

Abstract
The key molecular event in human cerebral proteinopathies, which include Alzheimer's, Parkinson's and Huntington's diseases, is the structural conversion of a specific host protein into a β-sheet-rich conformer. With regards to this common mechanism, it appears difficult to explain the outstanding infectious properties attributed to PrPSc, the hallmark of another intriguing family of cerebral proteinopathies known as transmissible spongiform encephalopathies (TSE) or prion diseases. The infectious PrPSc or "prion" is thought to be composed solely of a misfolded form of the otherwise harmless cellular prion protein (PrPc). To gain insight into this unique situation, we used the 263K scrapie hamster model to search for a putative PrPSc-associated factor that contributes to the infectivity of PrPSc amyloid. In a rigorously controlled set of experiments that included several bioassays, we showed that originally innocuous recombinant prion protein (recPrP) equivalent to PrPc is capable of initiating prion disease in hamsters when it is converted to a prion-like conformation (β-sheet-rich) in the presence of RNA purified from scrapie-associated fibril (SAF) preparations. Analysis of the recPrP-RNA infectious mixture reveals the presence of 2 populations of small RNAs of approximately 27 and 55 nucleotides. These unprecedented findings are discussed in light of the distinct relationship that may exist between this RNA material and the 2 biological properties, infectivity and strain features, attributed to prion amyloid.
Keywords: nature of prions, PMCA, prions, prion infectivity, PrPsc, recombinant PrP, RNA, SAF-derived RNA, scrapie infectivity
Introduction
Transmissible spongiform encephalopathies (TSEs), which include scrapie or bovine spongiform encephalopathy (BSE) in animals and Creutzfeldt-Jakob disease in humans, are caused by an unconventional type of infectious agent called prions. According to the prion hypothesis,1 TSE infectious agents are essentially (if not entirely) composed of host-encoded prion proteins (PrPs) of synaptic origin2 that have been converted into a pathological folding and aggregation-prone structure, which is known as PrPSc. This abnormal conformer is believed to be solely responsible for the infectivity known to be associated mainly with synaptosome-rich fractions of the infected brain.3-6 Although it is currently accepted by the scientific community, the prion concept has been unable to provide a satisfying explanation for all phenomena associated with infectious TSEs. More than a decade ago, findings from experimental BSE transmissions in mice alerted the scientific community to the possibility that TSEs could be transmitted in the absence of detectable PrPSc.7 Subsequent studies have also shown a poor correlation between infectivity and PrPSc under certain experimental conditions, suggesting that components other than PrPSc could be involved in the infectivity of TSE agents.8-13 Using various sophisticated approaches involving amplification methods for autocatalytic conversion of recombinant PrP, recent experiments conducted on wild-type animals have attempted to precisely define the composition of the PrP-associated infectious solution.14–16 All of these studies have indicated that co-factors are involved in the prion infectivity process, such as nucleic acids (RNA) and lipids, both of artificial and cellular origins. These cofactors seem to play a fundamental role as catalysts in the PrPc-PrPSc conversion reaction, allowing the specific architecture of PrPSc to cause infectivity.17 However, the mechanism of conversion remains uncertain, and the role of each component has yet to be defined.18-22 Moreover, it is important to note that the standard experimental conditions set up to generate PrPSc may not necessarily lead to infectious preparations.23
Here, we re-addressed the fundamental question of the biochemical nature of TSE agents by searching for a putative nucleic acid implicated in the infectivity process, assuming that to exert its function, the candidate molecule needed to be a component of the prion particle.24 Previous studies have demonstrated that nucleic acids can be isolated from the TSE agent. However, they did not reveal any role for these nucleic acids in the infectious phenomenon of prion diseases.25-27
Focusing our attention on this crucial point, we extracted highly infectious scrapie-associated fibrils (SAFs) from the brain of Syrian hamsters infected with the 263K scrapie agent. SAFs provide the molecular support for the infectious agent, which is composed of purified PrPSc amyloids of a large size range with multiple copies of the infectious unit.28 The BioRad method was selected over other PrPSc purification methods because it allows for the purification of a crude preparation of the prion agent (SAFs) and possibly other components29 with relatively low compaction forces. The idea was to keep the prion molecules as "loose" as possible to facilitate the next steps: intense PK digestion (500 μg/ml for 24 h) and phenol/chloroform extraction (Materials and Methods) with the aim of eliminating all proteins, including PrPSc, and purifying the associated nucleic acids.
The remaining nucleic acids in the SAF preparations, now without PrPSc, were incubated with recombinant PrP (recPrP) under conditions allowing for its conversion (from α-helices to β-sheets) to reconstitute an infectious preparation. In this report, we describe the induction of prion disease in Syrian hamsters inoculated with preparations of recombinant PrP oligomers co-incubated with RNA extracted from Scrapie-infected brains and include a partial characterization of these nucleic acids.
Results
β-sheet-rich recombinant PrP oligomers co-incubatedwith RNAs from SAFs are infectious
Infectious SAF preparations from 263K scrapie-infected hamster brain were exposed to harsh enzymatic and chemical treatments (Proteinase K [PK] digestion and phenol/chloroform extraction; see Materials and Methods) to eliminate all proteins, including PrPSc, as well as scrapie infectivity, and to extract putative nucleic acids associated with SAFs. The material obtained under these conditions was mixed with monomeric, oligomeric or fibrillar forms of recombinant hamster PrP and subsequently inoculated intracerebrally into hamsters to investigate whether the infectivity would be restored. Of the various mixtures, only one (inoculum 8) caused clinical disease in the recipient hamsters. Two out of 5 animals challenged with inoculum 8 showed clinical signs of scrapie at 151 and 246 days post infection (dpi) (Table 1). This particular preparation contained heat-converted β-sheet-rich oligomeric recPrP and the SAF preparation-nucleic acid extracts treated with DNase I. None of the animals in any of the other recipient groups challenged with either SAF-derived control samples (inocula 2, 6, 7 and 11) or negative controls (inocula 12 and 13) developed scrapie symptoms during a long observation period of 545 dpi (Table 1).
Table 1.
Bioassay results for the phenol/chloroform group: first and second experiments
| Inoculaa | Description | Scrapie after IC | Death from other causes | Residual infectivity | WB PrPsc | Incubation periodsb |
|---|---|---|---|---|---|---|
| 1 | BioRad purified scrapie-associated fibrils | 4/4 | 0/4 | N/A | + | 85, 85, 85, 85 |
| Experiment 1: SAF treated with PK and phenol/chloroform (duration: 545 days) | ||||||
| 2 | Phenol/chloroform aqueous phase (Ph/Ch) | 0/5 | 1/5φ | Noφ | + | 413φ |
| 6 | Ph/Ch + β-recPrP oligomers | 0/7 | 2/7 | No | − | 300*, 424* |
| 7 | Ph/Ch + α-recPrP | 0/5 | 1/5 | No | − | 310* |
| 8 | Ph/Ch + DNase I + β-recPrP oligomers | 2/5 | 1/5 | No | + | 151, 246, 452* |
| 11 | Ph/Ch + β-recPrP fibrils | 0/7 | 3/7 | No | - | 424*, 508*, 515* |
| 12 | α - recPrP monomers | 0/4c | 0/4 | No | 545 c | |
| 13 | β- recPrP oligomers | 0/2 | 1/2 | No | 410* | |
| Experiment 2: SAF treated with PK and phenol/chloroform (duration: 480 days) | ||||||
| A | Ph/Ch II | 0/7 | 0/7 | No | ||
| B | Ph/Ch II + DNase I | 0/17 | 1/17 | No | − | 279* |
| C | Ph/Ch II + DNase I + α- recPrP monomers | 0/5 | 0/5 | No | ||
| D | Ph/Ch II +DNase I + β-recPrP oligomers | 3/19 | 0/19 | No | + | 148, 178, 207 |
Note: a, Inoculations were performed while avoiding cross contamination; for example, infectious inoculum 8 was inoculated following the 2 negative inocula 6 and 7. b, Period of time between injection and death/sacrifice upon the appearance of severe clinical signs in the animal (ataxia, rachitis, tremor). c, All animals that did not develop symptoms were sacrificed 545 days post inoculation. Φ, Scrapie infection observed in one control animal that was attributed to environmental contamination. This animal (death at 413 days) lived in a cage with 3 animals of another experimental group injected with contaminated inoculum from an unsuccessful PrPSc removal protocol involving GndHCl. Scrapie in this animal was confirmed by WB and histopathology, which revealed a lesion profile identical to that of 263K (data not shown). *, These animals died of causes other than scrapie and did not show clinical TSE signs or cerebral PrPSc deposition.
Nucleic acid content and electron microscopy analysis of inoculated samples
To characterize the nucleic acid content of our samples, the Agilent 2100 Bioanalyzer was used. To obtain an accurate view of the nucleic acids present before and after DNase I digestion, we analyzed both infectious inoculum 8 and the solution from which it was derived, inoculum 2. This inoculum (inoculum 2) contained the complete nucleic acid extract from the SAF preparation that included a markedly heterogeneous mixture of DNA and RNA species with sizes of < 300 nucleotides (Fig. 1C). After DNase I treatment (inoculum 8), the Agilent profile was much cleaner, and only 2 distinct small peaks corresponding to distinct populations of RNA with sizes averaging ∼27 and ∼55 nucleotides could be observed (Fig. 1A). Approximate molar mass ratios were calculated for these 27- and 55-nt-long nucleic acids elements with regard to PrPSc levels (quantitations of RNA elements provided in the legends of Figures 1 and 2). Ratios of 1 molecule of ssRNA per 12-14 and 72-100 molecules of PrPSc for the 27 nt and 55 nt RNA species, respectively, were found.
Figure 1.
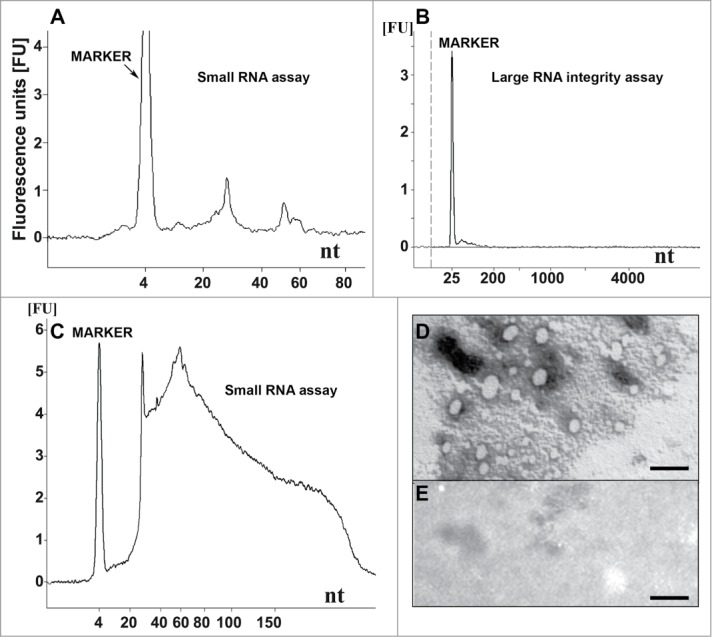
Nucleic acid analysis and formation of recPrP nanostructures. (A) Electropherogram of inoculum 8 (DNase I-digested inoculum 2) analyzed using a small RNA assay chip, showing 2 distinct populations of RNA with average lengths of approximately 27 and 55 nucleotides. Approximate quantities of oligonucleotides measured in each peak are 0.5 ng for the marker, 0.2 ng for the first peak (∼27 nt) and 0.07 ng for the second peak (∼55 nt). (B) Electropherogram of DNase I-digested inoculum 2 analyzed using a large RNA integrity assay chip, showing the absence of large cellular RNA and degradation products. (C) Inoculum 2 analyzed using a small RNA assay chip, showing the presence of cellular DNA (sizes ≤ 300 nucleotides). Approximately 37 ng of DNA is observed for 0.16 mg brain equivalent loaded. (D) Electron micrograph of inoculum 8 after negative staining, indicating the presence of ovoid and spherical aggregates of PrP with diameters of 7-40 nm. (E) Electron micrograph of control inoculum 7 (Ph/Ch + α-recPrP monomers), showing the absence of PrP aggregates (scale bar, 100 nm). For (A-C) 1 μl of sample was loaded onto the chip, which is approximately equal to 8 ng equivalent PrPSc and approximately 0.16 mg brain equivalent.
Figure 2.

Nucleic acid analysis from the second reconstitution experiment. Electropherogram of inoculum D, showing the same distribution of small RNA populations as in inoculum 8. Approximate quantities of each peak are 0.5 ng for the marker, 0.18 ng for the first peak and 0.05 ng for the second peak. A 1 μl sample was loaded onto the chip, which is approximately equal to 8 ng equivalent PrPSc and approximately 0.16 mg brain equivalent.
Another type of chip, the Agilent Large RNA Integrity Assay, which allows the quality of the nucleic acids to be evaluated, revealed the complete absence of contaminating large cellular RNA and/or DNA in inoculum 2 (Fig. 1B), indicating that the nucleic acid preparation was of good purity.
Electron microscopy (EM) analysis of the oligomeric recPrP in inoculum 8 displayed ovoid and spherical aggregates (diameter ∼7–40 nm; Fig. 1D). These supramolecular structures of recPrP were not observed in the control sample containing monomeric α-helical recPrP (inoculum 7; Fig. 1E). No filament-like structures were observed in any of the preparations, indicating that no fibrils were formed.
Confirmation of results in a second experiment—Bioassay and RNA analysis
To confirm the initial results, a similarly designed second experiment (lasting 480 days) was performed using independently prepared solutions (Table 1). Here, 3 animals out of 19 (16%) developed clinical signs of scrapie at 148, 178, and 207 dpi with inoculum D. This sample was prepared identically to the infectious inoculum 8 of the first experiment. None of the negative control animals (29 in total: inocula A, B and C) developed clinical signs of scrapie (Table 1). Analysis of inoculum D using the Agilent bioanalyzer also revealed 2 populations of small RNAs (in the same size range; i.e., 27 and 55 nt) that were completely consistent with the findings from the first experiment (Fig. 2).
Two new sets of experiments confirmed the absence of 263K contamination but failed to induce scrapie infection
Two other experiments (corresponding to second and third repeats of the initial experiment) were performed identically to the first experiment to further demonstrate the reproducibility of the method. These experiments were conducted in parallel in two separate laboratories (1. the laboratory of Jean-Philippe Deslys at the CEA, France and 2. the laboratory of Maurizio Pocchiari at the Istituto Superiore di Sanita (ISS), Italy) with material belonging to each laboratory (including new 263K brain homogenate). Apart from the positive 263K controls, none of the inoculated animals developed disease (sacrificed at ∼400 (CEA) and ∼300 (ISS) dpi) in either studies, confirming the efficacy of the phenol/chloroform protocol in eliminating all detectable infectivity associated with 263K SAF preparations. Strikingly, the quality and content of the DNase I-treated samples of both repeat experiments were markedly different (in one experiment, the RNA peaks were not present at all, and in the other, contaminating brain cellular RNA was observed; data not shown).
Phenol/chloroform extraction of nucleic acids from 263K SAF preparations: Absence of residual infectivity in the final product
Of key importance for the study described in this report was the reliable elimination of residual prion infectivity in the final nucleic acid extract. This extract was used to generate the infectious RNA-β-recPrP inocula (inoculum 8 and D) described above. RNA was purified from SAF-preparations obtained from a 20% 263K scrapie brain homogenate containing PrPSc and high titers of infectivity (2 × 109 LD50/g of brain). An alternative method consisting of a PK treatment and a 4 M GndHCl (details provided in Materials and Methods) treatment clearly failed to eliminate infectivity completely (Table 2) and was unable to modify the strain characteristics of this agent. For the phenol/chloroform extraction method, we addressed whether our nucleic acid extracts contained residual infectivity using three approaches to verify the findings. First, using serial Protein Misfolding Cyclic Amplification (sPMCA), we examined whether our method (extensive 24 hr PK and phenol/chloroform nucleic acid extraction) could completely eliminate the PrPSc seeding activity (the putative biochemical correlate to scrapie infectivity30) from purified 263K SAFs. For this part of the study, we extracted infectious PrPSc preparations from 263K brain homogenate using a distinct protocol,31 which yielded comparable amounts of PrPSc compared with the Bio-Rad protocol used to produce SAF-preparations for RNA extraction in this study (∼70 μg PrPSc per gram of brain tissue in comparison to 50–100 μg PrPSc per gram of brain tissue obtained using the Bio-Rad protocol). No residual PrPSc seeding activity could be detected after four rounds of PMCA (Fig. 3A, lanes 11 and 12) in the RNA extracts obtained using the phenol/chloroform extraction protocol. This observation indicated that the residual seeding activity, if any was present in these extracts, had to be lower than that present in 1 × 10-12 g of 263K scrapie brain tissue (Fig. 3A, lanes 2 and 3; theoretically, this value would correspond to an infectivity of less than 10-3 LD50). To exclude artifactual inhibition of PMCA by the phenol/chloroform-nucleic acid preparation, negative samples were spiked with minimal amounts of 263K-infected brain homogenate, and these samples amplified correctly (Fig. 3A, lanes 13 and 14).
Table 2.
Bioassay results for the guanidinium-HCl (GndHCl) group
| Inocula | Description | Scrapie after IC | Death from other causes | Residual infectivity | WB PrPsc | Incubation periodsa |
|---|---|---|---|---|---|---|
| 1 | BioRad-purified scrapie-associated fibrils | 4/4 | 0/4 | N/A | + | 85, 85, 85, 85 |
| Experiment 1: SAF treated with PK and Gnd-HCl (duration: 545 days) | ||||||
| 3 | GndHCl 4 M (GndHCl) | 5/5 | 0/5 | Yes | + | 207, 231, 231, 247, 372, |
| 4 | GndHCl + β-recPrP oligomers | 5/7 | 2/7 | Yes | + | 226, 226, 324, 324, 372, 399*, 457* |
| 5 | GndHCl α-recPrP monomers | 4/6 | 0/6 | Yes | + | 227, 231, 241, 437 |
| 9 | GndHCl + β-recPrP dimers | 6/7 | 0/7 | Yes | + | 123, 196, 207, 214, 214, 227 |
| 10 | GndHCl + β-recPrP fibrils | 2/5 | 1/5 | Yes | + | 269, 310, 505* |
Note: All groups of animals inoculated with preparations treated with GndHCl (4 M) developed scrapie due to the residual 263K scrapie agent.
Figure 3.
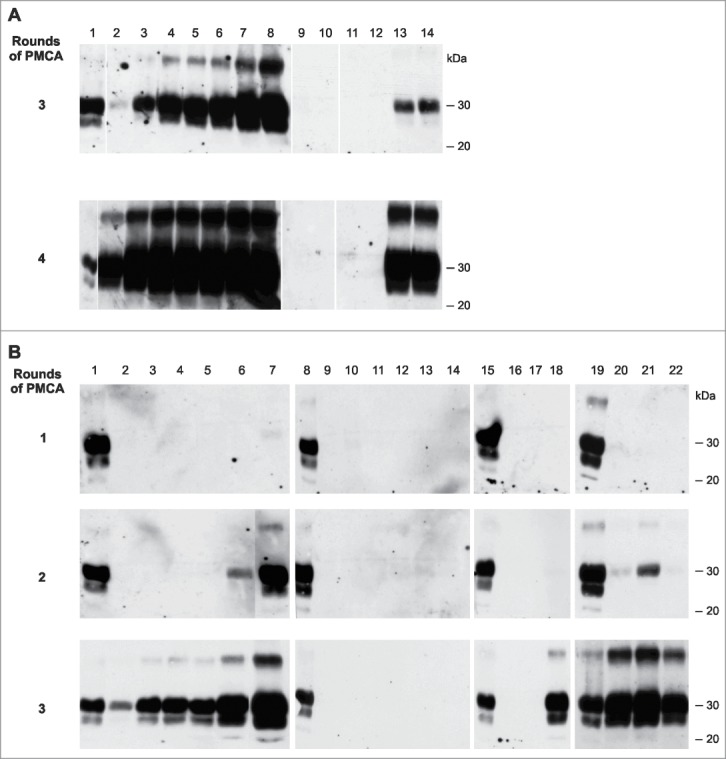
Testing for PrPSc seeding activity in phenol/chloroform extracts and bioassay samples (A) Testing phenol/chloroform extracts (Ph/Ch) from a crude preparation of 263K scrapie-associated fibrils that had been purified using a previously published protocol31 for residual PrPSc seeding activity using serial PMCA (the third and fourth PMCA rounds are shown). Lanes 1: PK digested 263K scrapie hamster brain homogenate containing 5 × 10−7 g of 263K hamster brain tissue. Lanes 2–8: Reference PMCA samples seeded with 1 × 10−12 (lanes 2 and 3), 1 × 10−11 (lanes 4 and 5), 1 × 10−10 (lanes 6 and 7) or 1 × 10−9 g 263K hamster brain tissue (lane 8). Lanes 9 and 10: Phenol/chloroform extracts isolated from uninfected hamster brain. Lanes 11 and 12: Ph/Ch extracts isolated from 263K scrapie hamster brain (corresponding to ∼500 ng 'equivalent PrPSc' per lane, i.e., to an approximately 2.5-fold amount of the intracerebral inoculum administered to hamsters, which was 200 ng 'equivalent PrPSc'). Lanes 13 and 14: Ph/Ch extract isolated from 263K scrapie hamster brain spiked with 1 × 10−11 g of 263K scrapie brain tissue following phenol/chloroform extraction. (B) Testing various bioassay inocula for the amount of residual or newly produced PrP seeding activity using serial PMCA. Lanes 1, 8, 15 and 19: PK-digested 263K scrapie hamster brain homogenate containing 5 × 10−7 g of 263K hamster brain tissue. Lanes 2–7: Reference PMCA samples seeded with 1 × 10−12 (lanes 2 and 3), 1 × 10−11 (lanes 4 and 5), 1 × 10−10 (lane 6) or 1 × 10−9 g 263K hamster brain tissue (lane 7). Lanes 9–14: Negative controls without seeding material (i.e., PMCA with normal hamster brain homogenate only). Lane 16: PMCA seeded with 10 μl of inoculum 6 (Ph/Ch + β-rec PrP oligomers). Lane 17: PMCA seeded with 6 μl of inoculum 7 (Ph/Ch + α-rec PrP monomers). Lane 18: PMCA seeded with 6 μl of inoculum 8 (Ph/Ch + DNase I + β-rec PrP oligomers). Lanes 20–22: Same samples as in lanes 16–18, spiked with 1 × 10−10 g of 263K scrapie brain tissue.
Second, we verified by sPMCA whether the various inocula from the original experiment, inocula 6 (Ph/Ch + β-rec PrP oligomers), 7 (Ph/Ch + α-recPrP monomers) and 8 (Ph/Ch + DNase I + β-rec PrP oligomers), all of which were made from the same starting solution of 263K-infected brain-derived nucleic acids (Ph/Ch), contained residual PrPSc seeding activity. Here we found that inoculum 6 and inoculum 7 contained no detectable residual seeding activity (i.e., limit of detection [LOD] = the seeding activity present in 1 × 10−12 g of 263K scrapie brain tissue; Fig. 3B, lanes 16 and 17). In contrast, PrP seeding activity was detected in the infectious inoculum 8, which corresponded approximately to the seeding activity present in 1 × 10−10 – 1 × 10−11 g of 263K scrapie brain tissue (Fig. 3B, lane 18, compared against the positive controls in lanes 2-7). These PMCA results were in perfect agreement with the results obtained in the bioassays: None (0/7) of the animals for inoculum 6, 0/5 for inoculum 7, and 2/5 for inoculum 8 developed clinical signs of scrapie (Table 1). The sPMCA experiments were performed twice with identical results. These results confirmed that the seeding activity (and corresponding bioassay infectivity) of inoculum 8 was not an artifact caused by residual PrPSc infectivity from the prepared SAF material.
In the third approach, we performed phenol/chloroform extraction and inoculated samples from the various phases of the extraction directly into Syrian hamsters to test for the presence of residual infectivity. The bioassay results confirmed the absence of infectivity (data not shown) in all fractions: 1) the aqueous phase containing the nucleic acids and 2) the interphase containing the denatured proteins, which provided final proof (by bioassay) that no residual infectivity was present in the nucleic acid brain extracts. More importantly, the phenol/chloroform extraction alone (without the 24 h PK treatment) proved to be sufficient to completely inactivate 263K infectivity in both the aqueous and interphase fractions, demonstrating the robustness of the method used.
RNA boosts the self-replication of seeding-active PrP
We performed protein misfolding cyclic amplification (PMCA) experiments to further investigate the involvement of RNA in the structural conversion of PrP (Fig. 4). PMCA-mediated amplification of seeding-active PK-resistant prion protein (PrPres) was suppressed either by treatment of the PMCA substrate (i.e., normal hamster brain homogenate [NBH], Fig. 4c), with Benzonase (Fig. 4) or RNAse A (Fig. 4C), 2 RNA-degrading enzymes, but was reconstituted by the addition of polyA RNA after Benzonase digestion (Fig. 4B). Remarkably, after nuclease treatments of the PrPSc seeds and NBHs, PrPres amplification was still possible, but only during the first round of PMCA (Fig. 4A and C). This result further indicated that RNA molecules are present within the original seeding material (i.e., 263K extracts) and that this material is protected from Benzonase and RNase A treatments, which could thus support the initial PrPres amplification. However, with progressing serial dilutions during PMCA and the use of NBHs devoid of RNA, original 263K seed material is diluted out and could not further support the conversion of PrPc into PrPres.
Figure 4.

In-vitro amplification of PrPSc is fully reconstituted by the addition of synthetic polyA RNA. Serial PMCA (5 rounds) was performed with nuclease-treated PrPSc seeds purified from 263K scrapie hamster brains and differently pretreated normal hamster brain homogenates (NBHs) as substrates. NBHs were digested with (A) Benzonase, (B) Benzonase subsequently spiked with PolyA RNA (200 μg/ml), or (C) RNAse A. The amount of original seeding material corresponded to an extract from 1 × 10−6 g 263K scrapie hamster brain homogenate. After each PMCA round, samples were diluted 1:5 in the corresponding substrates. P: PK digested 263K scrapie hamster brain homogenate containing 5 × 10−7 g of 263K hamster brain tissue, which served as a western blot positive control.
Neuropathological and immunohistochemical analyses
To confirm that the animals infected with inoculum 8 died from authentic prion disease, neuropathological and PrPSc immunohistochemical analyses were performed (Fig. 5). Histopathology revealed spongiosis typical of TSE infection (Fig. 5E). Compared to classical 263K-infected hamster brains, the 2 animals that had been identified as positive during the first experiment with inoculum 8 (animals 8.1 and 8.2) showed more intense spongiosis. In contrast, animals injected with similar inocula prepared using a GndHCl denaturation protocol (an alternative protein/PrPSc elimination method that did not work, i.e., did not completely eliminate infectivity; see Materials and Methods and Table 2 for details) developed vacuolar changes similar to those of the 263K strain.
Figure 5.

Histopathology and PrP-immunohistochemistry of brain sections from hamsters challenged with inoculum 8 (hamster 8.1: 151 dpi). Hematoxylin and eosin (H&E)-stained samples (2 columns on the left) and PrP immunohistochemistry using anti-PrP-antibody SAF32 (right column). (A-C) Control hamster, (D-F) hamster challenged with inoculum 8.1 (terminal phase, 76 dpi), (G-I) hamster challenged with SAFs of 263K scrapie agent (terminal phase, 76 dpi). The brain of the hamster challenged with inoculum 8.1 showed intense tissue damage of the hippocampus, extensive spongiosis, severe gliosis and neuronal loss (D, E). PrPSc immuno-detection (F) displayed a diffuse and widespread distribution that has not previously been observed in hamsters challenged with 263K SAFs (I).
The animals infected in the second experiment (injected with inoculum D) also showed higher spongiosis than the regular 263K strain (data not shown). However, after the second passage, the lesions were similar to those of 263K scrapie. Of note, the distinct histopathological lesions observed with the infectious recPrP preparations included neuronal loss, particularly in the pyramidal cells of the hippocampus (Fig. 5D). Clinically, all animals showed signs similar to those of the 263K strain.
The accumulations of PrPSc in the brain of animals that developed disease after challenge with samples 8 and D were analyzed by immunohistochemistry.32 In all passages of these inocula (inoculum 8.1: 1st [Fig. 6C, D], 2nd [Fig. 6E, F] and 3rd [Fig. 7C, D] passages; inoculum 8.2: 1st [Fig. 6G, H] and 2nd [Fig. 6I, J] passages, and inoculum D: 1st [Fig. 6K, L] and 2nd [Fig. 7E, F] passages) the animals showed a more pronounced PrPSc accumulation compared to the 263K-inoculated animals (Fig. 6 and 7). Notably, a unique PrPSc deposition pattern was observed in the molecular layer of the hippocampal dentate gyrus. This synapse-rich region showed immunoreactivity after incubation with each of the anti-PrP monoclonal antibodies used in this study (i.e., SAF32 and 3F4; Fig. 6). Compared to 263K scrapie, a much more intense PrPSc immunostaining was observed in the inner zone of the molecular layer that involved the proximal part of the granule cell dendrites. This PrPSc immunolabeling was particularly well visualized with the 3F4 anti-PrP antibody (Fig. 6 and 7). PrPSc immunostaining in the molecular layer was predominantly granular or punctate, i.e., of the synaptic type (Fig. 7 B, D and F), in agreement with the synaptic localization of prion proteins.2
Figure 6.

PrP-immunohistochemistry of hippocampus sections from infected hamsters using 3F4 and SAF32 anti-PrP antibodies. (A, B) 263K scrapie agent (IP: ∼80 dpi). (C, D) Inoculum 8.1 (1st passage; IP: 151 dpi). (E, F) Inoculum 8.1 (2nd passage; IP: 76 dpi). (G, H) Inoculum 8.2 (1st passage; IP: 246 dpi). I, J, Inoculum 8.2 (2nd passage; IP: 76 dpi). (K, L) Inoculum D (1st passage; IP: 148 dpi). The intensity of labeling for all passages was consistently stronger in the sections of animals infected with inocula 8.1, 8.2 and D compared with those of animals inoculated with 263K scrapie agent. A peculiar lamination pattern that had not previously been described in scrapie pathology can be observed in the molecular layer for inocula 8.1, 8.2 and D. This lamination is more clearly visible with the 3F4 antibody (arrows).
Figure 7.
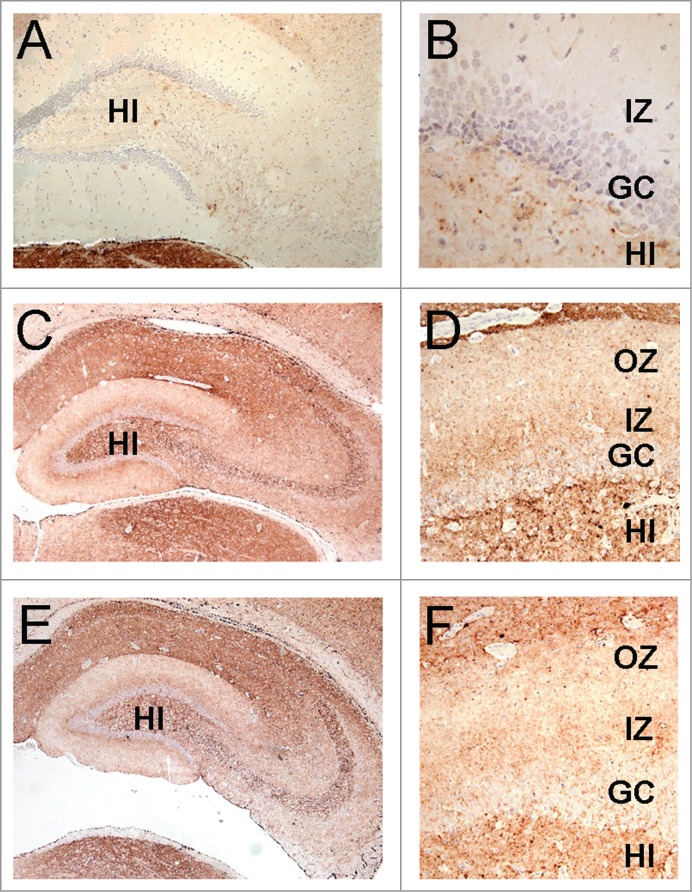
The lamination pattern observed in the hippocampal molecular layer corresponds to differential PrP labeling on synapses from specific areas. PrP-immunohistochemistry (3F4 antibody) of hippocampal sections from infected hamsters. (A, B) 263K scrapie agent (IP: ∼80 dpi). (C, D) inoculum 8.1 (3rd passage; IP: ∼80 dpi). (E, F) inoculum D (2nd passage; IP: ∼80 dpi). Lamination pattern corresponding to synaptic PrP immunostaining. (B, D, and F) Granular/punctate PrP depositions. HI (hilus), GC (granule cells), IZ (inner zone of the molecular layer), OZ (outer zone of the molecular layer). Magnification: (A) (x100), (B) (x400) (C and E) (x50), (D and F) (x200).
Electrophoretic mobility and physicochemical properties of PrPSc
Despite their immunohistopathological differences during the first passages, all infected animals exhibited a PrPSc western blot (WB) profile indistinguishable from that of the reference 263K scrapie strain (Fig. 8A and C) under normal WB conditions. More extensive investigations into the conformational stability of PrPSc during PK digestion after exposure to various pH values or guanidinium-HCl (GndHCl) concentrations and biophysical analyses of the PrPSc structure by Fourier-transform-infrared-spectroscopy33 did not reveal any detectable differences compared with 263K scrapie-associated PrPSc (data not shown).
Figure 8.
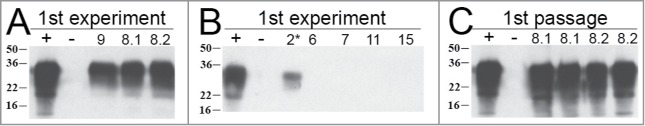
Detection of PrPSc in the brains of hamsters from the first and second passage (data from the first reconstitution experiment). (A) Brain PrPSc profile of infected animals from first reconstitution experiment. Inocula 9 (lane 3), 8.1 (lane 4; 151 dpi) and 8.2 (lane 5; 246 dpi). Lane 1: Positive control from 263K scrapie hamster; lane 2: control sample from healthy hamster. (B) Brain PrPSc profile of infected animals from first reconstitution experiment. Inocula 2* (lane 3; environmentally contaminated animal), 6 (lane 4), 7 (lane 5), 11 (lane 6) and 15 (lane 7). Positive control from 263K scrapie hamster; lane 2: control sample from healthy hamster. A selection of animals that died of causes not related to scrapie or deliberate infection are represented here. (C) First passage: WB profile of 2 distinct hamsters inoculated with BH of animal 8.1 (lanes 3 and 4); WB profile of 2 distinct hamsters inoculated with BH of animal 8.2 (lanes 5 and 6).
Discussion
It is worth noting that experiments in the prion field have periodically reported discrepancies between the amount of PrPSc (the sole protagonist according to the prion hypothesis) and the level of infectivity, such as recent studies that described in vivo robust scrapie infections in apparent absence of PrPSc,8,34,35 or the in vitro generation of PrPSc in the complete absence of infectivity.23 These results favor the persistent idea that a supplementary co-factor is necessary to induce infectivity, thereby justifying the investment in efforts to determine its identity.
In this study, we identified small RNAs in SAF preparations that were purified from 263K scrapie-infected hamster brains. Furthermore, we used bioassays, the 'gold standard' in prion infectivity investigations, to demonstrate that non-infectious recombinant PrP could become infectious after denaturation and conversion to a β-sheet rich form in the presence of RNA extracted from purified prions (SAFs). Experimentally, this finding was shown by the induction of scrapie in hamsters following the intracerebral injection of DNase I-treated inoculum 8. In contrast, inoculum 6, the parent of inoculum 8 (i.e., before DNase I treatment), was not infectious, most likely due to the large excess of residual contaminating DNA present in this sample (Fig. 1c) that prevented the de novo RNA-PrP interaction that generated infectivity in inoculum 8. We hypothesize that artificial infectious ribonucleoprotein complexes (263KRNA-β-sheeted recPrP) generated in vitro could trigger the in vivo formation of infectious 263KRNA-PrPSc complexes in hamster brains. When observed under electron microscopy, the size range of molecular complexes 263KRNA-β-sheeted recPrP was in agreement with those proposed for the most infectious prion protein particles.28 The fact that the RNA material was preserved from digestion during the ribonuclease treatment step of the SAF purification procedure strongly suggests a tight association with PrPSc. Moreover, the size ranges of the RNA elements (∼27 and ∼55 nucleotides) are in full agreement with previously reported data for nucleic acids extracted from highly purified PrPSc 25 and with the minimal length of the RNA co-factor required for efficient in vitro propagation of PrPres in PMCA reactions.36 Our own PMCA experiments with the 263K agent also supported this association. Indeed, the observation that Benzonase- or RNAse A-treated 263K scrapie preparations were able to effectively seed sPMCA reactions in NBH devoid of nucleic acids only for one round and that spiking Benzonase-digested NBH with polyA RNA resulted in full recovery of seeding activity for subsequent PMCA rounds was consistent with the notion that a protected RNA component was part of the hamster PrPSc.24,25,37
The ability of this “precursor” infectious agent (263KRNA-recPrPSc) to induce scrapie-like infection was validated in two separate bioassays by the development of a neurological disease that resembled typical experimental scrapie in a total of 5/24 (20% attack rate) wild-type hamsters. Neuropathology and PrP immunostaining of the animals inoculated with the reconstituted infectious agent (263KRNA-recPrPSc) showed qualitative differences compared with hamsters infected with the 263K strain. Indeed, for animals inoculated with inocula 8 and D, the lesions (spongiosis, neuronal loss and gliosis) appeared more severe, particularly during the first passage, but were less perceptible, if at all, during later passages. This behavioral change supports a type of adaptation mechanism from the initial inoculated configuration (263KRNA-recPrPSc) to the natural 263K strain (263KRNA-PrPSc), thus arguing against mere involuntary contamination with the 263K pathogen. Although the vacuolation profiles derived from inocula 8.1, 8.2 and D became similar to that of 263K upon second and third passage in hamsters, the intense neuronal loss in the CA3-CA4 hippocampal region and the appearance of PrPSc immunolabeling observed in the molecular layer of the hippocampal dentate gyrus provided further discriminatory features of the scrapie infectivity generated in vitro.
Interestingly, the patterns of PrPSc deposition that were only observed with inocula 8.1, 8.2 and D highlight the lamination of the hippocampus, mirroring normal cortico-hippocampal projections.38 In contrast, the granular/punctate aspect of PrP immunostaining observed with all infectious inocula designates PrP as member of the synaptic protein family.39, 40 Among the various sub-microscopic neuronal sites described, the PrP synaptic location appears crucial for improving our understanding of synaptolysis, the key event in the neurodegenerative process observed during TSE infection. 41
Further experiments are necessary to clarify whether our synthetic biomolecular complex directly produces authentic 263K or a transient “263K subvariant” that requires adaptation before reverting to its original configuration.
Obviously, the infectivity titers observed with inocula 8 and D were quite low. Therefore, in future studies, it will be important to attempt to optimize the reconstitution conditions by possibly 1) using higher concentrations of either the RNA or the recPrP or both, 2) inoculating higher doses into animals and 3) investigating the influence of other factors (e.g., salts, lipids or glycosaminoglycans). Indeed, it appears necessary to improve the reproducibility of our experimental approach, bearing in mind that two additional repeat experiments (one in a different laboratory: M. Pocchiari lab at ISS, Italy) performed with different sources of infected brain material (new batches of 263K brain homogenate were used) did not result in disease transmission by samples prepared identically to our inocula 8 and D. These negative results most likely reflected distinct properties of the new brain homogenates used (e.g., lower initial titer, number of freezing and thawing cycles) that ultimately could have interfered with the efficiency of RNA extraction or affected its quality. The two additional replication attempts did however confirm that 1) the phenol/chloroform extraction protocol eliminated scrapie infectivity (because no infectivity was detected) below the detection limit of the bioassay and 2) the quality of the extracted RNA is crucial (possibly requiring an adapted protocol for each new infectious material). Opportunely in a pilot study, a first attempt at cloning the aqueous phase RNA sequences resulted in cDNA clones with sizes in the corresponding range. Although the work is incomplete at the moment, it confirmed that these two RNA populations were present.
Taken together, we have used bioassays and PMCA to show that a 24 h PK treatment combined with a phenol/chloroform extraction effectively eliminates infectivity and PrPSc seeding activity of SAFs while leaving the associated nucleic acids intact, which subsequently can become infectious by interacting with recPrP oligomers under appropriate conditions (conversion of recombinant PrP monomers into β-sheeted recPrP oligomers using heat; this procedure is detailed in the Materials and Methods). However, the conclusion that PrP may play an important role does not completely preclude the involvement of other potential proteins. Indeed, it has been shown recently that a group of proteins and peptides could provide the necessary components for infectivity in the absence of PrPSc.29,42 By adapting the approach described in the current work, the analysis of RNA-protein interactions may provide a way to further specify the implication of these “other proteins” in prion infectivity. Recently, several attempts have been made to show that recombinant PrP can be converted from an innocuous state into an infectious one.14–16 These studies have not provided completely convincing data in favor of the protein-only hypothesis.43,44 Indeed, in all attempts, the infectious recombinant PrP preparations were obtained in the presence of additional components, possibly nucleic acids that were present either in normal brain homogenate,14 liver-purified RNA15 or partially purified PrPSc.16 These experiments relied on PMCA-based conversion, demonstrating that RNA was essential for transforming non-infectious native PrPc into an infectious complex.18,36,45 Of the plethora of co-factors tested in the PMCA reaction (lipids, heparin, plasminogen, RNA, polyglutamate, NADH), RNA seems to be the best candidate for producing an infectious product.46 However, this conclusion has been challenged in a study reporting that membrane lipids alone could provide cofactor activity.20 Nevertheless, this result requires confirmation due to the lack of complete RNA elimination in the control experiments.47
In our study, the infectious entity contains RNA extracted from the prion infectious agent itself (263KPrion), which was unlike the ubiquitous RNA of cellular or synthetic origin used by others in the studies described above.14-16 It is notable here that we generated diseases that closely resembled that of the initial 263K, in contrast to previous studies that reported the induction of 263K-distinct phenotypes. Future studies with other hamster strains will be important to specify whether the SAF preparation-purified RNA could play a role in the conversion process and strain stabilization.37
Although some questions remain unanswered, such as the identity of the 263K agent-extracted RNA sequences, the present work provides significant evidence that the biology and infectious properties of the TSE agent are not governed solely by the abnormal form of prion protein (PrPSc). The novel approach described in this manuscript offers a new way to investigate the etiological uncertainties of TSE. Knowledge of the exact molecular nature of the TSE infectious agent will allow researchers to develop new tools to accurately diagnose and/or detect the agent (i.e., for securing blood transfusion), along with exploring new therapeutic strategies against these devastating neurodegenerative diseases.
Note:
The initial experimental design and preliminary results of this study were posted on Nature Precedings in 2009 (Simoneau, et al. Small critical RNAs in the scrapie agent. Website: http://hdl.handle.net/10101/npre.2009.3344.1).
Materials and Methods
PrPSc purification from 263K-infected brains
Brain homogenate (20%) was prepared from 263K-infected hamster brains collected at the terminal stage of the disease (∼80 dpi; titer of ∼2 × 109, 50% lethal doses (LD50) per gram of brain tissue) and pooled for SAF purification. The homogenate was incubated with 20 μg/ml RNase A (Ambion) at 37°C for 2 hrs. Then, CaCl2 was added to a final concentration of 5 mM, and the homogenate was incubated for an additional 2 hrs with 20 U/ml of DNase I (Roche). PrPSc was then purified from this homogenate using the BioRad TeSeE purification kit according to the manufacturer's instructions (500 μl of homogenate per tube). This method yields a relatively crude (compared to other methods) and relatively less compacted preparation of PrPSc and other proteins.
Elimination of PrPSc and infectivity from scrapie-associated fibril (SAFs) preparations
Two markedly distinct approaches were used to attempt to eliminate PrPSc and infectivity while sparing any hypothetical associated co-factor. They are described in detail below.
1) Extensive PK digestion combined with phenol/chloroform extraction: SAF pellets (see PrPSc purification above) were resuspended in 50 μl of PBS containing 0.5% SDS and sonicated for 1 minute at maximal strength. The resuspended pellets from 10 tubes (total volume: 500 μl) were pooled and mixed with 2.5 ml of PBS/0.5% SDS containing 500 μg/ml of PK and incubated overnight at 37°C (∼24 hrs). The PK reaction was stopped by heating the samples to 70°C for 5 minutes. Nucleic acids from this solution were then extracted using phenol/chloroform/isoamyl alcohol (25:24:1) (# 77617 Sigma) and collecting the aqueous phase. Extreme caution was taken to avoid touching the meniscus containing the denatured proteins and potentially residual PrPSc. This solution was designated as solution A.
2) Extensive PK digestion combined with 4 M Gnd-HCl denaturation: SAF pellets were resuspended in 50 μl of 2X SB3-14 solution (0.24% SB3-14, 20 mM sodium phosphate pH 7.2, 32 mM NaCl) and sonicated in a cup horn sonicator for 1 minute at the highest setting. The resuspended pellets from 10 tubes (500 μl) were pooled together and mixed with 2500 μl of a solution composed of 992 μl of 2X SB3-14 solution, 1500 μl of 8 M GndHCl, 8 μl of 20 mg/ml PK (final concentrations of PK and GndHCl were ∼50 μg/ml and 4 M, respectively) and incubated overnight at 37°C (∼24 hrs). The PK reaction was stopped by incubating the sample at 70°C for 20 minutes and subsequently adding PMSF to a final concentration of 5 mM. This solution was designated as solution B1
A 1 μl/ml solution of SUPERase-In (Ambion, Life Technologies, France) was added to solutions A and B to suppress any potential RNAse activity, and from this stage on, all reagents and materials were prepared under RNase-free conditions.
Converting recombinant PrP in the presence of purified nucleic acids
Recombinant hamster PrP (PrP 23-231 AllPrion ref: P00025) was suspended in Milli-Q H2O at a concentration of 1 μg/μl, aliquoted and stored at -20°C. Beta-sheet conversion of this recombinant PrP during reconstitution (mixing of PrP and nucleic acids from solution A) was achieved using two methods: heating (72°C), which generated the PrP oligomers, and using 3 M urea at pH 5 to generate PrP fibrils (see below).48,49
Nucleic acid analysis using a Bioanalyzer 2100
Aliquots of the test solutions (1 μl each) from the PrPSc/infectivity elimination procedures (or from the inocula containing recPrP) were added to wells of a lab-on-a-chip device (2100 Bioanalyzer, Small RNA Assay and Large RNA integrity assay, Agilent Technologies) and analyzed according to the manufacturer's recommendations.
Electron microcopy
Samples of the inocula (5 μl each) prepared as described above were deposited onto grids and negatively stained as previously described.48
Histopathology
Formalin-fixed brains were embedded in paraffin wax and processed for histopathology and PrP immunocytochemistry as previously described.32 The anti-PrP antibodies used were SAF 32 or 3F4. Sections were analyzed by an experimenter who was blind to the treatment conditions.
Normalization of samples
Based on previous experience in our laboratory with the 263K scrapie hamster model, we estimated the quantity of PrPSc in the Bio-Rad pellets (see PrPSc purification) to be between 5 and 10 μg per pellet. We referred to these amounts of PrPSc as 'equivalent PrPSc'. The initial quantity was not of critical importance per se but was helpful as a reference measure for normalizing the various samples throughout the experiment and also as an indicator of how much recombinant PrP needed to be added to the different samples.
Attempt to reconstitute infectious prions: mixing solution A with β-sheeted oligomeric recombinant PrP
Solution A was dialyzed against 20 mM MOPS pH 7.2 (MOPS buffer) using Slide-A-Lyzer 2K MWCO dialysis cassettes (Pierce) according to the manufacturer's instructions. The buffer was replaced with fresh buffer after 3 hours, and the dialysis was continued overnight. The 'equivalent PrPSc' of the dialyzed solution was calculated (taking into account the initial volume prior to dialysis and the final volume after dialysis) and used with the following preparations:
To prepare inocula 6 (72°C, β-sheeted recPrP) and 8 (72°C, β-sheeted recPrP + DNase I), 4 μg of 'equivalent PrPSc' from MOPS dialyzed solution A was combined with MOPS buffer to a final volume of 400 μl. In the case of inoculum 8, 1 μl of 2 M CaCl2 (final concentration 4 mM) and 1 μl of DNase I were added and then incubated at 37°C for 60 minutes then at 75°C for 10 minutes (to inactivate the DNase I). A 4 μg sample (1 μg/μl) of recombinant hamster PrP (recPrP; AllPrion) in a total of 100 μl of 20 mM MOPS pH 7.2 was heated to 72°C for 20 minutes and immediately mixed with the freshly prepared 400 μl (performed separately for both inocula). Under these conditions, hamster recombinant PrP 23-231 forms β-sheeted PrP oligomers.48
Attempt to reconstitute infectious prions: mixing solution A with β-sheeted recombinant PrP fibrils
Solution A was dialyzed as described above against 0.2 M NaCl, 50 mM sodium acetate pH 5 (sodium acetate buffer).
To prepare inoculum 11, 4 μg of hamster recPrP and 1 μl of SUPERase-In were added to 140 μl of sodium acetate buffer containing 10 M urea. In parallel, 4 μg of 'equivalent PrPSc' from sodium acetate dialyzed solution A was brought to a final volume of 350 μl in sodium acetate buffer and then mixed with the 145 μl solution that had been freshly prepared. The final solution (∼500 μl) was incubated at 37°C for ∼48 hrs. Under these conditions, recPrP forms PrP fibrils.49 The solution was then dialyzed as described above against PBS before inoculation.
Bioassays in Syrian hamsters
All samples were kept at 4°C until injection day (no longer than 3 days). On the day of the injection, samples were diluted to a final 'equivalent PrPSc' of 4 ng/μl with either PBS or MOPS. Each hamster received a total of 50 μl in the right hemisphere of the brain. The order of injection was: inocula 1, 2, 7, 8, 6, 11, 12, 13. Several control solutions were injected into the hamsters. These were prepared as detailed above, usually with one of the components or one of the steps omitted. These include inocula 1, 2, 7, 12 and 13 (Table 1).
Protein Misfolding Cyclic Amplification (PMCA)
A detailed protocol for the PMCA method, which describes all steps to avoid sample contamination and achieve robust reproducibility, is published elsewhere.30 Briefly, 10% brain homogenate from uninfected Syrian hamsters, serving as a PrPc substrate, was prepared in conversion-buffer (PBS containing complete protease inhibitor cocktail [Roche], 4 mM EDTA and 1% Triton X-100, pH 7.4) and kept at –70°C until use. Samples in a total volume of 150 μl were pipetted into 0.5 ml Eppendorf safe-lock tubes. To analyze Solution A (Ph/Ch) and determine whether it contained residual PrPSc-associated seeding activity, 30 μl aliquots containing ∼500 ng “equivalent” PrPSc were mixed with 120 μl PMCA substrate. This solution represented an approximately 2.5-fold higher quantity of 'equivalent PrPSc' than was inoculated intracerebrally into hamsters (200 ng of 'equivalent PrPSc'). Tubes were subjected to cycles of 1 h incubation at 37°C followed by a 40 sec sonication pulse at approximately 200 Watts (Misonix 4000). One PMCA round consisted of 24 cycles. After each round, the samples were diluted 1:5, and a new round of PMCA cycles was performed.
PMCA samples were digested with 150 μg/ml Proteinase K at 55°C for 1 h and then centrifuged for 1 min at 13,000 rpm in a table top centrifuge. The supernatants were collected, and the PK digestion was stopped by adding the same volume of 2 × sample-loading buffer (Laemmli) and incubating for 10 min at 99°C. To ensure complete inactivation of Proteinase K, 2.5 mM PMSF was added after boiling. Samples (10 μl) were loaded onto 12% PAA gels, and proteins were separated using SDS polyacrylamide gel electrophoresis and blotted onto PVDF membranes. Blot membranes were probed with mAb 3F4, and the immunoreactive bands were visualized using CDP-star reagent and subsequent film exposure for 30 min.50
PrPSc purification for PMCA reaction
Purification of PrPSc from brain homogenate of intracerebrally infected Syrian hamsters with 263K scrapie (terminal stage of disease) was performed as previously published31 with the following modifications: 10 aliquots of 500 μl 20% scrapie brain homogenate or brain homogenate from uninfected animals were prepared in phosphate buffered saline (PBS). The homogenates were digested with RNAse A (20 μg/ml) at 37°C for 2 h, and after adding CaCl2 (5 mM) and 20 U/ml DNase I, a further incubation for 2 h at 37°C was performed prior to carrying out the purification procedure described previously. 31 This protocol produces a crude SAF extract with a high recovery of PrPSc (∼70 μg PrPSc per gram brain tissue). However, within this extract, PrPSc makes up only ∼25% of the total protein.
Nucleic acid depletion of normal hamster brain homogenates used as substrates in PMCA
Normal hamster brain homogenate (10% w/v) was digested with Benzonase (1,000 U/ml; Boehringer, Ingelheim) or RNase A (70 U/ml; Qiagen, Hilden) for 5 h at 37°C. Before reconstitution with PolyA RNA (200 μg/ml; Sigma-Aldrich, München), Benzonase was inactivated by the addition of 25 mM EDTA. Aliquots of the substrate were kept at −70°C until use.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to acknowledge our colleague Shannon Braithwaite (Canada) for helpful comments and English corrections and Derrick Gibbings (CNRS – IBMP Strasbourg, FRANCE) for his implication with the original cloning work.
Ethics Statement
All experiments involving animals in this study were approved by a Commissariat à l'Energie Atomique internal ethics committee in accordance with European Directive 86/609/EEC related to animal protection and welfare in research and were performed under veterinarian surveillance.
Note
1This method was not able to completely inactivate prion infectivity according to the bioassay (see the results of Table 2 for inoculum 3). These results, although presented, were therefore not discussed in the manuscript but merely used to compare with those of the first method (phenol/chloroform extraction), after which complete inactivation was demonstrated.
References
- 1. Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363-83; PMID:9811807; http://dx.doi.org/ 10.1073/pnas.95.23.13363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fournier JG. Cellular prion protein electron microscopy: attempts/limits and clues to a synaptic trait. Implications in neurodegeneration process. Cell Tissue Res 2008; 332:1-11; PMID:18236081; http://dx.doi.org/ 10.1007/s00441-007-0565-5 [DOI] [PubMed] [Google Scholar]
- 3. Somerville RA, Merz PA, Carp RI. Partial copurification of scrapie-associated fibrils and scrapie infectivity. Intervirology 1986; 25:48-55; PMID:2422138; http://dx.doi.org/ 10.1159/000149654 [DOI] [PubMed] [Google Scholar]
- 4. Merz PA, Kascsak RJ, Rubenstein R, Carp RI, Wisniewski HM. Antisera to scrapie-associated fibril protein and prion protein decorate scrapie-associated fibrils. J Virol 1987; 61:42-9; PMID:2878092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Safar J, Ceroni M, Piccardo P, Liberski PP, Miyazaki M, Gajdusek DC, et al. Subcellular distribution and physicochemical properties of scrapie-associated precursor protein and relationship with scrapie agent. Neurology 1990; 40:503-8; PMID:1969124; http://dx.doi.org/ 10.1212/WNL.40.3_Part_1.503 [DOI] [PubMed] [Google Scholar]
- 6. Merz PA, Somerville RA, Wisniewski HM, Manuelidis L, Manuelidis EE. Scrapie-associated fibrils in Creutzfeldt-Jakob disease. CA>Nature 1983; 306:474-6. [DOI] [PubMed] [Google Scholar]
- 7. Lasmezas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM, et al. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 1997; 275:402-5; PMID:8994041; http://dx.doi.org/ 10.1126/science.275.5298.402 [DOI] [PubMed] [Google Scholar]
- 8. Piccardo P, Manson JC, King D, Ghetti B, Barron RM. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci U S A 2007; 104:4712-7; PMID:17360589; http://dx.doi.org/ 10.1073/pnas.0609241104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barron RM, Campbell SL, King D, Bellon A, Chapman KE, Williamson RA, et al. High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of PrPSc in vivo. J Biol Chem 2007; 282:35878-86; PMID:17923484; http://dx.doi.org/ 10.1074/jbc.M704329200 [DOI] [PubMed] [Google Scholar]
- 10. Race R, Meade-White K, Raines A, Raymond GJ, Caughey B, Chesebro B. Subclinical scrapie infection in a resistant species: persistence, replication, and adaptation of infectivity during four passages. J Infect Dis 2002; 186 Suppl 2:S166-70; PMID:12424693; http://dx.doi.org/ 10.1086/344267 [DOI] [PubMed] [Google Scholar]
- 11. Balkema-Buschmann A, Eiden M, Hoffmann C, Kaatz M, Ziegler U, Keller M, et al. BSE infectivity in the absence of detectable PrP(Sc) accumulation in the tongue and nasal mucosa of terminally diseased cattle. J Gen Virol 2011; 92:467-76; PMID:20943888; http://dx.doi.org/ 10.1099/vir.0.025387-0 [DOI] [PubMed] [Google Scholar]
- 12. Manuelidis L, Fritch W, Xi YG. Evolution of a strain of CJD that induces BSE-like plaques. Science 1997; 277:94-8; PMID:9204907; http://dx.doi.org/ 10.1126/science.277.5322.94 [DOI] [PubMed] [Google Scholar]
- 13. Miyazawa K, Kipkorir T, Tittman S, Manuelidis L. Continuous production of prions after infectious particles are eliminated: implications for Alzheimer's disease. PLoS One 2012; 7:e35471; PMID:22509412; http://dx.doi.org/ 10.1371/journal.pone.0035471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol 2010; 119:177-87; PMID:20052481; http://dx.doi.org/ 10.1007/s00401-009-0633-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132-5; PMID:20110469; http://dx.doi.org/ 10.1126/science.1183748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 2010; 285:14083-7; PMID:20304915; http://dx.doi.org/ 10.1074/jbc.C110.113464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miller MB, Wang DW, Wang F, Noble GP, Ma J, Woods VL Jr., et al. Cofactor molecules induce structural transformation during infectious prion formation. Structure 2013; 21:2061-8; PMID:24120764; http://dx.doi.org/ 10.1016/j.str.2013.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature 2003; 425:717-20; PMID:14562104; http://dx.doi.org/ 10.1038/nature01979 [DOI] [PubMed] [Google Scholar]
- 19. Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J 2002; 21:1031-40; PMID:11867531; http://dx.doi.org/ 10.1093/emboj/21.5.1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 2010; 49:3928-34; PMID:20377181; http://dx.doi.org/ 10.1021/bi100370b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grassmann A, Wolf H, Hofmann J, Graham J, Vorberg I. Cellular aspects of prion replication in vitro. Viruses 2013; 5:374-405; PMID:23340381; http://dx.doi.org/ 10.3390/v5010374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abid K, Morales R, Soto C. Cellular factors implicated in prion replication. FEBS Lett 2010; 584:2409-14; PMID:20412808; http://dx.doi.org/ 10.1016/j.febslet.2010.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Timmes AG, Moore RA, Fischer ER, Priola SA. Recombinant prion protein refolded with lipid and RNA has the biochemical hallmarks of a prion but lacks in vivo infectivity. PLoS One 2013; 8:e71081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soto C. Prion hypothesis: the end of the controversy? Trends Biochem Sci 2011; 36:151-8; PMID:21130657; http://dx.doi.org/ 10.1016/j.tibs.2010.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Safar JG, Kellings K, Serban A, Groth D, Cleaver JE, Prusiner SB, et al. Search for a prion-specific nucleic acid. J Virol 2005; 79:10796-806; PMID:16051871; http://dx.doi.org/ 10.1128/JVI.79.16.10796-10806.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. German TL, McMillan BC, Castle BE, Dees C, Wade WF, Marsh RF. Comparison of RNA from healthy and scrapie-infected hamster brain. J Gen Virol 1985; 66 ( Pt 4):839-44; PMID:2580051; http://dx.doi.org/ 10.1099/0022-1317-66-4-839 [DOI] [PubMed] [Google Scholar]
- 27. Dees C, McMillan BC, Wade WF, German TL, Marsh RF. Characterization of nucleic acids in membrane vesicles from scrapie-infected hamster brain. J Virol 1985; 55:126-32; PMID:2409296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, et al. The most infectious prion protein particles. Nature 2005; 437:257-61; PMID:16148934; http://dx.doi.org/ 10.1038/nature03989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miyazawa K, Emmerling K, Manuelidis L. High CJD infectivity remains after prion protein is destroyed. J Cell Biochem 2011; 112:3630-7; PMID:21793041; http://dx.doi.org/ 10.1002/jcb.23286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pritzkow S, Wagenfuhr K, Daus ML, Boerner S, Lemmer K, Thomzig A, et al. Quantitative detection and biological propagation of scrapie seeding activity in vitro facilitate use of prions as model pathogens for disinfection. PLoS One 2011; 6:e20384; PMID:21647368; http://dx.doi.org/ 10.1371/journal.pone.0020384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beekes M, Baldauf E, Cassens S, Diringer H, Keyes P, Scott AC, et al. Protein gel blot mapping of disease-specific amyloid in various animal species and humans with transmissible spongiform encephalopathies using a high-yield purification method. J Gen Virol 1995; 76 ( Pt 10):2567-76; PMID:7595360; http://dx.doi.org/ 10.1099/0022-1317-76-10-2567 [DOI] [PubMed] [Google Scholar]
- 32. Herzog C, Sales N, Etchegaray N, Charbonnier A, Freire S, Dormont D, et al. Tissue distribution of bovine spongiform encephalopathy agent in primates after intravenous or oral infection. Lancet 2004; 363:422-8; PMID:14962521; http://dx.doi.org/ 10.1016/S0140-6736(04)15487-1 [DOI] [PubMed] [Google Scholar]
- 33. Thomzig A, Spassov S, Friedrich M, Naumann D, Beekes M. Discriminating scrapie and bovine spongiform encephalopathy isolates by infrared spectroscopy of pathological prion protein. J Biol Chem 2004; 279:33847-54; PMID:15155741; http://dx.doi.org/ 10.1074/jbc.M403730200 [DOI] [PubMed] [Google Scholar]
- 34. Lewis V, Haigh CL, Masters CL, Hill AF, Lawson VA, Collins SJ. Prion subcellular fractionation reveals infectivity spectrum, with a high titre-low PrPres level disparity. Mol Neurodegener 2012; 7:18; PMID:22534096; http://dx.doi.org/ 10.1186/1750-1326-7-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dobie K, Barron R. Dissociation between transmissible spongiform encephalopathy (TSE) infectivity and proteinase K-resistant PrP(Sc) levels in peripheral tissue from a murine transgenic model of TSE disease. J Virol 2013; 87:5895-903; PMID:23487470; http://dx.doi.org/ 10.1128/JVI.03469-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Geoghegan JC, Valdes PA, Orem NR, Deleault NR, Williamson RA, Harris BT, et al. Selective incorporation of polyanionic molecules into hamster prions. J Biol Chem 2007; 282:36341-53; PMID:17940287; http://dx.doi.org/ 10.1074/jbc.M704447200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Akowitz A, Manuelidis EE, Manuelidis L. Protected endogenous retroviral sequences copurify with infectivity in experimental Creutzfeldt-Jakob disease. Arch Virol 1993; 130:301-16; PMID:8517790; http://dx.doi.org/ 10.1007/BF01309662 [DOI] [PubMed] [Google Scholar]
- 38. Forster E, Zhao S, Frotscher M. Laminating the hippocampus. Nat Rev Neurosci 2006; 7:259-67; PMID:16543914; http://dx.doi.org/ 10.1038/nrn1882 [DOI] [PubMed] [Google Scholar]
- 39. Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci 2008; 31:439-77; PMID:18558863; http://dx.doi.org/ 10.1146/annurev.neuro.31.060407.125620 [DOI] [PubMed] [Google Scholar]
- 40. Fournier JG, Escaig-Haye F, Billette de Villemeur T, Robain O. Ultrastructural localization of cellular prion protein (PrPc) in synaptic boutons of normal hamster hippocampus. C R Acad Sci III 1995; 318:339-44; PMID:7788502 [PubMed] [Google Scholar]
- 41. Siskova Z, Perry VH, Asuni AA. Prion Protein Misfolding at the Synapse. In: Wyttenbach A, O'Connors V, eds. Folding for the Synapse: Springer, 2011:289-312. [Google Scholar]
- 42. Kipkorir T, Tittman S, Botsios S, Manuelidis L. Highly Infectious CJD Particles Lack Prion Protein but Contain Many Viral-Linked Peptides by LC-MS/MS. J Cell Biochem 2014; 115:2012-21; PMID:24933657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fournier JG. Is RNA Critical for the Production of TSE Infection? Front Psychiatry 2010; 1:24; PMID:21423435; http://dx.doi.org/ 10.3389/fpsyt.2010.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Manuelidis L. Infectious particles, stress, and induced prion amyloids: a unifying perspective. Virulence 2013; 4:373-83; PMID:23633671; http://dx.doi.org/ 10.4161/viru.24838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Silva JL, Lima LM, Foguel D, Cordeiro Y. Intriguing nucleic-acid-binding features of mammalian prion protein. Trends Biochem Sci 2008; 33:132-40; PMID:18243708; http://dx.doi.org/ 10.1016/j.tibs.2007.11.003 [DOI] [PubMed] [Google Scholar]
- 46. Supattapone S. Biochemistry. What makes a prion infectious? Science 2010; 327:1091-2; PMID:20185716; http://dx.doi.org/ 10.1126/science.1187790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Saa P, Sferrazza GF, Ottenberg G, Oelschlegel AM, Dorsey K, Lasmezas CI. Strain-specific role of RNAs in prion replication. J Virol 2012; 86:10494-504; PMID:22811520; http://dx.doi.org/ 10.1128/JVI.01286-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Simoneau S, Rezaei H, Sales N, Kaiser-Schulz G, Lefebvre-Roque M, Vidal C, et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 2007; 3:e125; PMID:17784787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, et al. Synthetic mammalian prions. Science 2004; 305:673-6; PMID:15286374; http://dx.doi.org/ 10.1126/science.1100195 [DOI] [PubMed] [Google Scholar]
- 50. Thomzig A, Kratzel C, Lenz G, Kruger D, Beekes M. Widespread PrPSc accumulation in muscles of hamsters orally infected with scrapie. EMBO Rep 2003; 4:530-3; PMID:12776740; http://dx.doi.org/ 10.1038/sj.embor.embor827 [DOI] [PMC free article] [PubMed] [Google Scholar]