

Abstract
RHOA is a member of RHO family small GTPases. Over the past 2 decades, numerous biochemical and cell biological studies on RHOA have demonstrated signalings such as activation of RHO-associated coiled-coil forming kinases through guanine nucleotide exchange and GTP hydrolysis, cellular responses such as actin fiber formation and myocin activation, biological consequences such as cell motility and cytokineses, etc. There have also been a plenty of active discussion on the roles of RHOA in tumorigenesis, primarily based on gain- and loss-of-function experiments. However, cell-type-specific functions of RHOA have only recently been delineated by conditional gene targeting strategies. Furthermore, very little information had been available on human cancer genetics until we and others recently reported frequent somatic RHOA mutations in a distinct subtype of T-cell-type malignant lymphoma called angioimmunoblastic T-cell lymphoma (AITL), and other T-cell lymphoma with AITL-like features. The RHOA mutations were very specific to these types of lymphoma among hematologic malignancies, and a single hotspot, glycine at the 17th position, was affected by the replacement with valine (G17V). Remarkably, G17V RHOA did not bind GTP, and moreover, it inhibited the GTP binding to wild-type RHOA. How G17V RHOA contributes to T-cell lymphomagenesis needs to be clarified.
Keywords: RHOA, mutation, lymphoma, T cell
Abbreviations
- AITL
angioimmunoblastic T-cell lymphoma
- PTCL-NOS
peripheral T-cell lymphoma, not otherwise specified
- TFH
follicular helper T cells
- SRF
serum response factor
RHOA, encoding a small GTPase, is a founding member of the RHO family consisting of 23 genes1 in mammals. The function of RHOA is regulated by guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs),2 like many other RHO family members (Fig. 1). There are a large number of GEFs and GAPs (each ≥ 80) that catalyze GDP to GTP exchange (activation) and GTP hydrolysis (inactivation), respectively, while 3 GDIs block spontaneous activation.2 GTP-bound active form of RHOA family proteins associate with over 50 effector molecules such as RHO-associated coiled-coil forming kinases (ROCK) and mammalian Diaphanous (mDia) to transduce various signalings.1 The best characterized cellular response to RHOA activation is actin polymerization and subsequent myocin activation, which plays an essential role for cytoskeleton formation, adhesion, migration, and cytokinesis.2
Figure 1.
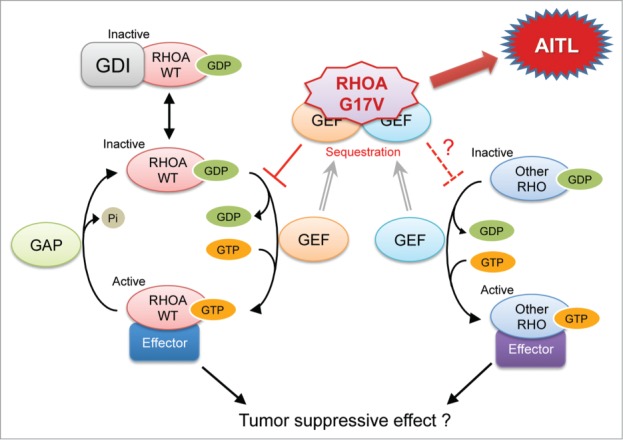
Normal RHOA signaling and potential influence of G17V mutant RHOA. WT, wild type; GDP, guanine diphosphate; GTP, guanine triphsphate; GDI, GDP-dissociation inhibitor; GAP, GTPase-activating protein; GEF, guanine nucleotide-exchange factor; AITL, angioimmunoblastic T-cell lymphoma.
Nevertheless, despite over 2 decades of extensive studies on RHOA, many questions are yet to be answered; for example, the combination between each GEF/GAP and each RHO family member, compensation by and redundancy between RHOB and RHOC and even more distantly related RHO family members, etc. Also, still unclear is how much of cellular responses is dependent on actomyocin regulation and how the response is specific to each cellular system. Indeed, because most of the characterizations have been led by cell culture-based experiments using model cells, physiologic functions of RHOA in specific types of cells are not well understood. Only recently, conditional gene targeting approaches have gradually been making an access to understanding cell-specific roles of RHOA.1,3
Another important issue is how RHOA is involved in tumorigenesis. Many experimental results supported the notion that activation of RHOA promotes tumorigenesis,4,5 with a particular emphasis on the increased cell motility. Despite a plenty of cell biological knowledge about functions of mutants, which were created due to biochemical contexts, as well as wild-type RHOA, only a handful of RHOA mutations had been reported in human cancers in just a sparse manner, and there had been no clue to the significance of those mutations in human diseases.
Recently, we and others found frequent RHOA mutations in specific subtypes of malignant lymphoma, a group of lymphocyte cancers. Malignant lymphoma is divided into Hodgkin and non-Hodgkin lymphomas, and non-Hodgkin lymphomas are further classified into B-cell, T-cell, and NK-cell lymphoma, each of which has precursor-cell type and peripheral-cell type. AITL and peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS) are major subtypes of peripheral T-cell lymphomas (PTCLs).6 Tumor cells of AITL show similar phenotypes to follicular helper T cells (TFH),7 and some PTCL-NOS cases show AITL-like features including TFH phenotypes8,9 (hereafter called AITL-like PTCL-NOS). Recurrent mutations in TET2,8,10,11 DNMT3A,10 and IDH212 had been found in AITL and PTCL-NOS. Each of these 3 genes encodes an epigenetic regulator and recurrent mutations of these genes are also identified in other hematologic malignancies, such as acute myeloid leukemias and myelodysplastic syndromes.13-15 By whole exome sequencing of PTCL samples, 3 groups found recurrent mutations of RHOA, and extended analyses commonly demonstrated that 53–68% of AITL and AITL-like PTCL-NOS had RHOA mutations, and that RHOA mutations were concentrated on a hotspot, Gly17, which is mostly replaced with valine (G17V).16-18
Based on the crystallographic structures of GDP- and GTPγS-bound RHOA of G14V mutant,19,20 O atom in the main chain of G17 of wild-type RHOA should interact with the guanine base of GDP/GTP via a water molecule. When G17 is replaced by valine, its bulky side chain leads to compromised binding of GDP/GTP. Indeed, no GTP-bound form of G17V RHOA was pulled down by rhotekin-beads. On the other hand, G17V RHOA bounds GEFs such as ECT217 and ARHGEF116 with higher affinities than wild-type RHOA. As predicted by these results, G17V RHOA inhibited GTP binding of wild-type RHOA.
Among well-characterized cellular responses by RHOA activation are enhanced transcription driven by serum response factor (SRF)21 and actin stress fiber formation.22 Incapability of G17V RHOA in GTP binding and its inhibitory effect on wild-type RHOA predicted dominant-negative nature of this mutant RHOA. Indeed, a reporter assay using an SRF-response element and immunostaining for F-actin in fibroblasts both demonstrated that G17V RHOA was functionally defective and inhibited the function of wild-type RHOA in a dominant-negative manner.16,17
We demonstrated that exogenously expressed wild-type RHOA suppressed the growth of a T-cell lymphoblastic cell line, Jurkat, but that G17V RHOA did not.17 The difference in the impact of wild-type and G17V RHOA on Jurkat cells were also demonstrated by other groups16,18; nevertheless, distinct growth advantage due to G17V RHOA expression was not shown by any groups. Therefore, the tumor suppressive role of wild-type RHOA was implicated, but the tumorigenicity of G17V RHOA was not, in terms of growth advantage. It is true that Jurkat is a T-lineage cell line, but it does not represent TFH or AITL cells, and the difficulty lies on the fact that there are no model cell systems for AITL.
Many questions remain (Fig. 1). The most fundamental one is what is the actual downstream signaling of RHOA in TFH, if wild-type RHOA functions in TFH as a tumor suppressor. It is also to be delineated whether cellular responses other than proliferation is also involved in the tumor suppressive role. Although actomyocin regulation by wild-type RHOA is generally considered to promote tumor progression, it is unknown whether the potential tumor suppressive function of wild-type RHOA is related to the actomyocin regulation. The second question is whether G17V RHOA suppresses other RHO members, and if so, which members are affected. RHOA gene knockout studies have demonstrated that expression of RHOB and/or RHOC is upregulated in a cell type-specific manner, and that these RHO members compensate the function of RHOA, depending on the cellular context. Therefore, it may be important whether RHOB/C is functioning and how much of their function is affected by G17V RHOA, and eventually, how much of the total RHOA/B/C function remains? In this regard, we found a significant increase in the ratio of cells with very low RHOA immunostaining in AITL and AITL-like PTCL-NOS having G17V RHOA mutation. It is known that nucleotide-unbound RHOA is unstable, and actually we observed that G17V RHOA protein levels were always much lower than wild-type RHOA when exogenously expressed in various cell lines. Therefore, very low RHOA levels detected by immunostaining could be explained if the G17V RHOA mutation is homozygous. Although we speculated that the mutation was heterozygous, only reason was the fact that the RHOA mutant allele frequencies were generally low (typically <0.2). This question may be important in correlation with the dominant-negative function of G17V RHOA, because the presence or absence of remaining wild-type RHOA could determine the efficiency of suppression of total RHOA/B/C by G17V RHOA.
In our study, all G17V RHOA mutation-carrying cases also had TET2 mutations. TET2 is one of the 3 dioxygenases (TET1-3) that convert methylcytosine in DNA to hydroxymethylcytosine. TET2 mutations cause truncation or functional impairment of the catalytic domains of TET2, and thus, lead to epigenetic dysregulation. Heavy overlap of the TET2 and RHOA mutations questions whether the combination of abnormal epigenetic regulation caused by the TET2 mutations and RHO signaling dysregulation by the G17V RHOA mutation is necessary for the development of lymphoma from TFH. Cooperation between abnormally up/down-regulated gene products and G17V mutant RHOA should be delineated in the future.
Finally, identification of very frequent RHOA mutations raises important potentials for diagnostics and therapeutics. We have already established a clinical testing laboratory-friendly method to detect G17V RHOA mutation by quantitative PCR with mutant-specific primers.23 On the other hand, developing therapeutics may not be simple, because the G17V mutant RHOA is a dominant-negative form at least in terms of the GTP-binding-based function. Identifying and targeting a synthetic lethality pathway for impaired RHOA function might be an attractive approach. Alternatively, if the G17V mutant RHOA actively acquires some new oncogenic function, this could also be considered as a target for the therapeutics.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Thumkeo D, Watanabe S, Narumiya S. Physiological roles of Rho and Rho effectors in mammals. Eur J Cell Biol 2013; 92:303-15; PMID:24183240; http://dx.doi.org/ 10.1016/j.ejcb.2013.09.002 [DOI] [PubMed] [Google Scholar]
- 2. Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 2005; 21:247-69; PMID:16212495; http://dx.doi.org/ 10.1146/annurev.cellbio.21.020604.150721 [DOI] [PubMed] [Google Scholar]
- 3. Zhou X, Zheng Y. Cell type-specific signaling function of RhoA GTPase: lessons from mouse gene targeting. J Biol Chem 2013; 288:36179-88; PMID:24202176; http://dx.doi.org/ 10.1074/jbc.R113.515486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Narumiya S, Tanji M, Ishizaki T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev 2009; 28:65-76; PMID:19160018; http://dx.doi.org/ 10.1007/s10555-008-9170-7 [DOI] [PubMed] [Google Scholar]
- 5. Karlsson R, Pedersen ED, Wang Z, Brakebusch C. Rho GTPase function in tumorigenesis. Biochim Biophys Acta 2009; 1796:91-8; PMID:19327386; http://dx.doi.org/ 10.1016/j.bbcan.2009.03.003 [DOI] [PubMed] [Google Scholar]
- 6. Swerdlow SH CE, H. N., AAAAA, BBBBB WHO classification of tuors of hematopoietic and lymphoid tissues. 4th ed Lyon, France: IARC Press. [Google Scholar]
- 7. de Leval L, Rickman DS, Thielen C, Reynies AD, Huang YL, Delsol G, Lamant L, Leroy K, Brière J, Molina T, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood 2007; 109:4952-63; PMID:17284527 [DOI] [PubMed] [Google Scholar]
- 8. Lemonnier F, Couronné L, Parrens M, Jaïs JP, Travert M, Lamant L, Tournillac O, Rousset T, Fabiani B, Cairns RA, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood 2012; 120:1466-9; PMID:22760778; http://dx.doi.org/ 10.1182/blood-2012-02-408542 [DOI] [PubMed] [Google Scholar]
- 9. Rodriguez-Pinilla SM, Atienza L, Murillo C, Pérez-Rodríguez A, Montes-Moreno S, Roncador G, Pérez-Seoane C, Domínguez P, Camacho FI, Piris MA. Peripheral T-cell lymphoma with follicular T-cell markers. Am J Surg Pathol 2008; 32:1787-99; PMID:18779728; http://dx.doi.org/ 10.1097/PAS.0b013e31817f123e [DOI] [PubMed] [Google Scholar]
- 10. Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med 2012; 366:95-6; PMID:22216861; http://dx.doi.org/ 10.1056/NEJMc1111708 [DOI] [PubMed] [Google Scholar]
- 11. Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011; 20:25-38; PMID:21723201; http://dx.doi.org/ 10.1016/j.ccr.2011.06.003 [DOI] [PubMed] [Google Scholar]
- 12. Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset P, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 2012; 119:1901-3; PMID:22215888; http://dx.doi.org/ 10.1182/blood-2011-11-391748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med 2009; 360:2289-301; PMID:19474426; http://dx.doi.org/ 10.1056/NEJMoa0810069 [DOI] [PubMed] [Google Scholar]
- 14. Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, Stevens-Linders E, van Hoogen P, van Kessel AG, Raymakers RA, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet 2009; 41: 838-42; PMID:19483684; http://dx.doi.org/ 10.1038/ng.391 [DOI] [PubMed] [Google Scholar]
- 15. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009; 361:1058-66; PMID:19657110; http://dx.doi.org/ 10.1056/NEJMoa0903840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palomero T, Couronné L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, Carpenter Z, Abate F, Allegretta M, Haydu JE, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet 2014; 46:166-70; PMID:24413734; http://dx.doi.org/ 10.1038/ng.2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, Muto H, Tsuyama N, Sato-Otsubo A, Okuno Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet 2014; 46:171-5; PMID:24413737; http://dx.doi.org/ 10.1038/ng.2872 [DOI] [PubMed] [Google Scholar]
- 18. Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, Kim SC, Lee B, Rho K, Lee JE, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet 2014; 46:371-5; PMID:24584070; http://dx.doi.org/ 10.1038/ng.2916 [DOI] [PubMed] [Google Scholar]
- 19. Ihara K, Muraguchi S, Kato M, Shimizu T, Shirakawa M, Kuroda S, Kaibuchi K, Hakoshima T. Crystal structure of human RhoA in a dominantly active form complexed with a GTP analogue. J Biol Chem 1998; 273:9656-66; PMID:9545299; http://dx.doi.org/ 10.1074/jbc.273.16.9656 [DOI] [PubMed] [Google Scholar]
- 20. Shimizu T, Ihara K, Maesaki R, Kuroda S, Kaibuchi K, Hakoshima T. An open conformation of switch I revealed by the crystal structure of a Mg2+-free form of RHOA complexed with GDP. Implications for the GDP/GTP exchange mechanism. J Biol Chem 2000; 275:18311-7; PMID:10748207; http://dx.doi.org/ 10.1074/jbc.M910274199 [DOI] [PubMed] [Google Scholar]
- 21. Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 1995; 81:1159-70; PMID:7600583; http://dx.doi.org/ 10.1016/S0092-8674(05)80020-0 [DOI] [PubMed] [Google Scholar]
- 22. Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992; 70:389-99; PMID:1643657; http://dx.doi.org/ 10.1016/0092-8674(92)90163-7 [DOI] [PubMed] [Google Scholar]
- 23. Nakamoto-Matsubara R, Sakata-Yanagimoto M, Enami T, Yoshida K, Yanagimoto S, Shiozawa Y, Nanmoku T, Satomi K, Muto H, Obara N, et al. Detection of the G17V RHOA mutation in angioimmunoblastic T-cell lymphoma and related lymphomas using quantitative allele-specific PCR. PLos One 2014; 9:3109714; PMID:25310466; http://dx.doi.org/ 10.1371/journal.pone.0109714 [DOI] [PMC free article] [PubMed] [Google Scholar]