°C to an OD600 of 0.4–0.6, 100 mg each of threonine, lysine–HCl, phenylalanine and cysteine, 50 mg each of leucine, isoleucine, valine and tryptophan and 120 mg dl-selenomethionine were added to the medium. The temperature was shifted to 25°C, and after 30 min 0.2 M IPTG was added to induce protein expression for 18 h. The protein was purified according to the protocol used for the wild-type enzyme. Both constructs were extended at their C-termini by the sequence Ala-Ala-Ala-Leu-Glu-His6, the additional residues representing a cloning artifact at the coding sequence–vector junction (NotI restriction site) and a His tag.
2.2. Crystallization
Crystals of both wild-type APRc105–231-His and its SeMet variant were grown in hanging drops set up by hand in Linbro crystallization plates. The well solution consisted of 24% PEG 4000 and 0.2 M Li2SO4 in 0.1 M sodium citrate buffer pH 5.8. Samples of wild-type APRc105–231-His were concentrated to 10 mg ml−1 in 20 mM HEPES buffer pH 7.4 also containing 0.2 M NaCl and 1 mM pepstatin A, an aspartic protease inhibitor. The concentration of SeMet APRc105–231-His was 4–5 mg ml−1 in the same buffer. Each drop consisted of 2 µl protein solution and 2 µl well solution and was equilibrated against 300 µl well solution. The same method was used to grow crystals of APRc110–231-His under a different condition. In this case, 6 mg ml−1 of sample in 20 mM HEPES pH 7.4 with 0.1 M NaCl was mixed with well solution consisting of 1.2 M dl-malic acid pH 7.0 with 2% 1,2-propanol in a 1:1(v:v) ratio.
2.3. Determination and refinement of the crystal structure
The structure of the SeMet variant of APRc105–231-His was solved using multiple-wavelength anomalous scattering (MAD) with diffraction data collected on beamline 22-ID of SER-CAT, Advanced Photon Source, Argonne, Illinois, USA. The MAD data were collected at the peak, edge and the low-energy side of the absorption edge (Table 1 ▸). Native data were collected to a higher resolution at a wavelength of 1.000 Å and were used for subsequent refinement. Diffraction data were integrated with DENZO and scaled with SCALEPACK, which are parts of the HKL-2000 package (Otwinowski & Minor, 1997 ▸). Each molecule of APRc105–231-His contained three methionine residues; based on the volume and symmetry of the unit cell it was expected that each asymmetric unit would contain 12 Se atoms. The heavy-atom substructure was determined with SOLVE (Terwilliger, 2003 ▸), with ten Se atoms being located. Refinement resulted in a figure of merit of 0.47 and a Z-score of 37.24. Buccaneer (Cowtan, 2006 ▸) was used for subsequent phasing and for initial model building. As predicted, four molecules could be located in the asymmetric unit, although a number of residues at both termini and in several loops could not be located in the automated procedure. These missing residues were fitted in cycles of rebuilding with Coot (Emsley & Cowtan, 2004 ▸) and refinement with REFMAC5 (Murshudov et al., 2011 ▸).
Table 1. Data collection and structure refinement.
Values in parentheses are for the outer resolution shell.
| APRc105231-His | |||||
|---|---|---|---|---|---|
| SeMet, peak | SeMet, edge | SeMet, low energy | Wild type | APRc110231-His | |
| Data collection | |||||
| Wavelength | 0.97923 | 0.97942 | 0.98254 | 1.0000 | 1.0000 |
| Space group | P3221 | P3221 | P3221 | P212121 | I4122 |
| Molecules in asymmetric unit | 4 | 4 | 4 | 4 | 2 |
| Unit-cell parameters | |||||
| a () | 101.99 | 102.03 | 101.99 | 50.10 | 105.29 |
| b () | 101.99 | 102.03 | 101.99 | 94.15 | 105.29 |
| c () | 127.10 | 127.14 | 127.06 | 118.64 | 91.00 |
| = () | 90 | 90 | 90 | 90 | 90 |
| () | 120 | 120 | 120 | 90 | 90 |
| Resolution () | 50.02.50 | 50.02.50 | 50.02.40 | 50.02.00 | 50.02.59 |
| R merge (%) | 6.8 (31.3) | 6.7 (56.8) | 5.4 (41.9) | 5.8 (51.0) | 6.5 (45.7) |
| No. of reflections (measured/unique) | 139753/25568 | 150123/25984 | 150258/26600 | 271435/36737 | 34463/7659 |
| I/(I) | 22.42 (3.16) | 19.92 (1.5) | 31.27 (2.67) | 30.56 (1.74) | 21.14 (3.16) |
| Completeness (%) | 94.7 (58.7) | 95.9 (60.3) | 87.6 (33.6) | 94.9 (64.0) | 92.5 (73.1) |
| Multiplicity | 5.5 (3.3) | 5.8 (3.4) | 5.6 (3.4) | 7.4 (3.5) | 4.5 (3.8) |
| Refinement | |||||
| Resolution () | 47.372.45 | 40.002.00 | 41.852.59 | ||
| No. of reflections (refinement/R free) | 26039/1386 | 35542/1140 | 7304/354 | ||
| R/R free (%) | 19.89/26.22 | 19.86/25.45 | 22.74/29.77 | ||
| No. of atoms | |||||
| Protein | 3892 | 4080 | 1832 | ||
| Ligand/ion | 15 | 1 | |||
| Water | 128 | 287 | 5 | ||
| R.m.s. deviations from ideal | |||||
| Bond lengths () | 0.016 | 0.015 | 0.011 | ||
| Bond angles () | 1.819 | 1.725 | 1.707 | ||
| Ramachandran plot (%) | |||||
| Favored | 88.9 | 91 | 77.7 | ||
| Allowed | 10.4 | 8.6 | 21.9 | ||
| Outliers | 0.7 | 0.4 | 0.5 | ||
| PDB code | 5c9b | 5c9b | 5c9d | ||
The structure of wild-type APRc105–231-His was determined by molecular replacement using Phaser (McCoy, 2007 ▸). Monomer A from the structure of SeMet APRc105–231-His was used as the search model. Four molecules were located with a Z-score of 33.1, and REFMAC5 was used for subsequent refinement. Monomer B of native APRc105–231-His with the six N-terminal residues removed was used to search for the shorter form APRc110–231-His using Phaser. Both monomers were found with a Z-score of 29.5. The solution was rebuilt with Coot and refined with REFMAC5. The results of refinement for all three structures are listed in Table 1 ▸.
3. Results
Three different crystal forms of the soluble domain of APRc were investigated in this study. The longer construct (APRc105–231-His; residues 105–231 followed by a His tag) was crystallized in two different space groups: trigonal P3221 for the SeMet variant and orthorhombic P212121 for the wild-type protein. Four protein molecules occupied the asymmetric unit in both crystal forms. The shorter construct APRc110–231-His (residues 110–231 followed by a His tag) crystallized in the tetragonal space group I4122 with two molecules in the asymmetric unit. In the following, the abbreviated term ‘APRc’ will refer to the wild-type longer construct only.
3.1. Structure solution and refinement
Extensive efforts to solve the structure of wild-type APRc105–231-His in the orthorhombic crystal form by molecular replacement did not succeed. The models used for molecular replacement included all retroviral proteases (Dunn et al., 2002 ▸), both as monomers and as dimers, as well as Saccharomyces cerevisiae Ddi1 (Sirkis et al., 2006 ▸), a protein domain that exhibits a retropepsin-like fold while not being enzymatically active. No detectable signal was present with any of these models, necessitating the preparation of a variant of APRc in which its three methionine residues (including the N-terminal methionine, which was an expression artifact) were replaced by selenomethionine (SeMet). The resulting trigonal crystals differed from those of wild-type APRc. The Se substructure, as determined by three-wavelength MAD (Table 1 ▸), yielded unambiguous positions for ten of the expected 12 Se atoms. The resulting phases enabled automated model building, which was followed by refinement and manual adjustment of the model, which consisted of four crystallographically independent molecules in the asymmetric unit.
The coordinates of the SeMet variant provided a starting point for molecular-replacement solution of the wild-type APRc in the orthorhombic crystals. Search models that consisted of only a monomer of APRc were successful in solving the structure, whereas models consisting of a dimer were not, indicating the possibility that the dimers were different. We found later that this was indeed the case (see below).
The shorter construct, APRc110–231-His, starting at residue 110 of the native sequence rather than at residue 105, yielded tetragonal crystals with only two molecules in the asymmetric unit. Its structure was solved by molecular replacement using monomer B of the wild-type APRc105–231-His structure as a starting model, whereas it was again not possible to solve the structure using a dimer as the model.
3.2. The fold of APRc
As predicted, the fold of the APRc monomer (both in the longer and the shorter form) resembles the canonical fold of retropepsins, which corresponds to the structural template for the family of aspartic proteases (Andreeva, 1991 ▸). As defined by the template, a monomer of retropepsin is formed by the duplication of four secondary-structure elements: a hairpin, a wide loop containing the catalytic aspartate, an α-helix and a second hairpin. All of the secondary-structure elements listed above are present in a monomer of APRc.
Overall, a monomer of APRc (Fig. 1 ▸) contains ten β-strands and two α-helices; the extent of the secondary-structure elements is well preserved in all ten crystallographically independent molecules of the protein, although minor differences in the locations of the starting and ending amino acids are noticed. Since the orthorhombic crystals yielded the highest resolution data and the chain of molecule A in this crystal form is complete, it is used for analysis of the secondary structure. A long N-terminal β-strand starts at Tyr105 of the longer construct of APRc (the first residue after the initiator methionine, with the latter being an expression artifact) and continues through Ala117. This strand starts at Glu110 of the shorter construct, again just after the N-terminal methionine, and also continues through Ala117. The following two strands, Tyr125–Val130 and Val133–Val139, form a β-hairpin. A short strand Ile146–Leu148 forms a central strand of a four-stranded β-sheet typical of all pepsin-like proteases. An α-helix that extends from Lys150 through Leu156 is followed by another β-hairpin consisting of the strands Arg167–Thr171 and Gly174–Val187. The tip of the hairpin loop is disordered in most of the ten APRc molecules. The last part of the latter strand forms another β-hairpin that includes the strand Glu191–Gly200, and both of these strands are part of the abovementioned four-stranded sheet. The next strand Ser207–Gly210 completes the sheet and is followed by an α-helix Met211–Glu215. The last β-hairpin is formed by strands Gly219–Asp223 and Leu226–Ala234 and forms the other strands of a six-stranded β-sheet that creates the dimer interface. It must be pointed out that the C-terminal strand extends past Tyr231, the last authentic residue present in the sequence of APRc, and includes part of the linker region leading to the disordered C-terminal His tag, which is not visible in the electron-density map.
Figure 1.

A cartoon-style stereoview showing the monomer of APRc in rainbow colors (changing smoothly from blue at the N-terminus to red at the C-terminus). Secondary-structure elements are indicated by ribbons and selected amino-acid side chains are shown in stick representation, with some of them labeled.
The fold of a monomer is similar in all three crystal forms, with the r.m.s.d. between the Cα atoms of molecules A in the orthorhombic and trigonal crystal forms being 1.23 Å for 125 atom pairs, and that between the 114 pairs of Cα atoms of molecules A in the orthorhombic and tetragonal cells being 0.86 Å. The largest deviations were in the poorly defined loop 168–177 and in the stretch 199–204.
Whereas the structure of the APRc monomer is closely related to those of the other retropepsins, the dimerization mode seen in the APRc crystals is completely different (Fig. 2 ▸). A dimer observed in all three crystal forms is made up of the directly interacting N-terminal strands of two molecules, extended to a six-stranded β-sheet by the C-terminal β-hairpin of each molecule. Whereas the topology of the dimer is the same in all three crystal forms, the angle between the two molecules is considerably different (Fig. 3 ▸). The differences are primarily owing to a rotation of the molecules around an axis parallel to and in the center of the intermolecular β-sheet.
Figure 2.

A superposition of monomers A of APRc (green) and HIV-1 PR (magenta), showing the different locations of the monomers B in their respective dimers (brown and magenta). The positions of the active-site aspartates (Asp140) in both monomers of APRc are shown as blue sticks and surfaces.
Figure 3.

A superposition of the dimers of APRc found in the three crystal forms of the protein. Monomers A were superimposed with SSM (Krissinel & Henrick, 2004 ▸) and the shift in the positions of monomers B emphasizes the differences in the angles between the two molecules forming the dimers. APRc105–231-His is shown in green, its SeMet variant is shown in blue and APRc110–231-His is shown in magenta.
3.3. A comparison with retropepsins
The structure of monomer A of the orthorhombic form of APRc was compared with the structures of several retropepsins using the program SSM (Krissinel & Henrick, 2004 ▸). For this purpose, we selected the structure of HIV-1 PR (PDB entry 5hvp; Fitzgerald et al., 1990 ▸), as well as the structures of Feline immunodeficiency virus (FIV) PR (PDB entry 1fiv; Wlodawer et al., 1995 ▸), Equine infectious anemia virus (EIAV) PR (PDB entry 2fmb; Kervinen et al., 1998 ▸) and Xenotropic murine leukemia virus-related virus (XMRV) PR (PDB entry 3nr6; Li, DiMaio et al., 2011 ▸). We also used for comparisons the structures of the central domain of yeast Ddi1 (PDB entry 2i1a; Sirkis et al., 2006 ▸) and the monomeric retropepsin encoded by Mason–Pfizer monkey virus (M-PMV; PDB entry 3sqf; Khatib et al., 2011 ▸).
The structures of XMRV PR and Ddi1 have already established the existence of a distinctive group of aspartic proteases with the fold of retropepsins but with structural features of the dimer interface resembling those of pepsin-like enzymes. The presence of a β-hairpin at the C-terminus of APRc clearly places this protein in the same group as XMRV PR and Ddi1. All three C-terminal hairpins are superimposed very well in the structures of monomers, while the N-termini adopt a different conformation in each structure (Fig. 4 ▸ a). Superposition of the APRc monomer with the monomers of HIV, EIAV and FIV PRs reveals similar differences in the mutual orientation of the N- and C-termini of APRc and the other three retropepsins, as previously described for XMRV PR (Li, DiMaio et al., 2011 ▸; Fig. 4 ▸ b). The loop containing residues 159–166 assumes a different conformation in APRc than in all of the other structures that are being compared (Fig. 4 ▸).
Figure 4.

Superposition of the monomers of APRc with several retropepsins. (a) APRc is shown in green with the N- and C-termini in cyan and red, respectively, XMRV PR in orange with the N- and C-termini in blue and yellow, respectively, and Ddi1 in raspberry with the N- and C-termini in blue and brown, respectively. The second α-helix in the monomers of APRc and Ddi1 is shown in gray and pink, respectively. (b) APRc is shown in green with the N- and C-termini in cyan and red, respectively, HIV-1 PR is in magenta, EIAV is in yellow and FIV PR is in purple. The N- and C-termini of the latter three proteins are shown in blue and pink, respectively. The second α-helix in the monomers of APRc and EIAV PR is shown in gray and orange, respectively.
Similarities and differences between APRc and selected retropepsins can be described in more detail by using pairwise comparisons. A molecule of HIV-1 PR, the most widely studied retropepsin, consists of 99 amino-acid residues, 90 of which can be aligned with their counterparts in APRc with an r.m.s.d. of 2.14 Å (based on Cα positions) despite a very low sequence identity of only 16.7%. The APRc monomer contains two full helices compared with one in HIV PR. Since helix 150–156 in APRc has no equivalent in HIV-1 PR, these residues, as well as their continuation through Thr166, did not align well (Fig. 5 ▸ a). The second APRc helix (211–215) aligned quite well with its counterpart in HIV-1 PR. The most profound difference is the presence of the C-terminal hairpin in APRc, in contrast to a single C-terminal β-strand in HIV-1 PR. The penultimate β-strand of APRc and the beginning of the N-terminal strand of APRc (starting at Ile115) topologically resemble the positions of the respective strands in HIV-1 PR, although their directionality is different (Fig. 4 ▸ b). The dimer of APRc is very different from its counterpart in HIV-1 PR (Fig. 2 ▸).
Figure 5.


A superposition of molecules A of APRc (green, with the N-and C-termini in cyan and red, respectively, and the second α-helix in gray) with other retropepsins. (a) HIV-1 PR in magenta, with the N- and C-termini in blue and pink, respectively; (b) EIAV PR in yellow, with the N- and C-termini in blue and pink, respectively, and the second α-helix in orange; (c) XMRV PR in orange, with the N- and C-termini in blue and pink, respectively; and (d) Ddi1 in wheat, with the N- and C-termini in blue and pink, respectively, and the second α-helix in brown.
The slightly larger FIV PR consists of 116 residues (with the three N-terminal residues not modeled), 84 of which align with their counterparts in APRc with an r.m.s.d. of 1.55 Å. Again, the largest deviations involve residues 150–166, as well as the termini, although the sequence identity is higher at 27.4%. The FIV PR monomer contains one helical turn and one full helix at the structural positions corresponding to the structural template (Fig. 4 ▸ b). The other interesting feature is the presence of a bulge in the loop containing residues 200–204 in APRc, which is topologically equivalent to the relevant structural fragment in FIV PR (residues 92–96; Fig. 6 ▸; Wlodawer et al., 1995 ▸). As was suggested by these authors, such an insertion in close proximity to the flap region can modulate the dynamic properties of the flap and the specificity of APRc.
Figure 6.

A bulge containing residues 200–204 in APRc (green) superimposed on the homologous segments in FIV PR (residues 92–97, purple) and HIV PR (residues 78–81, orange). The flaps in APRc and FIV PR are shown in the respective colors.
Similar to EIAV PR (104 amino acids), which differs from the other two retropepsins in having two full α-helices in a monomer and thus completing the set of secondary-structure elements required by the structural template of the family of aspartic proteases, APRc also has two helices (Fig. 5 ▸ b), as opposed to one full helix in HIV PR (Fig. 5 ▸ a) and one helix and a helical turn in FIV PR. The core of its monomer superimposes well on the core of APRc, with an r.m.s.d. of 1.88 Å for 94 pairs of Cα atoms. The two α-helices present in these structures superimposed particularly well (Fig. 5 ▸ b), whereas the largest differences were for residues 157–166 of APRc (corresponding to 42–50 in EIAV PR). The extent of sequence identity is even lower at only 14% and is limited to the active site and its vicinity.
XMRV PR is longer (124 amino acids) and differs in some topological details from the other retropepsins, especially regarding the strands forming the dimer interface. The Cα atoms superimpose with an r.m.s.d. of 1.94 Å for 92 pairs (sequence identity 20.6%). Similar to FIV PR, only one full helix and a helical turn are present in the monomer of XMRV PR (Fig. 5 ▸ c). The N-termini in these two proteases (the residues preceding His123 in APRc and Glu15 in XMRV PR) follow a completely different path. On the other hand, the C-terminal β-hairpins superimpose very well, although both of the β-strands comprising a hairpin in XMRV PR are shorter than in APRc. Residues 116–124 in XMRV PR following the second β-strand in the hairpin adopt a different path from their counterparts in APRc (however, some of the corresponding residues in the latter protein form the extension of the C-terminal strand but represent a cloning artifact; Figs. 5 ▸ c and 4 ▸ a).
The retropepsin-like domain of Ddi1 can be superimposed on APRc with an r.m.s.d. of 2.32 Å for 91 aligned Cα atoms, despite higher sequence identity (27.5%). As in the case of EIAV PR, the two helices present in both Ddi1 and APRc superimpose well, whereas the much longer C-terminal sequence in Ddi1 forms a three-stranded β-sheet in which the third strand occupies a topologically equivalent position to the N-terminal strand of APRc (Fig. 5 ▸ d). The wide loop leading to the flap in Ddi1 includes a helical turn (residues 238–242) that is structurally equivalent to the helical turn comprising residues 160–162 in APRc. Although this loop in Ddi1 is topologically similar to its counterparts in other retropepsins, it differs from the unique conformation of this loop found in APRc.
The monomeric M-PMV PR can be superimposed on APRc with an r.m.s.d. of 1.80 Å for 86 Cα atoms. Both the N- and C-termini of M-PMV PR are missing in the crystal structure; therefore, direct comparison of the dimerization interfaces between APRc and this enzyme is not possible. M-PMV PR has only one full helix and a helical turn in a monomer, similar to the FIV and XMRV PRs.
4. Discussion
As predicted based mainly on the presence of the canonical DTG motif in the primary structure of APRc, the fold of this protein does indeed follow the structural template of the family of aspartic proteases. In particular, the fold of APRc closely resembles the fold of retropepsins and, to a lesser extent, of a single domain of eukaryotic aspartic proteases. The overall fold of the monomer is preserved even though the identity of the sequences is below 28% compared with all retropepsins with known three-dimensional structure. Although both constructs of APRc that were investigated in this study formed topologically similar dimers, the observed quaternary structure cannot correspond to an active enzyme, since the catalytic aspartate residues of each monomer are not in close proximity, as is the case for pepsin-like aspartic proteases. The dimer interface is formed by both the N- and C-terminal β-strands, which, however, do not form an interdigitated sheet as seen in most retropepsin dimers (with the exception of XMRV PR and Ddi1). This unexpected dimer may be an artifact of the expression of the recombinant form of the enzyme with an extended C-terminus, a crystallization artifact, or a combination of both these factors.
Although the observed quaternary structure of APRc does not explain its mode of proven enzymatic activity (Cruz et al., 2014 ▸), the structure of the monomer provides comprehensive insights into the striking conservation of a folding domain among proteins with highly divergent primary amino-acid structures and with very diverse origins, consistent with the view that secondary and tertiary structures are generally conserved in evolution (Tang et al., 1978 ▸). Interestingly, the main differences between the monomers of APRc and retropepsins are accommodated by the lengths of the surface loops and by the lengths and conformations of the segments connecting these structural elements, which have also been previously established as the regions with highest variability among retropepsins (Dunn et al., 2002 ▸; Wlodawer et al., 1995 ▸). APRc shares the additional helix C1 (residues 150–156) with EIAV PR (Gustchina et al., 1996 ▸) and Ddi1 (Sirkis et al., 2006 ▸), which is usually either absent or substituted by a single helical turn in the corresponding segment in most retropepsins (Dunn et al., 2002 ▸). Two analogs of this helix are also found at appropriate positions in the N- and C-terminal domains of pepsin-type proteases (Gustchina et al., 1996 ▸). It is quite remarkable that this structural feature is shared between a prokaryotic, a retroviral (EIAV PR) and a eukaryotic (DdiI) retropepsin.
The region comprising an extra helix and the following wide loop is usually among the most variable regions in retropepsins. APRc is no exception to this rule. Furthermore, this wide loop (residues 157–166) is longer, resembling the FIV and EIAV PRs, in contrast to the shorter segments observed in XMRV PR (Li, Gustchina et al., 2011 ▸), and has a distinctively different conformation as well as topological juxtaposition compared with other retropepsins (Fig. 7 ▸). The unique structure of this segment has been referred to in several publications as an area which confers different immunological properties to various retropepsins (Gustchina & Weber, 1991 ▸; Wlodawer et al., 1995 ▸). This hypothesis was confirmed by recent studies of the antibody complexes of the allergen Bla g 2, which has the fold of a pepsin-like aspartic protease. These studies have shown that Lys61 in Bla g 2 (a structural homolog of Arg41 from the corresponding loop in HIV PR; Fig. 7 ▸) is the key residue for antigen–antibody recognition (Li et al., 2008 ▸).
Figure 7.
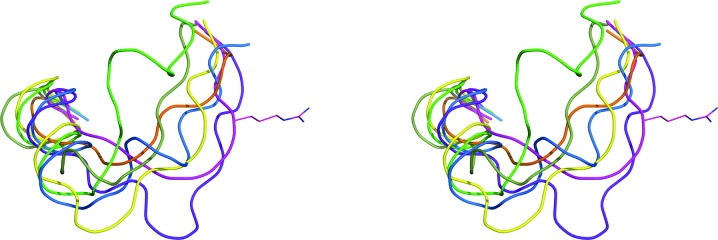
The most variable region in retropepsins. Superposition of APRc (green) with HIV-1 PR (magenta), FIV PR (purple), EIAV PR (yellow), Ddi1 (blue), M-PMV PR (green) and XMRV PR (orange). The side chain of Arg41 in HIV-1 PR is shown in stick representation.
Moreover, the region 200–203 in APRc shows some similarity to the bulge formed by residues 93–95 in FIV PR (Wlodawer et al., 1995 ▸), which is, to date, a feature uniquely found in the latter enzyme. On the other hand, the loop 187–191 is not as long as the equivalent region in FIV PR (75–85) (or in RSV PR), but has a similar size as those in EIAV, HIV-1 and XMRV PRs (Wlodawer et al., 1995 ▸).
The dimer of APRc observed in the crystals is entirely different from the canonical dimer of retroviral enzymes and is incapable of forming a proper active site (Fig. 2 ▸). The dimerization interface is formed by the N- and C-termini of two monomers, but they are not interdigitated as in the other retropepsins. Owing to the presence of a C-terminal β-hairpin in each monomer, upon dimerization they form a six-stranded β-sheet as in pepsin-like enzymes, with the difference that in the crystal structure of APRc two monomers interact via their N-termini (Fig. 3 ▸), whereas in pepsin the two domains interact via a C-terminal hairpin (Fig. 8 ▸). We speculate that an active dimer could possibly be created upon interaction with substrates or inhibitors, or that a more extensive search for new truncated constructs of this protein could yield different crystal forms of APRc that would contain a potentially active dimer.
Figure 8.

A comparison of the dimerization interface in APRc (N-terminus, cyan; C-terminus, red), XMRV PR (yellow) and Ddi1 (green) with pepsin (N-terminus, blue; C-terminus, pink).
A comparison of APRc with two other proteins that have C-terminal hairpins, XMRV PR and Ddi1, clearly indicates that APRc bears the closest resemblance of the three to the pepsin-like enzymes in its topological arrangement of both termini in the dimerization β-sheet, their orientation and their coplanarity with the pepsin interface (Fig. 8 ▸). This observation supports the concept that APRc may represent a putative common ancestor of monomeric and dimeric aspartic proteases, as well as hinting at the possible existence of a different evolutionary pathway for these enzymes (Li, Gustchina et al., 2011 ▸). Further studies are still required to elucidate the structural basis of the enzymatic activity of APRc.
Supplementary Material
PDB reference: APRc105–231-His, SeMet, 5c9b
PDB reference: APRc105–231-His, 5c9f
PDB reference: APRc110–231-His, 5c9d
Acknowledgments
We acknowledge the use of beamline 22-ID of the Southeast Regional Collaborative Access Team (SER-CAT), located at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38. This project has been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health under contract HHSN261200800001E and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This work was funded in part by Fundo Europeu de Desenvolvimento Regional (FEDER) Funds through the Operational Competitiveness Programme (COMPETE), by National Funds through the Fundação para a Ciência e a Tecnologia (FCT) under the project PTDC/SAU-MII/107942/2008 (to IS) and in part by NIH 1R21-AI111086 (to JM). PC is the recipient of a FCT PhD grant SFRH/BD/96769/2013. The authors would like to acknowledge grant PEst-C/SAU/LA0001/2013-2014. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products or organizations imply endorsement by the US Government.
References
- Andreeva, N. (1991). Structure and Function of the Aspartic Proteinases, edited by B. M. Dunn, pp. 559–572. New York: Plenum.
- Cascella, M., Micheletti, C., Rothlisberger, U. & Carloni, P. (2005). J. Am. Chem. Soc. 127, 3734–3742. [DOI] [PubMed]
- Cowtan, K. (2006). Acta Cryst. D62, 1002–1011. [DOI] [PubMed]
- Cruz, R., Huesgen, P., Riley, S. P., Wlodawer, A., Faro, C., Overall, C. M., Martinez, J. J. & Simões, I. (2014). PLoS Pathog. 10, e1004324. [DOI] [PMC free article] [PubMed]
- Dash, C., Kulkarni, A., Dunn, B. & Rao, M. (2003). Crit. Rev. Biochem. Mol. Biol. 38, 89–119. [DOI] [PubMed]
- Dunn, B. M. (2002). Chem. Rev. 102, 4431–4458. [DOI] [PubMed]
- Dunn, B. M., Goodenow, M. M., Gustchina, A. & Wlodawer, A. (2002). Genome Biol. 3, REVIEWS3006. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fitzgerald, P. M. D., McKeever, B. M., VanMiddlesworth, J. F., Springer, J. P., Heimbach, J. C., Leu, C.-T., Herber, W. K., Dixon, R. A. F. & Darke, P. L. (1990). J. Biol. Chem. 265, 14209–14219. [PubMed]
- Gustchina, A., Kervinen, J., Powell, D. J., Zdanov, A., Kay, J. & Wlodawer, A. (1996). Protein Sci. 5, 1453–1465. [DOI] [PMC free article] [PubMed]
- Gustchina, A. & Weber, I. T. (1991). Proteins, 10, 325–339. [DOI] [PubMed]
- Kervinen, J., Lubkowski, J., Zdanov, A., Bhatt, D., Dunn, B. M., Hui, K. Y., Powell, D. J., Kay, J., Wlodawer, A. & Gustchina, A. (1998). Protein Sci. 7, 2314–2323. [DOI] [PMC free article] [PubMed]
- Khatib, F., DiMaio, F., Cooper, S., Kazmierczyk, M., Gilski, M., Krzywda, S., Zabranska, H., Pichova, I., Thompson, J., Popović, Z., Jaskolski, M. & Baker, D. (2011). Nature Struct. Mol. Biol. 18, 1175–1177. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Li, M., DiMaio, F., Zhou, D., Gustchina, A., Lubkowski, J., Dauter, Z., Baker, D. & Wlodawer, A. (2011). Nature Struct. Mol. Biol. 18, 227–229. [DOI] [PMC free article] [PubMed]
- Li, M., Gustchina, A., Alexandratos, J., Wlodawer, A., Wünschmann, S., Kepley, C. L., Chapman, M. D. & Pomés, A. (2008). J. Biol. Chem. 283, 22806–22814. [DOI] [PMC free article] [PubMed]
- Li, M., Gustchina, A., Matuz, K., Tozser, J., Namwong, S., Goldfarb, N. E., Dunn, B. M. & Wlodawer, A. (2011). FEBS J. 278, 4413–4424. [DOI] [PMC free article] [PubMed]
- McCoy, A. J. (2007). Acta Cryst. D63, 32–41. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Rao, J. K. M., Erickson, J. W. & Wlodawer, A. (1991). Biochemistry, 30, 4663–4671. [DOI] [PubMed]
- Rawlings, N. D. & Barrett, A. J. (2000). Nucleic Acids Res. 28, 323–325. [DOI] [PMC free article] [PubMed]
- Rawlings, N. D. & Barrett, A. J. (2013). Handbook of Proteolytic Enzymes, 3rd ed., edited by N. D. Rawlings & G. Salvesen, pp. 3–19. New York: Academic Press.
- Rawlings, N. D. & Bateman, A. (2009). BMC Genomics, 10, 437. [DOI] [PMC free article] [PubMed]
- Simões, I., Faro, R., Bur, D., Kay, J. & Faro, C. (2011). FEBS J. 278, 3177–3186. [DOI] [PubMed]
- Sirkis, R., Gerst, J. E. & Fass, D. (2006). J. Mol. Biol. 364, 376–387. [DOI] [PubMed]
- Tang, J., James, M. N. G., Hsu, I. N., Jenkins, J. A. & Blundell, T. L. (1978). Nature (London), 271, 618–621. [DOI] [PubMed]
- Terwilliger, T. C. (2003). Methods Enzymol. 374, 22–37. [DOI] [PubMed]
- Wlodawer, A. & Gustchina, A. (2000). Biochim. Biophys. Acta, 1477, 16–34. [DOI] [PubMed]
- Wlodawer, A., Gustchina, A. & James, M. N. G. (2013). Handbook of Proteolytic Enzymes, 3rd ed., edited by N. D. Rawlings & G. Salvesen, pp. 19–26. New York: Academic Press.
- Wlodawer, A., Gustchina, A., Reshetnikova, L., Lubkowski, J., Zdanov, A., Hui, K. Y., Angleton, E. L., Farmerie, W. G., Goodenow, M. M., Bhatt, D., Zhang, L. & Dunn, B. M. (1995). Nature Struct. Mol. Biol. 2, 480–488. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: APRc105–231-His, SeMet, 5c9b
PDB reference: APRc105–231-His, 5c9f
PDB reference: APRc110–231-His, 5c9d