

Abstract
The number of known fungal proteins capable of switching between alternative stable conformations is steadily increasing, suggesting that a prion-like mechanism may be broadly utilized as a means to propagate altered cellular states. To gain insight into the mechanisms by which cells regulate prion formation and toxicity we examined the role of the yeast ribosome-associated complex (RAC) in modulating both the formation of the [PSI+] prion – an alternative conformer of Sup35 protein – and the toxicity of aggregation-prone polypeptides. The Hsp40 RAC chaperone Zuo1 anchors the RAC to ribosomes and stimulates the ATPase activity of the Hsp70 chaperone Ssb. We found that cells lacking Zuo1 are sensitive to over-expression of some aggregation-prone proteins, including the Sup35 prion domain, suggesting that co-translational protein misfolding increases in Δzuo1 strains. Consistent with this finding, Δzuo1 cells exhibit higher frequencies of spontaneous and induced prion formation. Cells expressing mutant forms of Zuo1 lacking either a C-terminal charged region required for ribosome association, or the J-domain responsible for Ssb ATPase stimulation, exhibit similarly high frequencies of prion formation. Our findings are consistent with a role for the RAC in chaperoning nascent Sup35 to regulate folding of the N-terminal prion domain as it emerges from the ribosome.
Keywords: chaperone, nascent chain, prion, protein misfolding, ribosome, yeast
Abbreviations
- RAC
ribosome-associated complex
- Hsp
heat shock protein
- NAC
Nascent Chain Associated Complex
Introduction
Prions are proteins that adopt self-propagating, transmissible conformations. Mammalian prions underlie the transmissible spongiform encephalopathies, a set of invariably fatal neurodegenerative disorders associated with misfolding of the mammalian prion protein, PrP.1 In the budding yeast Saccharomyces cerevisiae several different proteins can propagate alternative conformations via a prion-like mechanism and thus serve as epigenetic switches that produce heritable phenotypes.2 Although it has been suggested that yeast prions represent disease states,3–6 the evolution of prion domains in a variety of yeast proteins and the occurrence of prions in wild yeast populations raise the possibility that such a mechanism may also have important biological roles.7,8 A well-studied example is the [PSI+] prion, which is a self-propagating amyloid form of the translation termination factor Sup35.9–11 Folding of Sup35 into a prion conformation compromises its translation termination activity, resulting in elevated levels of nonsense suppression. Read-through of stop codons in [PSI+] cells uncovers cryptic genetic variation by creating novel proteins with C-terminal extensions, which can enhance cell fitness in some environments.12,13
Other yeast prions have similarly been shown to produce beneficial phenotypes, commonly in the form of enhanced resistance to environmental stresses. For example, the [MOD+] prion leads to intracellular accumulation of ergosterol and thereby increases resistance to antifungal agents that target ergosterol synthesis.14 The [ISP+] prion, which has an antisuppressor phenotype that can oppose nonsense suppression, enhances resistance to drugs that target translation.15 The [MOT3+] prion produces resistance to certain cell wall stressors, while the [SWI+] prion confers resistance to microtubule disruption.7 Yeast populations in which members contain different ensembles of prions (and thus a high degree of phenotypic variation) might be better equipped to survive in rapidly fluctuating environments. In the case of [PSI+], beneficial prion-conferred traits can ultimately become fixed (prion-independent) by genetic changes, enabling adaptation to the new environment.13
Given this potential for prion-mediated adaptation to environmental stress, it is perhaps not surprising that the rates of prion formation (and in some instances, prion loss) increase substantially during a diversity of environmental stress conditions.16–20 How prion formation is induced under such conditions is poorly understood. One regulatory mechanism might involve the requirement for the presence of other glutamine/asparagine-rich protein aggregates in prion induction.21 Such aggregates, termed [PIN+] factors (because they confer the Pin+ phenotype, for [PSI+] inducibility), are thought to serve as templates to inefficiently cross-seed de novo prion formation.22 Although the Pin+ phenotype commonly results from [RNQ+], the prion form of the Rnq1 protein,23–25 other proteins can also function as [PIN+] factors and their levels can influence de novo prion formation. For example, Lsb2 is a [PIN+] factor23 that promotes [PSI+] induction and whose levels are dramatically increased by thermal stress and decreased by proteasomal degradation.26
Templated remodeling of Sup35 to a prion conformation in [PSI+] cells can occur after Sup35 is fully synthesized (post-translational conversion).27 Thus, [PIN+] factor-templated post-translational conversion can likely occur as well during de novo prion formation. However, the earliest point in the life of a prion protein at which conversion to a prion conformation could potentially occur is during its synthesis (co-translational conversion). Indeed, co-translational switching could be highly amenable to regulatory control. Nascent polypeptides are especially vulnerable to misfolding because they emerge linearly from the ribosome and thus cannot complete folding until they are fully synthesized. Localized crowding of nascent polypeptides along polysomes further enhances the probability of misfolding and aggregation, necessitating co-translational stabilization by chaperones for a substantial fraction of nascent chains in both prokaryotes and eukaryotes.28–30 In eukaryotes, 2 ribosome-associated complexes interact with nascent chains: the Nascent Chain Associated Complex (NAC), and the Ribosome-Associated Complex (RAC). The NAC is a heterodimeric complex that associates with ribosomes near the polypeptide exit tunnel.31 Although the function of the NAC is poorly defined, it is thought to have roles in co-translational protein folding32 and in protein targeting to mitochondria33 and the endoplasmic reticulum.34 The RAC consists of the Heat Shock Protein (Hsp) 70/40 pair Ssz1/Zuo1,35 which is anchored to the ribosome in close proximity to the polypeptide exit tunnel via a charged C-terminal region of Zuo1.36,37 The RAC stimulates the ATPase activity of ribosome-associated Hsp70 Ssb chaperones to protect nascent chains from misfolding and aggregation.
Deletion of Ssb leads to elevated [PSI+] prion formation,38 although it is unclear whether this phenotype reflects enhanced co-translational prion formation because Ssb has both cytosolic and ribosome-associated activity. We therefore sought to determine whether the RAC influences prion formation and the toxicity of aggregation-prone proteins in S. cerevisiae. We found that the RAC helps to mitigate potentially toxic protein misfolding and antagonizes co-translational switching of Sup35 into [PSI+] prion conformations. Modulation of RAC function could thereby serve as a mechanism to regulate co-translational prion formation in response to environmental stimuli.
Results
The RAC Antagonizes Induced and Spontaneous Formation of the [PSI+] Prion
The amyloidogenic N-terminal domain of nascent Sup35 is the first region to emerge from the ribosomal exit tunnel and thus nascent Sup35 molecules could potentially adopt prionogenic conformations early in their synthesis. Indeed, in the absence of mechanisms to protect nascent polypeptides from misfolding, the locally high concentration of nascent Sup35 along polysomes would likely enhance prion nucleation. We reasoned that ribosome-associated chaperones, such as the RAC and Ssb, are likely to antagonize co-translational [PSI+] prion formation. We therefore assessed the frequency of prion induction following transient overexpression of the Sup35 N and M domains (Sup35NM) in cells lacking RAC function (Fig. 1A). Zuo1 is central to RAC function as it anchors the RAC onto the ribosome and stimulates Ssb activity via its J domain; deletion of Zuo1 therefore eliminates RAC function on the ribosome.35–37 Sup35 conversion to a [PSI+] conformation enhances nonsense suppression of the premature stop codon in the ade1–14 allele and is thus accompanied by conversion to an Ade+ phenotype. In wildtype cells, approximately 2% of the population had converted to Ade+ following Sup35NM over-expression for 36 hours. However, a significant (approximately 2.5-fold) increase in prion induction was observed in cells lacking Zuo1, consistent with a role for the RAC in antagonizing [PSI+] prion formation.
Figure 1.

Frequency of induced and spontaneous [psi−] to [PSI+] conversion for cells lacking RAC function compared with wildtype cells. (A) Induced [psi−] to [PSI+] conversion was measured following transient overexpression of the Sup35 N and M domains for Δzuo1 and wildtype strains containing the ade1–14 reporter. Cells were transformed with a plasmid encoding Sup35NM driven by the GAL1 promoter. Following growth in medium containing raffinose cultures were diluted into medium containing galactose (to induce expression), grown, then diluted and plated on YEPD (to determine total numbers of cells) and synthetic medium lacking adenine (to determine the number of Ade+ cells). Percentage Ade+ was calculated by dividing the number of Ade+ cells by the total number of cells and expressing as a percentage. In each case, a subset of Ade+ colonies was tested for curability to [psi−] by guanidine hydrochloride (GuHCl) to confirm that the vast majority of Ade+ colonies were [PSI+]. Error bars represent 95% confidence intervals. **** p < 0.0001 (2-proportion z-test). (B) Spontaneous prion formation was measured in cells with both the ade1–14 and ura3–14 nonsense suppression reporters. Cells that were [psi−] were plated to obtain single colonies, which were excised from the plate and resuspended in sterile H2O. A small volume was diluted and plated on YEPD to determine total numbers of cells, while the remainder of each resuspended colony was plated on synthetic medium lacking adenine and uracil and incubated for 14 d to determine the number of [PSI+] cells. Only colonies that were curable to [psi−] by GuHCl were counted as [PSI+]. Conversion rates were calculated by fluctuation analysis39,41 with error bars representing the 95% confidence intervals (n=10). (C) Relative abundance of Ssa1/2 (Ssa), Ssb1/2 (Ssb), Hsp104, Sis1 and Ydj1 in wildtype and Δzuo1 strains expressing each chaperone as a carboxyl-terminal GFP fusion from its endogenous promoter and natural chromosomal location. Median GFP fluorescence intensity (50000 cells per strain) was assessed by flow cytometry and normalized to fluorescence intensities from wildtype or Δzuo1 strains expressing GFP fused to Glyceraldehyde-3-phosphate dehydrogenase to account for variation due to intrinsic differences between the wildtype or Δzuo1 strains that may systematically influence fluorescence intensity. Error bars represent coefficient of variation.
Is the anti-prionogenic function of the RAC an artifact of Sup35NM overexpression or does the RAC also suppress prion formation at physiological Sup35 expression levels? Because spontaneous prion formation in the absence of Sup35NM over-expression is a low frequency event,39 large numbers of cells must be plated to identify [PSI+] cells and thus spontaneous mutations affecting the ade1–14 reporter system could potentially confound the results. We therefore employed 2 nonsense suppression reporters to determine whether functional RAC is required for suppression of spontaneous prion formation: the ade1–14 allele and the ura3–14 allele.40 We used Luria-Delbruck fluctuation analysis to estimate [PSI+] formation rates with 95% confidence intervals39,41 by measuring conversion to Ade+ Ura+ in single colonies grown from individual [psi−] cells (Fig. 1B). In wildtype cells, the rate of prion formation was determined to be 2.68 × 10−7 with a confidence interval of (1.33–5.40) × 10−7, in agreement with previous findings.39 However, cells lacking RAC function exhibit a prion formation rate nearly 7-fold greater than in wildtype cells (1.83 × 10−6 with a confidence interval of (1.24–2.71) × 10−6). Thus, the RAC significantly suppresses both induced and spontaneous [PSI+] prion formation.
Loss of RAC function could enhance prion formation by leaving nascent Sup35 unprotected, or it could influence prion formation indirectly through causing changes in the levels of other chaperones that affect prion formation and propagation. To address the latter possibility we assessed the relative abundance of Ssa, Ssb, Hsp104, Ydj1 and Sis1 by using flow cytometry to quantitatively determine green fluorescent protein (GFP) fluorescence intensity in wildtype and Δzuo1 strains expressing each chaperone as a carboxyl-terminal GFP fusion from its endogenous promoter and natural chromosomal location42 (Fig. 1C). The use of GFP as a precise and quantitative reporter for gene expression is well documented in the literature,43,44 and the relative abundances we determined are in good agreement with those determined using epitope tags45 (correlation coefficient = 0.92). We confirmed our ability to detect changes in chaperone expression using this approach by comparing GFP intensity at 30°C and following heat shock (Ssa, Hsp104, Ydj1 and Sis1 exhibit increased expression after heat shock while Ssb exhibits a modest decrease; not shown). Importantly, we found no substantial differences in the abundance of these chaperones between wildtype and Δzuo1 strains (Fig. 1C). Thus, the suppression of prion formation we observe in cells lacking RAC function is not due to changes in the expression of these chaperones.
The Absence of the RAC Reduces the [PIN+] Requirement in de novo [PSI+] Formation
In wildtype cells, de novo conversion to [PSI+] is enhanced by the presence of other glutamine/asparagine-rich protein aggregates termed [PIN+] factors,21 which likely function as templates to cross-seed the de novo formation of [PSI+].22 The Pin+ phenotype commonly results from the [RNQ+] prion.23–25 Given the enhanced levels of prion formation in strains lacking RAC function, we asked whether the absence of the RAC bypasses the [PIN+] requirement in spontaneous de novo [PSI+] formation. We first compared the rate of spontaneous prion formation in strains that had been cured of [RNQ+] by treatment with guanidine hydrochloride (GuHCl), an agent that eliminates most yeast prions, including [PSI+] and [RNQ+], through inhibition of the molecular chaperone Hsp104.46,47 Given the sensitivity of RAC and Ssb mutants to cations including GuHCl,48,49 we used a somewhat lower concentration (2.5 mM) than is typically used for prion curing (3–5 mM). Cells were plated on YEPD+GuHCl, incubated until single colonies formed, then individual colonies restreaked onto fresh YEPD+GuHCl plates to obtain single colonies. This process was then repeated once more. Following GuHCl treatment, the rate of conversion to Ade+ Ura+ was still more than 5-fold greater in cells lacking RAC function than in wildtype cells (Fig. 2A). However, given that the rate of wildtype conversion was only reduced by approximately 30% following GuHCl treatment, we were concerned that full curing may not have occurred and some cells may have remained [RNQ+]. To address this possibility, we assessed the aggregation state of Rnq1 by transforming GuHCl−treated and untreated cells with a plasmid expressing Rnq1 fused to GFP in which expression is driven by the CuSO4-inducible Cup1 promoter. We induced moderate Rnq1-GFP expression for 90 minutes and then examined the cells by fluorescence confocal microscopy (Fig. 2B, left and center panels). In untreated [RNQ+] cells, Rnq1-GFP is recruited to existing prion aggregates and appears as many small cytosolic foci. In cultures treated with GuHCl, most cells showed diffuse fluorescence without foci, suggesting they are [rnq−]. Occasional GuHCl−treated cells (<10%) exhibited a small number of distinct foci (typically 1–3 per cell, and with more Δzuo1 cells showing foci than wildtype cells). This pattern was clearly distinguishable from that seen in the untreated [RNQ+] cells, and could either represent non-heritable (non-prion) aggregates,50 a different [RNQ+] variant that arose after curing of the initial [RNQ+] variant, or the persistence of the original [RNQ+] variant with altered morphology due to incomplete curing (similar to the “diffuse+dot” cells described previously).51 To address the latter possibility, we unequivocally eliminated [RNQ+] by deleting the Rnq1 gene in wildtype and Δzuo1 strains, then treated the Δrnq1 cells with GuHCl to eliminate any other prions that may have been present. We then transformed the Δrnq1 cells with the plasmid expressing Rnq1-GFP, induced moderate Rnq1-GFP expression for 90 minutes and then examined the cells by fluorescence confocal microscopy (Fig. 2B, right panels). While most cells showed diffuse fluorescence without foci, we still observed occasional cells (<2%) with a small number of distinct foci (again occurring more frequently in Δzuo1 cells than in wildtype cells), confirming that Rnq1-GFP foci can readily appear in some [rnq−] cells.
Figure 2.
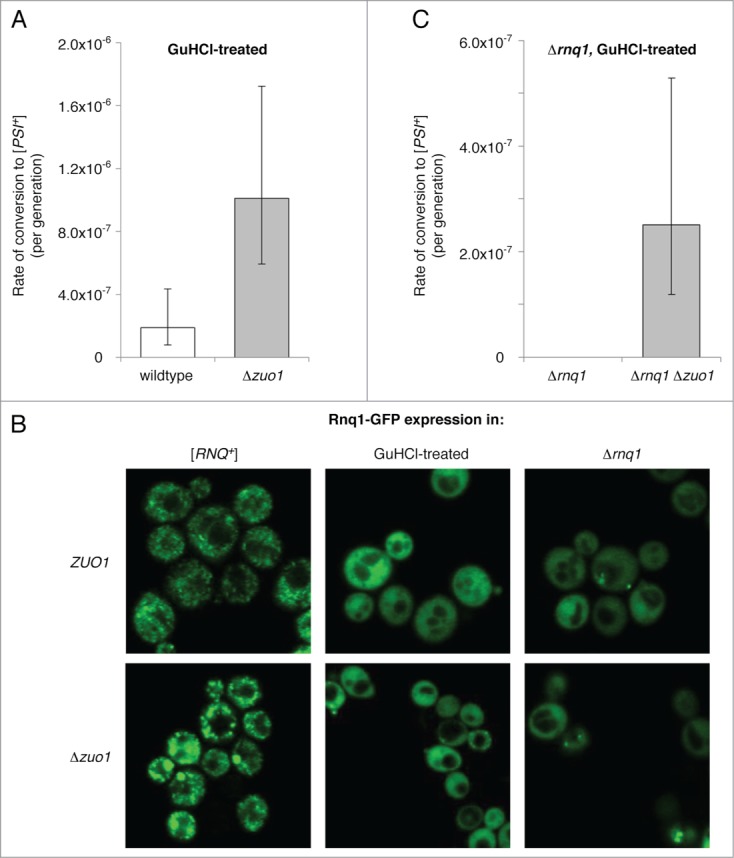
Loss of RAC function bypasses the [PIN+] requirement in de novo [PSI+] formation. (A) Rates of spontaneous prion formation in wildtype and Δzuo1 strains that were cured to [rnq−] by treatment with GuHCl. Prion formation rates and 95% confidence intervals were calculated as for Figure 1B. (B) The aggregation state of Rnq1 was assessed in cells with (top panels) and without (bottom panels) RAC function in [RNQ+] cells (left), GuHCl−treated cells (center), and Δrnq1 cells (right) by expression of Rnq1-GFP from the inducible Cup1 promoter followed by confocal microscopy. Strains transformed with p316 Cup1pr-Rnq1-GFP were diluted into synthetic medium lacking uracil and supplemented with CuSO4 (25 µM) and incubated at 30°C for 90 minutes prior to imaging. (C) Rates of spontaneous prion formation in Δrnq1 and Δrnq1 Δzuo1 strains that were cured to [pin−] by treatment with GuHCl. Prion formation rates and 95% confidence intervals were calculated as for Figure 1B.
While the aforementioned experiments indicate that loss of RAC function likely reduces the [PIN+] requirement in spontaneous de novo [PSI+] formation, the possibility remained that [RNQ+] arose spontaneously in some Δzuo1 cells to enable [PSI+] formation. We therefore measured spontaneous prion formation rates in Δrnq1 and Δrnq1 Δzuo1 strains that had been treated with GuHCl to eliminate any other prions that may have been present. We observed no spontaneous [PSI+] formation in GuHCl−passaged Δrnq1 cells that had functional RAC, suggesting that some of the GuHCl−cured cells in the previous experiment were indeed still [PIN+]. However, loss of RAC function enables spontaneous prion formation to occur at measurable rates even in GuHCl−passaged Δrnq1 cells (Fig. 2C), albeit with a >7-fold reduction in rate compared to [RNQ+] cells. Although it is probable that loss of RAC function enhanced the spontaneous appearance of other (non-Rnq1) [PIN+] factors in these cured Δrnq1 cells, prion formation templated by such spontaneous [PIN+] factors is unlikely to account for all [PSI+] appearance we observed. Even in the absence of RAC function, spontaneous [PSI+] formation is a low frequency event in [PIN+] cells (approximately 1.83 × 10−6; Fig. 1B). Spontaneous [PIN+] factor appearance also occurs at a low frequency and most commonly results from the appearance of [RNQ+] in previously [rnq−] cells.52 In our experiment, the rate of [PIN+] factor-templated [PSI+] formation was limited first by the frequency of spontaneous [PIN+] factor appearance, and then by the low frequency of spontaneous [PSI+] appearance among those rare [PIN+] cells. Given that we see only a ˜7-fold reduction in [PSI+] formation rates upon deletion of Rnq1 in cells lacking RAC function, our data strongly suggest that the absence of the RAC partially bypasses the [PIN+] requirement in de novo [PSI+] formation.
RAC Suppression of Prion Induction Requires Zuo1-Dependent Association with Ribosomes and Stimulation of Hsp70
The RAC is anchored to the ribosome via a charged region within Zuo1 and it stimulates the ATPase activity of Ssb via the J domain of Zuo1.35,36 Neither the peptide binding domain nor the ATPase activity of Ssz1 is required for RAC function in vivo.53,54 Thus, the importance of ribosome-association and Ssb stimulation for the anti-prionogenic function of the RAC can be assessed by deleting those 2 functional regions of Zuo1. Previous studies have shown that variants of Zuo1 with deletions of those regions are expressed at similar levels to wildtype Zuo1 but are not functional in vivo; specifically, deletion of the charged region eliminates the association of Zuo1 with ribosomes, whereas deletion of the J domain abolishes function without affecting ribosome association.36 To determine whether RAC suppression of prion induction requires its association with ribosomes and stimulation of Ssb, we constructed plasmids expressing wildtype Zuo1 or Zuo1 variants lacking either the J domain (zuo1Δ111–165) or the charged region (zuo1Δ284–363; Fig. 3A). We then measured the frequency of prion induction following transient overexpression of Sup35NM in a Δzuo1 strain expressing each Zuo1 construct (Fig. 3B). While expression of wildtype Zuo1 in a Δzuo1 strain reduced prion induction frequencies to wildtype levels, variants of Zuo1 lacking the charged region or the J domain were incapable of counteracting prion formation. These data are consistent with a model in which the ability of the RAC to associate with ribosomes and stimulate Ssb is essential for its function in suppressing prion formation.
Figure 3.

The ability of Zuo1 to associate with ribosomes and stimulate Ssb is required for suppression of prion formation. (A) Wildtype Zuo1, a mutant lacking the J domain required for Ssb ATPase activation (zuo1Δ111–165), and a mutant lacking the charged region required for ribosome association (zuo1Δ284–363) were examined for their ability to block prion formation in vivo. (B) Frequency of induced [psi−] to [PSI+] conversion following transient over-expression of the Sup35 N and M domains for Δzuo1 and wildtype strains ectopically expressing different Zuo1 constructs. Percentage Ade+ was calculated as described for Figure 1A. Error bars represent 95% confidence intervals. **** p < 0.0001 (2-proportion z-test).
Loss of RAC Function Enhances Toxicity Associated with Expression of Aggregation-Prone Glutamine/Asparagine Rich Proteins
We next asked whether loss of RAC function more generally enhances the toxicity of proteins that may be vulnerable to co-translational misfolding. We thus assessed the fitness consequences of ectopically expressing 3 different aggregation-prone sequences in wildtype and Δzuo1 cells: polyglutamine (62Q),55 the Sup35 NM domains, or the Rnq1 protein51 (Fig. 4A). Because polyglutamine toxicity requires the [RNQ+] prion and can be modulated by the [PSI+] prion,56–58 we conducted our analysis in strains that were [RNQ+] and either [psi−] or [PSI+]. Although previous studies have demonstrated growth inhibition associated with over-expressing Sup35NM in [psi−][PIN+] cells,21 under the growth conditions we used none of the aggregation-prone proteins resulted in substantial toxicity in wildtype [psi−] cells. Fluorescence microscopy confirmed the expression of the aggregation-prone proteins (not shown), and thus the lack of toxicity we observed may simply reflect lower expression levels under the growth conditions we used. Indeed, we did observe some growth inhibition upon expression of Sup35NM in wildtype [PSI+] cells (Fig. 4B, top panels), consistent with previous reports.21,59 However, in cells lacking RAC function, expression of either 62Q or Sup35NM substantially inhibits growth, and toxicity is exacerbated by [PSI+] (Fig. 4B, bottom panels). Expression of Rnq1, which has a C-terminal prion domain and was expressed at lower levels than 62Q and Sup35NM, does not substantially inhibit growth under these conditions. These data suggest that the RAC can protect cells against the potentially toxic consequences of expressing some aggregation-prone Q/N-rich proteins.
Figure 4.

Expression of the glutamine/asparagine-rich proteins 62Q or Sup35NM has little effect on wildtype cells but substantially inhibits the growth of Δzuo1 cells. (A) Schematic of the proteins that were expressed as GFP fusions driven by the inducible Cup1 promoter. (B) Overexpression of 62Q and Sup35NM from the Cup1 promoter on multicopy plasmids reduces the growth of Δzuo1 cells regardless of the [PSI+] prion status of the cell. Over-expression of Rnq1, which has a C-terminal prion domain, from the Cup1 promoter on a centromeric plasmid has little effect on the growth of Δzuo1 cells. Error bars represent the standard error of the mean (n = 25−50).
Sup35NM Over-Expression Is Lethal in the Absence of Rnq1 and RAC Function
As [RNQ+] enhances toxicity of Q/N-rich proteins,56,58 strains lacking Rnq1 – which are thus incapable of switching to [RNQ+] – should resist toxicity. Indeed, a screen of non-essential gene deletion strains previously identified Δrnq1 as a suppressor of polyglutamine aggregation and toxicity.58 We thus sought to determine whether the absence of Rnq1 might partially alleviate the heightened toxicity associated with expression of Sup35NM in cells lacking RAC function. To our surprise, we found that overexpression of Sup35NM is lethal in cells lacking both Zuo1 and Rnq1 (Fig. 5), even though it is tolerated in cells lacking only Rnq1 (Fig. 5) or in cells lacking only Zuo1 (Fig. 4 and Fig. 5).
Figure 5.

Overexpression of Sup35NM is lethal in [psi−] cells lacking both Rnq1 and Zuo1, but not in [psi−] cells lacking Rnq1 only nor in [psi−][rnq−] cells lacking Zuo1 only. Sup35NM with a C-terminal GFP fusion was expressed from the inducible Gal1 promoter on a high copy plasmid (pRS426 Gal1pr-Sup35NM-GFP) by plating cells onto medium lacking uracil and supplemented with 2% galactose (or with 2% glucose to repress expression). The [PSI+] prion partially restores growth in Δrnq1 Δzuo1 cells.
Previous studies have shown that toxicity due to overexpression of Sup35NM can result from depletion of soluble Sup35, causing translation termination defects. While it is not obvious how Rnq1 would alleviate toxicity due to this mechanism, an alternative (but not mutually exclusive) explanation may be that in the absence of RAC function, vulnerable nascent polypeptides can adopt multiple misfolded conformations, some of which are toxic and some of which are benign. In the case of Sup35NM, Rnq1 may shift the spectrum of such misfolded conformations toward more benign structures, such as the [PSI+] prion conformations, thus minimizing the extent to which toxic misfolded species accumulate. We therefore hypothesized that the [PSI+] prion – which could serve as a template to convert nascent Sup35 to more benign prion conformations – should rescue the Δrnq1 Δzuo1 strain from lethality of Sup35NM over-expression. Our results suggest this may be the case (Fig. 5): the [PSI+] prion enables Δrnq1 Δzuo1 cells to survive (albeit marginally) overexpression of Sup35NM that is lethal to [psi−] cells. It is noteworthy that the [psi−] Δrnq1 Δzuo1 strain grows poorly even in the absence of Sup35NM over-expression, such that any cytotoxicity caused by induction of Sup35NM overexpression may be sufficient to prevent growth. Although the apparent growth rescue is marginal, it occurred reproducibly over multiple repeats of the experiment.
Discussion
The ability of some proteins to adopt alternative conformations that confer heritable and potentially beneficial phenotypes may serve as a mechanism to facilitate adaptation to new environments.13,18,19 Rates of prion formation increase dramatically when cells experience various types of environmental stress,16 but the mechanisms by which prion formation is induced are not well understood. To begin to address this question we have examined the earliest point at which conversion to a prion conformation can occur: during the synthesis of a prion protein. We found that the RAC, which functions on the ribosome to stabilize nascent polypeptides, significantly influences the frequency of prion formation. Disruption of RAC function enhances the toxicity of aggregation-prone proteins and increases the frequency of both spontaneous and induced prion formation. The RAC is therefore well poised to serve as a mechanism to regulate co-translational prion formation.
It is unclear to what extent conversion of Sup35 to a prion conformation during de novo prion formation occurs co-translationally versus post-translationally. Fully synthesized soluble Sup35 is indeed capable of accessing prion conformations post-translationally, as forms of Sup35 that are blocked from adopting a prion conformation co-translationally by an N-terminal GST fusion can readily convert to prion conformations after cleavage of the GST tag.27 In [psi−] cells, conversion of Sup35 to prion forms typically depends upon cross-seeding by [PIN+] factors to initiate [PSI+] prion formation,21,22 and the presence or abundance of various [PIN+] factors may thus serve as a regulatory mechanism in prionogenesis. For example, the [PIN+] factor Lsb2, which interacts with Sup35 at the actin cytoskeleton during prionogenesis, is transiently induced to high levels during heat stress and then rapidly degraded by the ubiquitin proteasome system.23,26 Thus, the regulation of Lsb2 abundance could facilitate stress-induced prion formation.
Co-translational prion formation could be subject to additional regulatory mechanisms. There is no a priori reason to believe that [PIN+] factors only act on fully synthesized Sup35 and cannot cross-seed de novo prion formation co-translationally; indeed, in the absence of RAC function we found a >7-fold reduction in the rate of spontaneous prion formation in Δrnq1 cells compared to [RNQ+] cells, suggesting that [PIN+] factors greatly enhance the rate of co-translational conversion to [PSI+]. However, our data suggest that [PIN+] may not be an absolute requirement for prionogenesis when RAC function is absent (Fig. 2), although we cannot exclude the possibility that spontaneous non-Rnq1 [PIN+] factor appearance precedes [PSI+] formation in cells lacking RAC function. Elimination of the [PIN+] requirement for de novo [PSI+] formation has been shown to occur when the levels of some other chaperones are manipulated; for example, simultaneous overexpression of Sup35 and either the Hsp70 Ssa1, or the Hsp70 nucleotide exchange factor (NEF) Sse1, can bypass the requirement for [PIN+].60 It is worth noting that Sse1 serves as a NEF for Ssb1,61 a prion antagonist38,62,63 whose ATPase activity is stimulated by RAC. However, Sse1 is also a NEF for the prion agonist Ssa1,61 and the [PIN+]-independent [PSI+] formation enabled by Sse1 overexpression is thus likely to be mediated through its association with Ssa1.60 The partial [PIN+] bypass we observe may be due instead to lack of protection of nascent Sup35 by RAC and Ssb. The N-terminal prion domain of Sup35 is the first to emerge from the ribosomal exit tunnel during its translation, and the high concentration of nascent Sup35 along polysomes may further enhance aggregation and prion nucleation. Without protection by the RAC and Ssb, nascent Sup35 is thus particularly vulnerable to misfolding, as suggested by the increased toxicity of Sup35 over-expression (Fig. 4B) and the increased rates prion formation (Fig. 1) in cells lacking RAC function. These observations are consistent with previous studies that demonstrated suppression of Sup35 polymerization into amyloid by purified Zuo1, Ssz1 and Ssb in vitro64 and identified nascent Sup35 as a substrate of the RAC/Ssb chaperones in vivo.30 In addition, cells depleted of Ssb also exhibit elevated rates of prion formation.38 Indeed, here we have shown that the RAC must be capable of ribosome association and Ssb activation to suppress prion formation (Fig. 3). The presence or absence of RAC function on the ribosome therefore significantly impacts the frequency of prion formation and the toxicity of aggregation-prone polypeptides.
Under what circumstances might the RAC dissociate from ribosomes to facilitate co-translational prion formation? Intriguingly, several studies have found that both members of the RAC – Zuo1 and Ssz1 – can activate the pleiotropic drug resistance (PDR) pathway.65–68 The PDR pathway enhances resistance to various drugs and environmental toxins. Activation of the transcription factor Pdr1 by Zuo1 or Ssz1 also results in premature growth arrest upon glucose depletion during the diauxic shift, presumably facilitating adaptation to the utilization of non-fermentable carbon sources.67 Importantly, activation of Pdr1 by Zuo1 requires unfolding of the C-terminal domain of Zuo1 and its dissociation from the ribosome.68 Thus, the presence of environmental stresses, such as toxins or nutrient depletion, can trigger RAC dissociation from the ribosome to activate a transcriptional response to the stress. Our results suggest that the lack of RAC function on the ribosome under these conditions would enhance prion formation rates. This model is consistent with the increased rates of prion formation observed during various environmental stresses,16 and thus co-translational prion formation may represent the earliest mechanism to couple stress responses with prion induction. The RAC may not be the sole regulator of co-translational prion formation, however. Oxidative stress can trigger [PSI+] prion formation through methionine oxidation of Sup35,17 which is normally suppressed by 2 ribosome-associated peroxiredoxins, Tsa1 and Tsa2.69 The [PSI+] prion enhances fitness in a tsa1 tsa2 double mutant strain, and it was proposed that prion formation may confer an adaptive advantage under conditions of oxidative stress when these cellular antioxidant systems become overwhelmed.69 The co-localization of Tsa1 and Tsa2 with ribosomes suggests the potential for co-translational protection of nascent Sup35 against methionine oxidation, akin to RAC protection of nascent Sup35 against misfolding.
While the loss of RAC function on the ribosome may facilitate prion formation during environmental stress, it is not without cost. Moderate overexpression of some aggregation prone proteins, such as the Sup35NM domains and polyglutamine, is toxic in cells lacking RAC function but not in those with the RAC intact (Fig. 4). Thus, translation of proteins with aggregation-prone domains may be perilous during periods in which Zuo1 and Ssz1 dissociate from ribosomes to activate Pdr1. The mechanism by which Sup35NM and polyglutamine over-expression produce toxicity in the absence of RAC function is unclear from our data. Previous studies have shown that overproduction of Sup35NM and expression of expanded polyglutamine in yeast can cause cytotoxicity through sequestration of the termination factors Sup35 and Sup45.57,70 Thus, it is possible that the absence of RAC function enhances sequestration of essential factors by Sup35NM and polyglutamine. Regardless of the mechanism, the potential for cytotoxicity may be partially abrogated by the reduction in protein synthesis that occurs during the kinds of stresses that may trigger RAC dissociation from ribosomes. For example, activation of Pdr1 by Zuo1 and Ssz1 at the diauxic shift leads to premature growth arrest,67 which is accompanied by a general down-regulation of translation.71 Other kinds of stress responses, such as the heat shock response, similarly lead to a reduction in global translation while simultaneously increasing expression of chaperones and other protein quality control components.72,73 Together, the depression of translation and increased abundance of chaperones and protein degradation machinery could help to mitigate the cost of leaving nascent chains unprotected when RAC dissociates from ribosomes.
Our observation that overexpression of the Sup35NM domains is lethal in [psi−] cells lacking both Rnq1 and Zuo1 is intriguing (Fig. 5). This conditional synthetic lethality suggests the possibility of some functional overlap between the 2 proteins when cells are subjected to protein misfolding stress. We have shown that the RAC alleviates the toxicity of expressing aggregation-prone proteins, suggesting that RAC function may reduce sequestration of essential factors by overexpressed proteins or, alternatively, indicative of a role for RAC in preventing nascent chains from adopting toxic, non-native conformations. Similarly, Rnq1, through its [PIN+] factor activity, can template the folding of Sup35NM into relatively benign [PSI+] amyloid conformations21,22 and thus funnel excess Sup35NM away from adopting forms that could enhance sequestration of essential factors or potentially toxic misfolded forms. Consistent with this model, [PSI+] cells are immune to the lethality of Sup35NM over-expression (Fig. 5) as the existing [PSI+] prion aggregates can fulfill the templating function. We therefore propose that the RAC and [PIN+] factors can both serve as mechanisms to reduce the formation of toxic nascent chain conformations (Fig. 6). In the absence of RAC function on the ribosome, misfolding of nascent Sup35 is enhanced. [PIN+] factors such as [RNQ+] can help to divert the spectrum of misfolded conformations toward amyloid forms, thus elevating prion formation rates (Fig. 6, center panel). In [pin−] cells, [PSI+] formation rates are elevated to a lesser extent because the range of Sup35 conformations is not restrained by templating, and the accumulation of toxic misfolded species when Sup35NM expression levels are increased results in lethality (Fig. 6, right panel). Because we found that Rnq1 prevents lethality of Sup35NM overexpression in Δzuo1 cells even after curing to [rnq−] by treatment with GuHCl, our model suggests that soluble Rnq1 can readily switch to [RNQ+] at an appreciable frequency in the absence of RAC function. Alternatively, perhaps even non-prion conformations of Rnq1 can divert Sup35NM from misfolding into toxic forms. Indeed, a previous study found that stimulation of spontaneous de novo [PSI+] formation in cells lacking the ubiquitin conjugating enzyme Ubc4 is dependent upon the Rnq1 protein but not the [RNQ+] prion.74 Future studies will be aimed at further characterizing the role of Rnq1 in co-translational prion formation and examining the contributions of other ribosome-associated factors, such as the NAC, in prionogenesis.
Figure 6.
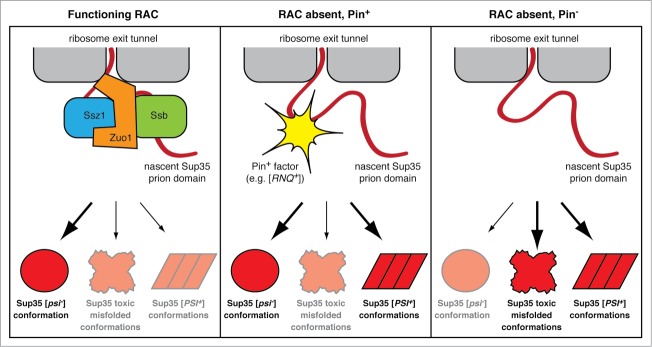
A model for the function of the RAC in antagonizing prion formation. (Left panel) RAC-mediated Ssb chaperoning of nascent Sup35 minimizes its co-translational conversion into non-native conformations, including toxic misfolded species and [PSI+] prion conformations. (Center panel) In the absence of RAC function, the N-terminal prion domain of Sup35 is more vulnerable to misfolding. [PIN+] factors, such as the [RNQ+] prion, can serve as templates to cross-seed Sup35 folding into [PSI+] prion conformations and thereby reduce the propensity of Sup35 to adopt more toxic conformations. (Right panel) In the absence of RAC function and without [PIN+] factors, Sup35 is free to misfold into a wide spectrum of conformations, including prion forms and toxic misfolded forms.
Materials and Methods
Yeast Strains and Plasmids
All yeast strains used were either W303 or S288C derivatives containing the ade1–14 allele as a reporter for [PSI+]. The zuo1::HIS3 and ssz1::LYS2 alleles have been described previously.35,36 Strains expressing chaperone-GFP fusions were generated by crossing the zuo1::KANr allele derived from the deletion collection75 (or the HIS3::KANr allele as a wildtype control) into GFP-tagged strains from the GFP collection,42 sporulating and selecting for KANr His+ haploids. Increased [PSI+] prion formation in the Δzuo GFP-tagged strains was confirmed using the induced prion formation assay. The Δzuo1 [RNQ+] strong [PSI+] and Δssz1 [rnq−][psi−] strains were generously provided by E. Craig. For prion formation assays, the Δzuo1 [RNQ+] strong [PSI+] strain was cured to [psi−] by over-expression of Hsp104 as described previously.24 The Δssz1 [rnq−][psi−] strain was crossed with a [RNQ+][psi−] strain, sporulated and then haploid Δssz1 [RNQ+][psi−] progeny selected. To construct Δrnq1, the RNQ1 ORF was replaced with a hygromycin B-resistance cassette (hph) by transformation of a strong [PSI+] W303 strain with a PCR product encoding hph with 50 base pairs of homology to the sequences immediately upstream and downstream of the RNQ1 ORF (primers P1 and P2, Table 1), followed by selection of transformants on YEPD+Hygromycin B (200 µg/ml; Corning cellgro). The replacement of RNQ1 with hph was confirmed by PCR from yeast lysates with 2 primer sets: one set that amplifies hph from the RNQ1 locus (to confirm correct integration of hph at the RNQ1 locus; primers P3 and P4, Table 1), and another set that amplifies part of the RNQ1 promoter region and ORF if the RNQ1 ORF is still present (to confirm loss of the RNQ1 ORF; primers P3 and P5, Table 1). The Δrnq1 Δzuo1 double mutant was constructed by crossing the Δrnq1 strain with a Δzuo1 strong [PSI+] strain, sporulating and selecting for Δrnq1 Δzuo1 ade1–14 strong [PSI+] haploids. The Δrnq1 Δzuo1 haploid strain was cured to [psi−] by passaging 3 times on YEPD supplemented with 2.5 mM guanidine hydrochloride (ACROS Organics).
Table 1.
Primers used in this study
| Primer | Sequence (5′→3′) |
|---|---|
| P1 | CATTAAAAGAACGTACATATAGCGATACAAACGTATAGCAAAGATCTGAAGGTCGACGGATCCCCGGGTT |
| P2 | TATATAAACAAATACGTAAACAAAGGATAGAAGGCGAACTGAATCATCGTTCGATGAATTCGAGCTCGTT |
| P3 | GATAGCGGTTCCATTGTCGT |
| P4 | CTGCAGCGAGGAGCCGTAAT |
| P5 | CCTTGGGAATTGTTACCTGAC |
| P6 | GCTACTCGAGGGATGGACGCAAAGAAGTTTAATAATC |
| P7 | GCTAGGATCCGTTTTTTCTCCTTGACGTTAAAGTATAG |
| P8 | GGTAGGTAGCGGCCGCAAATCGTAGCGCGATGAGAC |
| P9 | GCTAGGATCCCATACCGCCAAAATTTCCAA |
| P10 | CATCGGCAACAAAATCACATGATCTGAAACGCAACTTAGACAAACCC |
| P11 | GGGTTTGTCTAAGTTGCGTTTCAGATCATGTGATTTTGTTGCCGATG |
| P12 | GCATCACCAAAGTAGTCAGCTTCATCTTCACTGACAGCTCTTTCAAC |
| P13 | GTTGAAAGAGCTGTCAGTGAAGATGAAGCTGACTACTTTGGTGATGC |
For prion induction assays, strains were transformed with a plasmid encoding the Sup35 N and M domains with a C-terminal GFP fusion under the control of the GAL1 promoter (pRS426 Gal1pr-Sup35NM-GFP). To construct this plasmid, a 628 base pair fragment of the GAL1 promoter was PCR amplified from genomic DNA with primers (P6 and P7) that introduced XhoI and BamHI restriction sites (Table 1). The digested product was used to replace the XhoI-BamHI fragment encompassing the Cup1 promoter from an existing plasmid pRS426 Cup1 Sup35NM-GFP.55
Plasmid pRS315 ZUO1pr-ZUO1 (encoding wildtype Zuo1) was constructed by PCR amplifying a 2106 base pair product from W303 genomic DNA, including the ZUO1 ORF as well as 478 base pairs upstream of the start codon and 300 base pairs downstream of the stop codon using a forward primer (P8) with flanking sequence including a NotI restriction site and a reverse primer (P9) with flanking sequence including a BamHI site (Table 1). Following digestion with NotI- and BamHI (New England Biolabs), this fragment was ligated into NotI- and BamHI-digested CEN/ARS vector pRS315. Plasmids encoding Zuo1 truncations were constructed using an overlap extension PCR strategy. For pRS315 ZUO1pr-zuo1Δ111–165, a 846 base pair product was PCR amplified from W303 genomic DNA, including 478 base pairs upstream of the start codon and the ZUO1 sequence encoding positions 1–110, followed by 22 base pairs of ZUO1 sequence that encodes the region immediately C-terminal of the J domain (primers P8 and P10, Table 1). A second PCR product of 1142 base pairs was amplified that encompassed the ZUO1 sequence encoding amino acid positions 166–433, as well as 300 base pairs downstream of the stop codon (primers P11 and P9, Table 1). The two PCR products were used as templates in an overlap extension PCR including the outside primers (P8 and P9) with NotI and BamHI restriction sites to produce a 1941 base pair product, which was digested and ligated into NotI- and BamHI-digested CEN/ARS vector pRS315. For pRS315 ZUO1pr-zuo1Δ284–363, a 1366 base pair product was PCR amplified from W303 genomic DNA, including 478 base pairs upstream of the start codon and the ZUO1 sequence encoding positions 1–283, followed by 23 base pairs of ZUO1 sequence that encodes the region immediately C-terminal of the deleted region (primers P8 and P12). A second PCR product of 547 base pairs was amplified that encompassed the ZUO1 sequence encoding amino acid positions 364–433, as well as 300 base pairs downstream of the stop codon (primers P13 and P9, Table 1). The two PCR products were used as templates in an overlap extension PCR including the outside primers (P8 and P9) with NotI and BamHI restriction sites to produce a 1866 base pair product, which was digested and ligated into NotI- and BamHI-digested CEN/ARS vector pRS315. All plasmids were confirmed by DNA sequencing.
Plasmids pRS426 Cup1pr-Sup35NM-GFP,55 pRS426 Cup1pr-62Q-M-GFP55 and pRS316 Cup1pr-RNQ1-GFP51 were generous gifts from J. Weissman and J. Hines, and have been described previously.
Spontaneous Prion Formation Assay
Rare spontaneous prion formation events were measured by employing both the ade1–14 reporter as well as the ura3–14 reporter.40 These reporters contain a premature stop codon that can be suppressed by [PSI+] to enable growth on medium lacking adenine or uracil. W303 [psi−] Ade− Ura− strains (ade1–14, ura3–1) were transformed with the [PSI+] reporter plasmid pLEU2ura3–14.40 Transformants were resuspended in sterile H2O, diluted, plated on selection medium and incubated to obtain single colonies of approximately equal diameter for all strains (the fluctuation analysis relies on the total cell count being similar in all parallel cultures; the average number of cells per colony differed by <7% for WT and Δzuo1 strains). For each strain, 10 single colonies were excised from the agar per fluctuation analysis and resuspended in 100 µl sterile H2O. A small volume was diluted and plated on YEPD to estimate total viable cell count, and the remainder was plated on synthetic medium lacking adenine and uracil then incubated at 30°C for 14 d to select for [PSI+] cells. Only colonies that were curable to [psi−] by guanidine hydrochloride (ACROS Organics) were counted as [PSI+]. The rate of Ade+ Ura+ conversion along with 95% confidence intervals was estimated by employing Luria-Delbruck fluctuation analysis using Maximum Likelihood Estimates39,41 (Fluctuation analysis programs and source code, Shaver and Sniegowski, University of Pennsylvania, [http://www.bio.upenn.edu/people/paul-sniegowski]). The fluctuation analysis is a classical technique that produces a Luria-Delbrück distribution of colony counts from parallel replicates.76 Maximum likelihood estimates of the probable number of [PSI+] cells per replicate can be calculated from this distribution and transformed into a precise estimate of [PSI+] appearance rate from the total cell count per replicate. Results of a single representative fluctuation analysis are shown, but each analysis was repeated at least 3 times.
Induced Prion Formation Assay
W303 [psi−] ade1–14 strains were transformed with pRS426 Gal1pr-Sup35NM-GFP. For each replicate, a single colony was used to inoculate 3 ml of synthetic medium lacking uracil and supplemented with 2% raffinose (ACROS Organics). Cultures were grown at 30°C overnight then diluted 100-fold into medium lacking uracil and supplemented with 2% galactose (ACROS Organics). Following 24–36 hours growth at 30°C, dilutions were plated on YEPD to estimate total viable cell count and on synthetic medium lacking adenine to select for [PSI+] cells. The number of Ade+ colonies on each plate was determined following growth at 30°C for 14 d and the percentage Ade+ cells in each culture was then calculated by dividing the number of Ade+ colonies by the total viable cell count after accounting for the dilution factors. Four independent cultures were assessed for each strain, giving a total of ∼200–400 Ade+ colonies counted from ∼8000 cells assessed per strain. A subset of colonies from each plate was tested for curability to Ade− by treatment with guanidine hydrochloride (ACROS Organics) to ensure Ade+ colonies were [PSI+]. P-values were determined using a 2-proportion z-test, and error bars represent 95% confidence intervals.
Determination of Relative Protein Abundance by Flow Cytometry
GFP-tagged strains were inoculated from single colonies and grown overnight in liquid YEPD. Overnight cultures were washed in sterile H2O, diluted into synthetic defined complete medium at an optical density (600 nm) of 0.1 then incubated with shaking for 90 minutes at either 30°C or 39°C. For each strain, GFP fluorescence intensity was measured for 50000 cells using a BD FACS Canto II flow cytometer and analyzed using the FITC ‘height’ parameter and the %rCV function to determine coefficients of variation (represented as error bars). Median GFP fluorescence intensities for chaperone-GFP fusion strains were normalized to fluorescence intensities from wildtype or Δzuo1 strains expressing GFP fused to Glyceraldehyde-3-phosphate dehydrogenase to account for potential variation due to intrinsic differences between the wildtype or Δzuo1 strains that may systematically influence fluorescence intensity (e.g. cell size differences, etc.)
Yeast Growth Assays
For quantitative growth measurements, strains were transformed with plasmids pRS426, pRS426 Cup1pr-Sup35NM-GFP, pRS426 Cup1pr-62Q-M-GFP and pRS316 Cup1pr-RNQ1-GFP.51,55 Dilutions of overnight cultures were plated onto medium lacking uracil and supplemented with 100 µM CuSO4 (Sigma-Aldrich). Plates were incubated at 30°C for 2–4 d and then photographed. Colony area (pixels^2) for 25–50 isolated, single colonies was measured using the “area” parameter within the “measure” function of the colony counter plugin for ImageJ software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, [http://imagej.nih.gov/ij/, 1997–2014]; and Vieira, B., Colony Counter, University of Lisbon, Portugal, [http://rsb.info.nih.gov/ij/plugins/colony-counter.html]). Mean colony sizes (n=25−50) were plotted with error bars indicating the standard error of the mean.
For qualitative growth assays, overnight cultures of strains transformed with pRS426 Gal1pr-Sup35NM-GFP were diluted to an optical density (600 nm) of 0.2 then subjected to 5-fold serial dilutions. Cells were then spotted onto medium lacking uracil and supplemented with either glucose (to repress expression of Sup35NM-GFP; Fisher Chemical) or galactose (to induce expression of Sup35NM-GFP; ACROS Organics). Plates were incubated at 30°C for 3–5 d
Confocal Microscopy
Cells were transformed with pRS316 Cup1pr-RNQ1-GFP51 and overnight cultures diluted into medium lacking uracil and supplemented with 25 µM CuSO4 (Sigma-Aldrich) to induce expression of Rnq1-GFP. Cells were incubated at 30°C for 90 minutes and then examined by confocal microscopy using a Nikon C2 microscope with NIS-Elements AR 4.20.00 64-bit software. All strains were observed via laser-excitation of GFPs using the FITC filter set with a 90µm or 150µm pinhole, varying in gain (75–100 units), offset (−10 – 0 units), and laser power (2.25 – 4.00 units). Cells were transferred onto 3% agarose on a slide and viewed at 60x magnification.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to E. Craig (University of Wisconsin, Madison), J. Hines (Lafayette College), J. Weissman (University of California, San Francisco), and S. Liebman (University of Nevada, Reno) for generous gifts of yeast strains and plasmids, and T. Kress (The College of New Jersey) for helpful discussion and feedback on the manuscript.
Funding
This work was supported by a Cottrell College Science Award from the Research Corporation for Science Advancement and by Ursinus College.
REFERENCES
- 1.Poggiolini I, Saverioni D, Parchi P. Prion protein misfolding, strains, and neurotoxicity: an update from studies on mammalian prions. Int J Cell Biol 2013; 2013:e910314; PMID:24454379; http://dx.doi.org/ 10.1155/2013/910314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crow ET, Li L. Newly identified prions in budding yeast, and their possible functions. Semin Cell Dev Biol 2011; 22:452–9; PMID:21397710; http://dx.doi.org/ 10.1016/j.semcdb.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wickner RB, Edskes HK, Bateman D, Kelly AC, Gorkovskiy A. The yeast prions [PSI+] and [URE3] are molecular degenerative diseases. Prion 2011; 5:258–62; PMID:22052353; http://dx.doi.org/ 10.4161/pri.17748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wickner RB, Edskes HK, Kryndushkin D, McGlinchey R, Bateman D, Kelly A. Prion diseases of yeast: amyloid structure and biology. Semin Cell Dev Biol 2011; 22:469–75; PMID:21345375; http://dx.doi.org/ 10.1016/j.semcdb.2011.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGlinchey RP, Kryndushkin D, Wickner RB. Suicidal [PSI+] is a lethal yeast prion. Proc Natl Acad Sci U S A 2011; 108:5337–41; PMID:21402947; http://dx.doi.org/ 10.1073/pnas.1102762108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bateman DA, Wickner RB. [PSI+] Prion transmission barriers protect saccharomyces cerevisiae from infection: intraspecies “species barriers.” Genetics 2012; 190:569–79; PMID:22095075; http://dx.doi.org/ 10.1534/genetics.111.136655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009; 137:146–58; PMID:19345193; http://dx.doi.org/ 10.1016/j.cell.2009.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halfmann R, Jarosz DF, Jones SK, Chang A, Lancaster AK, Lindquist S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 2012; 482:363–8; PMID:22337056; http://dx.doi.org/ 10.1038/nature10875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wickner RB. ; [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 1994; 264:566–9; PMID:7909170; http://dx.doi.org/ 10.1126/science.7909170 [DOI] [PubMed] [Google Scholar]
- 10.Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 1996; 273:622–6; PMID:8662547; http://dx.doi.org/ 10.1126/science.273.5275.622 [DOI] [PubMed] [Google Scholar]
- 11.Sparrer HE, Santoso A, Szoka FC, Weissman JS. Evidence for the prion hypothesis: induction of the yeast [PSI+] factor by in vitro-converted sup35 protein. Science 2000; 289:595–9; PMID:10915616; http://dx.doi.org/ 10.1126/science.289.5479.595 [DOI] [PubMed] [Google Scholar]
- 12.True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 2000; 407:477–83; PMID:11028992; http://dx.doi.org/ 10.1038/35035005 [DOI] [PubMed] [Google Scholar]
- 13.True HL, Berlin I, Lindquist SL. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature 2004; 431:184–7; PMID:15311209; http://dx.doi.org/ 10.1038/nature02885 [DOI] [PubMed] [Google Scholar]
- 14.Suzuki G, Shimazu N, Tanaka M. A yeast prion, mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 2012; 336:355–9; PMID:22517861; http://dx.doi.org/ 10.1126/science.1219491 [DOI] [PubMed] [Google Scholar]
- 15.Rogoza T, Goginashvili A, Rodionova S, Ivanov M, Viktorovskaya O, Rubel A, Volkov K, Mironova L. Non-mendelian determinant ; [ISP+] in yeast is a nuclear-residing prion form of the global transcriptional regulator Sfp1. Proc Natl Acad Sci 2010; 107:10573–7; PMID:20498075; http://dx.doi.org/ 10.1073/pnas.1005949107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tyedmers J, Madariaga ML, Lindquist S. Prion switching in response to environmental stress. PLoS Biol 2008; 6:e294; PMID:19067491; http://dx.doi.org/ 10.1371/journal.pbio.0060294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sideri TC, Koloteva-Levine N, Tuite MF, Grant CM. Methionine oxidation of Sup35 protein induces formation of the [PSI+] prion in a yeast peroxiredoxin mutant. J Biol Chem 2011; 286:38924–31; PMID:21832086; http://dx.doi.org/ 10.1074/jbc.M111.272419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halfmann R, Lindquist S. Epigenetics in the extreme: prions and the inheritance of environmentally acquired traits. Science 2010; 330:629–32; PMID:21030648; http://dx.doi.org/ 10.1126/science.1191081 [DOI] [PubMed] [Google Scholar]
- 19.Suzuki G, Tanaka M. Active conversion to the prion state as a molecular switch for cellular adaptation to environmental stress. BioEssays 2013; 35:12–6; PMID:23175284; http://dx.doi.org/ 10.1002/bies.201200121 [DOI] [PubMed] [Google Scholar]
- 20.Hines JK, Craig EA. The sensitive ; [SWI+] prion. Prion 2011; 5:164–8; PMID:21811098; http://dx.doi.org/ 10.4161/pri.5.3.16895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in saccharomyces cerevisiae. Genetics 1997; 147:507–19; PMID:9335589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc Natl Acad Sci U S A 2004; 101:12934–9; PMID:15326312; http://dx.doi.org/ 10.1073/pnas.0404968101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN+]. Cell 2001; 106:171–82; PMID:11511345; http://dx.doi.org/ 10.1016/S0092-8674(01)00427-5 [DOI] [PubMed] [Google Scholar]
- 24.Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell 2001; 106:183–94; PMID:11511346; http://dx.doi.org/ 10.1016/S0092-8674(01)00440-8 [DOI] [PubMed] [Google Scholar]
- 25.Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell 2000; 5:163–72; PMID:10678178; http://dx.doi.org/ 10.1016/S1097-2765(00)80412-8 [DOI] [PubMed] [Google Scholar]
- 26.Chernova TA, Romanyuk AV, Karpova TS, Shanks JR, Ali M, Moffatt N, Howie RL, O'Dell A, McNally JG, Liebman SW, et al.. Prion induction by the short-lived, stress-induced protein Lsb2 is regulated by ubiquitination and association with the actin cytoskeleton. Mol Cell 2011; 43:242–52; PMID:21777813; http://dx.doi.org/ 10.1016/j.molcel.2011.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satpute-Krishnan P, Serio TR. Prion protein remodelling confers an immediate phenotypic switch. Nature 2005; 437:262–5; PMID:16148935; http://dx.doi.org/ 10.1038/nature03981 [DOI] [PubMed] [Google Scholar]
- 28.Deuerling E, Patzelt H, Vorderwülbecke S, Rauch T, Kramer G, Schaffitzel E, Mogk A, Schulze-Specking A, Langen H, Bukau B. Trigger factor and DnaK possess overlapping substrate pools and binding specificities. Mol Microbiol 2003; 47:1317–28; PMID:12603737; http://dx.doi.org/ 10.1046/j.1365-2958.2003.03370.x [DOI] [PubMed] [Google Scholar]
- 29.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci 2005; 62:670–84; PMID:15770419; http://dx.doi.org/ 10.1007/s00018-004-4464-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willmund F, del Alamo M, Pechmann S, Chen T, Albanèse V, Dammer EB, Peng J, Frydman J. The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell 2013; 152:196–209; PMID:23332755; http://dx.doi.org/ 10.1016/j.cell.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pech M, Spreter T, Beckmann R, Beatrix B. Dual binding mode of the nascent polypeptide-associated complex reveals a novel universal adapter site on the ribosome. J Biol Chem 2010; 285:19679–87; PMID:20410297; http://dx.doi.org/ 10.1074/jbc.M109.092536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wegrzyn RD, Deuerling E. Molecular guardians for newborn proteins: ribosome-associated chaperones and their role in protein folding. Cell Mol Life Sci CMLS 2005; 62:2727–38; PMID:16231086; http://dx.doi.org/ 10.1007/s00018-005-5292-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yogev O, Karniely S, Pines O. Translation-coupled translocation of yeast fumarase into mitochondria in vivo. J Biol Chem 2007; 282:29222–9; PMID:17666392; http://dx.doi.org/ 10.1074/jbc.M704201200 [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Berndt U, Gölz H, Tais A, Oellerer S, Wölfle T, Fitzke E, Rospert S. NAC functions as a modulator of SRP during the early steps of protein targeting to the endoplasmic reticulum. Mol Biol Cell 2012; 23:3027–40; PMID:22740632; http://dx.doi.org/ 10.1091/mbc.E12-02-0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gautschi M, Lilie H, Fünfschilling U, Mun A, Ross S, Lithgow T, Rücknagel P, Rospert S. RAC, a stable ribosome-associated complex in yeast formed by the DnaK-DnaJ homologs Ssz1p and zuotin. Proc Natl Acad Sci U S A 2001; 98:3762–7; PMID:11274393; http://dx.doi.org/ 10.1073/pnas.071057198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yan W, Schilke B, Pfund C, Walter W, Kim S, Craig EA. Zuotin, a ribosome-associated DnaJ molecular chaperone. EMBO J 1998; 17:4809–17; PMID:9707440; http://dx.doi.org/ 10.1093/emboj/17.16.4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leidig C, Bange G, Kopp J, Amlacher S, Aravind A, Wickles S, Witte G, Hurt E, Beckmann R, Sinning I. Structural characterization of a eukaryotic chaperone–the ribosome-associated complex. Nat Struct Mol Biol 2013; 20:23+; PMID:23202586; http://dx.doi.org/ 10.1038/nsmb.2447 [DOI] [PubMed] [Google Scholar]
- 38.Chernoff YO, Newnam GP, Kumar J, Allen K, Zink AD. Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone ssb in formation, stability, and toxicity of the [PSI] prion. Mol Cell Biol 1999; 19:8103–12; PMID:10567536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lancaster AK, Bardill JP, True HL, Masel J. The spontaneous appearance rate of the yeast prion [PSI+] and its implications for the evolution of the evolvability properties of the [PSI+] system. Genetics 2010; 184:393–400; PMID:19917766; http://dx.doi.org/ 10.1534/genetics.109.110213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manogaran AL, Kirkland KT, Liebman SW. An engineered nonsense URA3 allele provides a versatile system to detect the presence, absence and appearance of the [PSI+] prion in saccharomyces cerevisiae. Yeast Chichester Engl 2006; 23:141–7; PMID:16491470; http://dx.doi.org/ 10.1002/yea.1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaver AC, Sniegowski PD. Spontaneously Arising mutL Mutators in Evolving Escherichia coli Populations Are the Result of Changes in Repeat Length. J Bacteriol 2003; 185:6076–82; PMID:14526019; http://dx.doi.org/ 10.1128/JB.185.20.6076-6082.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huh W-K, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature 2003; 425:686–91; PMID:14562095; http://dx.doi.org/ 10.1038/nature02026 [DOI] [PubMed] [Google Scholar]
- 43.Soboleski MR, Oaks J, Halford WP. Green fluorescent protein is a quantitative reporter of gene expression in individual eukaryotic cells. FASEB J Off Publ Fed Am Soc Exp Biol 2005; 19:440–2; PMID:15640280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Newman JRS, Ghaemmaghami S, Ihmels J, Breslow DK, Noble M, DeRisi JL, Weissman JS. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 2006; 441:840–6; PMID:16699522; http://dx.doi.org/ 10.1038/nature04785 [DOI] [PubMed] [Google Scholar]
- 45.Ghaemmaghami S, Huh W-K, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature 2003; 425:737–41; PMID:14562106; http://dx.doi.org/ 10.1038/nature02046 [DOI] [PubMed] [Google Scholar]
- 46.Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol 2001; 40:1357–69; PMID:11442834; http://dx.doi.org/ 10.1046/j.1365-2958.2001.02478.x [DOI] [PubMed] [Google Scholar]
- 47.Jung G, Masison DC. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr Microbiol 2001; 43:7–10; PMID:11375656; http://dx.doi.org/ 10.1007/s002840010251 [DOI] [PubMed] [Google Scholar]
- 48.Jones GW, Song Y, Masison DC. Deletion of the Hsp70 chaperone gene SSB causes hypersensitivity to guanidine toxicity and curing of the [PSI+] prion by increasing guanidine uptake in yeast. Mol Genet Genomics MGG 2003; 269:304–11; PMID:12684878; http://dx.doi.org/ 10.1007/s00438-003-0838-y [DOI] [PubMed] [Google Scholar]
- 49.Kim S-Y, Craig EA. Broad sensitivity of saccharomyces cerevisiae lacking ribosome-associated chaperone ssb or zuo1 to cations, including aminoglycosides. Eukaryot Cell 2005; 4:82–9; PMID:15643063; http://dx.doi.org/ 10.1128/EC.4.1.82-89.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du Z, Li L. Investigating the interactions of yeast prions - [SWI+], [PSI+] and [PIN+]. Genetics 2014; 197:685–700; PMID:24727082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aron R, Higurashi T, Sahi C, Craig EA. J-protein co-chaperone Sis1 required for generation of ; [RNQ+] seeds necessary for prion propagation. EMBO J 2007; 26:3794–803; PMID:17673909; http://dx.doi.org/ 10.1038/sj.emboj.7601811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Derkatch IL, Bradley ME, Masse SVL, Zadorsky SP, Polozkov GV, Inge‐Vechtomov SG, Liebman SW. Dependence and independence of [PSI+] and [PIN+]: a two‐prion system in yeast? EMBO J 2000; 19:1942–52; PMID:10790361; http://dx.doi.org/ 10.1093/emboj/19.9.1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang P, Gautschi M, Walter W, Rospert S, Craig EA. The Hsp70 Ssz1 modulates the function of the ribosome-associated J-protein Zuo1. Nat Struct Mol Biol 2005; 12:497–504; PMID:15908962; http://dx.doi.org/ 10.1038/nsmb942 [DOI] [PubMed] [Google Scholar]
- 54.Hundley H, Eisenman H, Walter W, Evans T, Hotokezaka Y, Wiedmann M, Craig E. The in vivo function of the ribosome-associated Hsp70, Ssz1, does not require its putative peptide-binding domain. Proc Natl Acad Sci U S A 2002; 99:4203–8; PMID:11929993; http://dx.doi.org/ 10.1073/pnas.062048399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osherovich LZ, Cox BS, Tuite MF, Weissman JS. Dissection and design of yeast prions. PLoS Biol 2004; 2:e86; PMID:15045026; http://dx.doi.org/ 10.1371/journal.pbio.0020086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duennwald ML, Jagadish S, Giorgini F, Muchowski PJ, Lindquist S. A network of protein interactions determines polyglutamine toxicity. Proc Natl Acad Sci 2006; 103:11051–6; PMID:16832049; http://dx.doi.org/ 10.1073/pnas.0604548103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gong H, Romanova NV, Allen KD, Chandramowlishwaran P, Gokhale K, Newnam GP, Mieczkowski P, Sherman MY, Chernoff YO. Polyglutamine toxicity is controlled by prion composition and gene dosage in yeast. PLoS Genet 2012; 8:e1002634; PMID:22536159; http://dx.doi.org/ 10.1371/journal.pgen.1002634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntingtin toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol 2002; 157:997–1004; PMID:12058016; http://dx.doi.org/ 10.1083/jcb.200112104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ter-Avanesyan MD, Kushnirov VV, Dagkesamanskaya AR, Didichenko SA, Chernoff YO, Inge-Vechtomov SG, Smirnov VN. Deletion analysis of the SUP35 gene of the yeast saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol Microbiol 1993; 7:683–92; PMID:8469113; http://dx.doi.org/ 10.1111/j.1365-2958.1993.tb01159.x [DOI] [PubMed] [Google Scholar]
- 60.Sadlish H, Rampelt H, Shorter J, Wegrzyn RD, Andréasson C, Lindquist S, Bukau B. Hsp110 chaperones regulate prion formation and propagation in S. cerevisiae by two discrete activities. PLoS ONE 2008; 3:e1763; PMID:18335038; http://dx.doi.org/ 10.1371/journal.pone.0001763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dragovic Z, Broadley SA, Shomura Y, Bracher A, Hartl FU. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J 2006; 25:2519–28; PMID:16688212; http://dx.doi.org/ 10.1038/sj.emboj.7601138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chacinska A, Szczesniak B, Kochneva-Pervukhova NV, Kushnirov VV, Ter-Avanesyan MD, Boguta M. Ssb1 chaperone is a [PSI+] prion-curing factor. Curr Genet 2001; 39:62–7; PMID:11405097; http://dx.doi.org/ 10.1007/s002940000180 [DOI] [PubMed] [Google Scholar]
- 63.Allen Kd, Wegrzyn RD, Chernova TA, Müller S, Newnam GP, Winslett PA, Wittich KB, Wilkinson Kd, Chernoff YO. Hsp70 chaperones as modulators of prion life cycle novel effects of ssa and ssb on the saccharomyces cerevisiae prion [PSI+]. Genetics 2005; 169:1227–42; PMID:15545639; http://dx.doi.org/ 10.1534/genetics.104.037168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shorter J, Lindquist S. Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of sup35 prions. EMBO J 2008; 27:2712–24; PMID:18833196; http://dx.doi.org/ 10.1038/emboj.2008.194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hallstrom TC, Katzmann DJ, Torres RJ, Sharp WJ, Moye-Rowley WS. Regulation of transcription factor Pdr1p function by an Hsp70 protein in saccharomyces cerevisiae. Mol Cell Biol 1998; 18:1147–55; PMID:9488429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eisenman HC, Craig EA. Activation of pleiotropic drug resistance by the J-protein and Hsp70-related proteins, Zuo1 and Ssz1. Mol Microbiol 2004; 53:335–44; PMID:15225326; http://dx.doi.org/ 10.1111/j.1365-2958.2004.04134.x [DOI] [PubMed] [Google Scholar]
- 67.Prunuske AJ, Waltner JK, Kuhn P, Gu B, Craig EA. Role for the molecular chaperones Zuo1 and Ssz1 in quorum sensing via activation of the transcription factor Pdr1. Proc Natl Acad Sci 2012; 109:472–7; PMID:22203981; http://dx.doi.org/ 10.1073/pnas.1119184109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ducett JK, Peterson FC, Hoover LA, Prunuske AJ, Volkman BF, Craig EA. Unfolding of the C-terminal domain of the J-protein Zuo1 releases autoinhibition and activates Pdr1-dependent transcription. J Mol Biol 2013; 425:19–31; PMID:23036859; http://dx.doi.org/ 10.1016/j.jmb.2012.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sideri TC, Stojanovski K, Tuite MF, Grant CM. Ribosome-associated peroxiredoxins suppress oxidative stress-induced de novo formation of the [PSI+] prion in yeast. Proc Natl Acad Sci U S A 2010; 107:6394–9; PMID:20308573; http://dx.doi.org/ 10.1073/pnas.1000347107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vishveshwara N, Bradley ME, Liebman SW. Sequestration of essential proteins causes prion associated toxicity in yeast. Mol Microbiol 2009; 73:1101–14; PMID:19682262; http://dx.doi.org/ 10.1111/j.1365-2958.2009.06836.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fuge EK, Braun EL, Werner-Washburne M. Protein synthesis in long-term stationary-phase cultures of saccharomyces cerevisiae. J Bacteriol 1994; 176:5802–13; PMID:8083172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hand SC, Hardewig I. Downregulation of cellular metabolism during environmental stress: mechanisms and implications. Annu Rev Physiol 1996; 58:539–63; PMID:8815808; http://dx.doi.org/ 10.1146/annurev.ph.58.030196.002543 [DOI] [PubMed] [Google Scholar]
- 73.Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA. Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell 2001; 12:323–37; PMID:11179418; http://dx.doi.org/ 10.1091/mbc.12.2.323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Allen KD, Chernova TA, Tennant EP, Wilkinson KD, Chernoff YO. Effects of ubiquitin system alterations on the formation and loss of a yeast prion. J Biol Chem 2007; 282:3004–13; PMID:17142456; http://dx.doi.org/ 10.1074/jbc.M609597200 [DOI] [PubMed] [Google Scholar]
- 75.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, et al.. Functional profiling of the saccharomyces cerevisiae genome. Nature 2002; 418:387–91; PMID:12140549; http://dx.doi.org/ 10.1038/nature00935 [DOI] [PubMed] [Google Scholar]
- 76.Rosche WA, Foster PL. Determining mutation rates in bacterial populations. Methods San Diego Calif 2000; 20:4–17; PMID:10610800; http://dx.doi.org/ 10.1006/meth.1999.0901 [DOI] [PMC free article] [PubMed] [Google Scholar]