

Abstract
The P-Rex family are Dbl-type guanine-nucleotide exchange factors for Rac family small G proteins. They are distinguished from other Rac-GEFs through their synergistic mode of activation by the lipid second messenger phosphatidyl inositol (3,4,5) trisphosphate and the Gβγ subunits of heterotrimeric G proteins, thus acting as coincidence detectors for phosphoinositide 3-kinase and G protein coupled receptor signaling. Work in genetically-modified mice has shown that P-Rex1 has physiological importance in the inflammatory response and the migration of melanoblasts during development, whereas P-Rex2 controls the dendrite morphology of cerebellar Purkinje neurons as well as glucose homeostasis in liver and adipose tissue. Deregulation of P-Rex1 and P-Rex2 expression occurs in many types of cancer, and P-Rex2 is frequently mutated in melanoma. Both GEFs promote tumor growth or metastasis. This review critically evaluates the P-Rex literature and tools available and highlights exciting recent developments and open questions.
Keywords: guanine-nucleotide exchange factors, GEFs, P-Rex1, P-Rex2, P-Rex2b, PREX1, PREX2, PREX2A, Rac, Rho family
Abbreviations
- aa
amino acid
- BRET
bioluminescence resonance energy transfer
- coIP
coimmunoprecipitation
- EST
expressed sequence tag
- GEF
guanine-nucleotide exchange factor
- HDAC
histone deacetylase
- iDHPH
isolated Dbl homology (DH) /pleckstrin homology (PH) domain tandem
- GPCR
G protein coupled receptor
- HRG
heregulin
- IP4P
inositol polyphosphate 4-phosphatase
- HLMVEC
human lung microvascular endothelial cells
- HMEC
human microvascular endothelial cells
- HUVEC
human umbilical vein endothelial cells
- miR
microRNA
- MEFs
mouse embryonic fibroblasts
- NRG
neuregulin
- PI3K
phosphoinositide 3-kinase
- PIP3
phosphatidyl inositol (3, 4, 5) trisphosphate
- PKA
cAMP-dependent kinase
- P-Rex
PIP3-dependent Rac exchanger
- PTX
pertussis toxin
- RTK
receptor tyrosine kinase
- S1P
sphingosine 1-phosphate
Rac family small G proteins (Rac1, Rac2, Rac3 and RhoG; a branch of the Rho family) control a myriad of essential cell responses, including adhesion, migration, reactive oxygen species formation and gene expression.1-3 They can be activated by 2 types of guanine-nucleotide exchange factors (GEFs), Dbl or DOCK, which are characterized by the structure of their catalytic domains. Rac-GEFs promote the release of GDP from Rac, allowing excess free cellular GTP to bind, and thus induce the active conformation of Rac which is able to engage downstream targets and stimulate cell responses 4-6 (Fig. 1).
Figure 1.
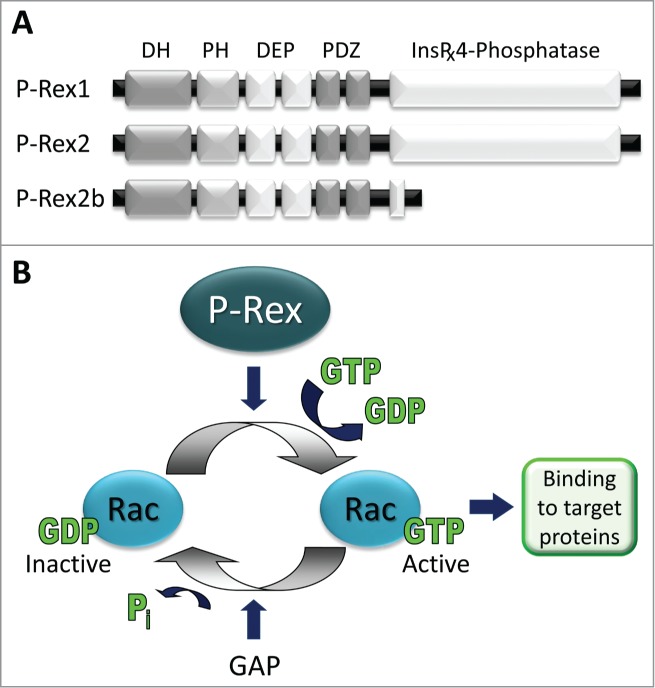
Domain structure and function. (A) Domain structure. P-Rex1 and P-Rex2 are typical Dbl-type Rho-GEFs4,5 and have identical domain structure. The N-terminal DH domain, which confers Rac-GEF activity, is followed by a PH domain which binds PIP3, followed by 2 DEP and 2 PDZ protein interaction domains and weak homology over their C-terminal half to inositol polyphosphate 4-phosphatase (IP4P) which harbours no phosphatase activity.7,8 P-Rex2b is a splice variant of P-Rex2.9 (B) The Rac-GEF activity of P-Rex family proteins promotes the release of GDP from Rac (Rac1, Rac2, Rac3, RhoG), allowing excess free cellular GTP to bind, and thus induces the active conformation of Rac which is able to engage downstream targets and stimulate cell responses.7-9,19,22,34 In vitro, but likely not is cells, P-Rex can also activate some CDC42-like small G proteins.7-9,22 P-Rex1 is strongly expressed in leukocytes and neurons but also in platelets, endothelial cells and melanoblasts.7,8,14-19 P-Rex2 strongly expressed in Purkinje neurons and in non-haematopoietic cells of the lung, but also in liver and adipose tissue.8,11,12 P-Rex2b is expressed in endothelial cells and in the heart.9,23 Both P-Rex1 and P-Rex2 are strongly overexpressed and/or mutated in several types of cancers.
The P-Rex family are Dbl-type Rac-GEFs. The founding member, PIP3-dependent Rac exchanger 1, P-Rex1 (PREX1), was discovered during a search for factors that activate Rac when stimulated by the lipid second messenger phosphatidyl inositol (3,4,5) trisphosphate (PIP3) which is generated by class I phosphoinositide 3-kinase (PI3K).7 P-Rex2 (PREX2) and P-Rex2b were discovered on the basis of their sequence homology with P-Rex1.8,9 P-Rex family proteins differ from other Dbl-type Rac-GEFs such as Vav, Tiam or Trio 4-6 in their domain structure and mechanisms of regulation. They have a number of physiological functions that are described in section “Physiological Function”, an emerging role in metabolic disease that is discussed in section “Insulin Resistance and Type 2 Diabetes”, and important roles in cancer progression that are described in section “Cancer”.
Genes and Proteins
The coding sequence of the 2 P-Rex genes is 49% identical and conserved throughout vertebrates but absent from invertebrates.8 In humans, the P-Rex1 gene (PREX1; NM_020820) is located in chromosome 20 (20q13.13), near a region associated with type 2 diabetes, and the P-Rex2 gene (PREX2; NM_024870) on chromosome 8 (8q13.2), in a region linked to aggressive cancers and metastasis. PREX1 encodes the protein P-Rex1 (185 kDa, 1659 amino acids (aa); NP_065871), and PREX2 encodes 2 proteins, full-length P-Rex2 (also known as P-Rex2a; 183 kDa, 1606 aa; NP_079146), and the splice variant P-Rex2b (112 kDa, 979 aa; NP_079446) which lacks the C-terminal half (Fig. 1). The possibility of 2 additional P-Rex1 isoforms was predicted from database entries, and Western blots for isoform-specific epitopes suggested that one of these, a version truncated C-terminally by 10 kDa, might be expressed in breast cancer cells.10 However, this protein appeared to co-migrate with full-length P-Rex1, and the underlying predictive database entry (BC-009948) was composed of 2 expressed sequence tags (ESTs) which show incomplete RNA processing at an exon/intron splice junction rather than alternative splicing. In addition, the possibility of a third isoform of P-Rex2, P-Rex2c, was recently proposed, based on mouse database entry AK138884 and the observation of a 120 kDa band in several tissues by P-Rex2 Western blot.11 However, that database entry (and derived human entry B4DFX0) was an incomplete cDNA clone which terminates prematurely in the middle of an exon and does not encode a 120 kDa product. It seems likely that the observed 120 kDa band is a proteolytic fragment of P-Rex2, in analogy with a P-Rex1 product of comparable size that is often seen in P-Rex1 expressing tissues and almost certainly a proteolytic fragment according to mass spectrometric analysis.7,12 Therefore, until firm identification of further isoforms through cloning, mass spectrometry and expression studies, P-Rex remains a 3-member protein family (Fig. 1).
Protein Domain Structure
P-Rex1 and P-Rex2 proteins are 56% identical in their aa sequence and share common domain architecture.7,8 They consist of an N-terminal Dbl homology (DH) which confers their Rac-GEF catalytic activity in tandem with a pleckstrin homology (PH) domain, as is characteristic for Dbl-type Rho-GEFs,5,6 followed by 2 DEP and 2 PDZ protein interaction domains and weak homology over their C-terminal half to inositol polyphosphate 4-phosphatase (IP4P) (Fig. 1). Although the IP4P domain contains the minimal residues required for phosphatase activity, P-Rex proteins appear to harbor no catalytic phosphatase activity.7,13 The functions of the various domains, as far as known, are discussed in sections “Regulation of Activity” and “Binding Proteins”. NMR or crystallographic structures are not yet available, except for a so-far unpublished database deposition of a crystal structure of the isolated N-terminal PDZ domain of P-Rex1 (PDB ID: 3QIK).
Tissue Distribution and Regulation of Expression
P-Rex1 was originally purified from pig neutrophil cytosol by column chromatography on the basis of its ability to activate recombinant Rac1 in the presence of PIP3. The purification, which began with 90 L of porcine blood and ended with 0.5 mg of purified protein, showed that P-Rex1 is highly abundant in neutrophils, making up 0.1% of the cytosolic protein.7,14 P-Rex1 is also strongly expressed in other peripheral blood leukocytes such as macrophages 15 and in several other cell types including platelets,16,17 endothelial cells 18 and neurons.19 Tissue-wise, P-Rex1 is expressed strongly throughout the brain, and less strongly in several other tissues, including bone marrow, thymus, spleen, lymph nodes and lung.7,12,19 P-Rex2 mRNA is detectable in many tissues,8 but the distribution of the protein is more restricted, being strongest in brain and lung, weaker in liver and adipose tissue, and just detectable in skeletal muscle, thymus and spleen.11,12 Notably, P-Rex2 is not expressed in leukocytes.20–22 Within the brain, P-Rex2 is predominantly found in the cerebellum, in particular in the cerebellar Purkinje neuron, but is also detectable in the frontal cortex, striatum, amygdala and hippocampus.12 It is currently unknown through which mechanism the tissue distribution of P-Rex2 protein is more restricted than its mRNA. The expression of P-Rex2b mRNA seems limited to heart and endothelial cells,9,23 and endogenous P-Rex2b protein has to date only been studied in endothelial cells.23 Both P-Rex1 and P-Rex2 are strongly deregulated in many types of cancer (see section “Cancer”).
The mechanisms that regulate P-Rex expression have to date only been characterized in the context of their deregulation in cancer cells. Two types of epigenetic regulation have been identified. Histone deacetylases (HDACs) repress PREX1 transcription by deacetylating histones associated with the PREX1 promoter,24 and micro RNA (miR) 338–3p represses the expression of PREX2 by interacting with its 3'UTR.25,26 Both mechanisms, described in detail here-below, repress P-Rex in normal cells, and loss of repression results in upregulation of P-Rex in cancer cells.
HDAC inhibitors or HDAC siRNA induce P-Rex1 expression in immortalized prostate epithelial cells and non-metastatic prostate cancer cell lines, whereas overexpression of HDACs reduces P-Rex1 levels in metastatic prostate cancer cells.24 HDACs interact with a region 190–98 bp 5' of the PREX1 coding region which was identified as the minimal promoter by dual luciferase assay and as a binding site for the transcription factor Sp1. Mutagenesis of this binding site as well as Sp1 inhibition or siRNA showed that Sp1 drives P-Rex1 expression and is required for HDAC-mediated repression. In metastatic prostate cancer cells, repression is lost by a reduction in HDAC binding to the PREX1 promoter (despite normal Sp1 binding), which results in increased acetylation of PREX1 promoter-associated histone H4 despite normal global levels. It was proposed that such PREX1 locus-specific H4 acetylation might induce an open chromatin conformation which favors transcription. Several questions remain: what controls HDAC association with Sp1 to repress P-Rex1 in normal cells; how is this deregulated in metastatic cancer cells; what controls the association of Sp1 with the PREX1 promoter; how important is HDAC/Sp1 dependent regulation of P-Rex1 in other cell types; and finally, does a similar mechanism apply for P-Rex2?
Microarray and bioinformatic analysis recently suggested that PREX2 might be a target of miR-338–3p, a microRNA which is downregulated in human metastatic neuroblastoma 25 and in gastric cancer.26 Mutational analysis combined with dual luciferase assays identified 3 adjacent regions in the 3' UTR of PREX2 as miR-338–3p binding sites and confirmed PREX2 to be a miR-338–3p target.25 Overexpression of miR-338–3p mimics in a range of human neuroblastoma cell lines led to several-fold reductions in P-Rex2 protein levels, whereas downregulation of miR-338–3p through antisense oligonucleotides led to equivalent increases. P-Rex2 siRNA inhibited soft agar colony formation and matrigel invasion induced by miR-338–3p antisense,25 suggesting that P-Rex2 could be a major target of miR-338–3p. Similarly, overexpression of miR-338–3p mimics reduced P-Rex2 levels in the gastric cancer cell line BGC-823, although in these cells, knockdown of miR-338–3p was insufficient to affect P-Rex2 levels or cell responses.26 Usually, miRs are modulators of gene expression rather than all-or-nothing switches. Therefore, it seems likely that the substantial miR-dependent changes in P-Rex2 expression observed in neuroblastoma and gastric cancer cells may be a special feature of some cancer cells, whereas unidentified mechanisms such as transcription factors are likely to dominate P-Rex2 expression in normal tissues.
Substrate Specificity
In vitro, full length P-Rex1 or the isolated DH/PH domain tandem (iDHPH) can activate all Rac-like Rho family small G proteins – the ubiquitous Rac1, haematopoietic Rac2, neuronal Rac3 and the more distantly related widely expressed RhoG – as well as some that are Cdc42-like (Cdc42 and TC10, but not TCL), but none that are Rho-like (RhoA, RhoB or RhoC).7,22,27 In vivo, P-Rex1 substrate specificity is more restricted, which is not uncommon in GEFs and usually attributed to local substrate availability and binding affinities playing larger roles in vivo than in vitro.28 In vivo, P-Rex1 activates Rac1, Rac2, Rac3 and RhoG, whereas there is no evidence for activation of Cdc42. Which particular isoform of Rac is activated by P-Rex1 in vivo depends both on cell type and upstream signal.
In mouse neutrophils, where endogenous Rac1 and Rac2 play non-redundant roles in ROS formation, cell spreading and motility,29 P-Rex1 preferentially activates Rac2 upon GPCR stimulation,20,21,30 but Rac1 upon E-selectin engagement.31 In contrast, in macrophages, which also express both Rac1 and Rac2, P-Rex1 activates Rac1 upon GPCR stimulation.15 Recently, RhoG was identified as a major in vivo target of P-Rex1 in neutrophil GPCR signaling.22 The similar effects of P-Rex1 and RhoG-deficiency led to the suggestion that P-Rex1-dependent Rac activity may be downstream of RhoG, potentially mediated by DOCK2/Elmo.22 However, while the phenotypes of P-Rex1−/− and RhoG−/− neutrophils are compellingly similar, they are not identical. P-Rex1 deficiency impairs GPCR-stimulated actin polymerization as well as actin polarization, whereas RhoG deficiency only affects the latter.21,22,30 Furthermore, P-Rex1−/− mice show a defect in peritoneal neutrophil recruitment during inflammation whereas RhoG−/− mice do not.21,30,32 Hence, the hypothesis that P-Rex1 controls Rac largely indirectly in neutrophil GPCR signaling, in a hierarchical pathway through RhoG and DOCK2, remains to be confirmed by double knockout and reconstitution studies and must also be investigated for other pathways and in other cell types (see also section “Leukocytes and Inflammation”).
In neurons, endogenous Rac1 and Rac3 play non-redundant roles in neuritogenesis,33 and P-Rex1 has variously been reported to induce activation of either isoform in NGF-stimulated neuronal PC12 cells.19,34 However, P-Rex1 activity toward Rac3 has to date only been investigated using overexpressed Rac protein and therefore requires confirmation.34
Lack of P-Rex1-dependent Cdc42 activity under conditions that stimulate its Rac-GEF activity has been seen in a range of cell types, including neutrophils,35 PC12 cells 19 and overexpressing Sf9 insect cells.7 Similarly, P-Rex1 did not activate RhoA.17,19,36 Moreover, overexpression of P-Rex1 in endothelial cells,7 myeloid cells,37 fibroblasts 38 or neurons 19 induces cell morphologies typical for active Rac (spreading, lamellipodia formation, membrane ruffling) rather than Cdc42 (filopodia and microspike formation) or RhoA (stress fibers).3,39 However, as none of these studies included controls for Cdc42 activation, such as Cdc42-GEF expression, in vivo Cdc42-GEF activity of P-Rex1 cannot be categorically excluded, although on balance, it seems unlikely.
Like P-Rex1, P-Rex2 can activate Rac1 in vitro and in vivo, and is unable to activate Cdc42 in vivo, but unlike P-Rex1, its iDHPH domains seem unable to activate Cdc42 even in vitro.8,40 P-Rex2b activates endogenous Rac1 in sphingosine 1-phosphate (S1P)-stimulated endothelial cells 23 and overexpressed Rac1 but not Cdc42 in HEK-293 cells,9 but it remains to be tested in vitro. Curiously, analysis of Rac1 binding to P-Rex2 mutants suggested that the PH domain rather than the DH domain might confer substrate recognition and specificity, as its iPH is sufficient for binding.40 However, if such Rac binding to the PH domain occurs, it is unlikely that the Rac molecule which binds to the PH domain is the same as that activated by the DH domain, as iDH of P-Rex2 is sufficient for Gβγ-stimulated Rac1-GEF activity (Fig. 2), similar to the iDH domain of P-Rex1 being sufficient for its Gβγ-stimulated Rac-GEF activity.41
Figure 2.

Synergistic activation by PIP3 and Gβγ. (A) P-Rex proteins have low basal activity and are stimulated by the release of intramolecular inhibition. The Rac-GEF activity of P-Rex1 is directly stimulated by the lipid second messenger PIP3 which is generated by PI3K activity and by the Gβγ subunits of heterotrimeric G proteins.7,42 Direct stimulation of P-Rex1 Rac-GEF activity by PIP3 occurs through the PH domain, and stimulation by Gβγ occurs through the DH domain,37,42 although Gβγ stimulation requires the presence of further domains in vivo.43 The left-hand panel was adapted from ref. 7, the others from ref. 42. (B) PIP3 or Gβγ alone are sufficient to stimulate P-Rex Rac-GFEF activity, but they can also activate P-Rex GEFs synergistically,7,42,43 both in vitro and in vivo. This enables P-Rex family Rac-GEFs to act as coincidence detectors for the concomitant stimulation of a range of different types of PI3K-coupled receptors and GPCRs.7-9,23,43,51,75 In addition to Rac-GEF activity, PIP3 and Gβγ, also synergistically stimulate the membrane localization of P-Rex1.37 (C) The iDH domain of P-Rex2 is sufficient for Gβγ-stimulated Rac1-GEF activity. Activation of GST-Rac1 by EE-tagged full-length P-Rex2 (black) or isolated DH domain of P-Rex2 (iDH) (gray) is stimulated by EE-Gβ1γ2 or by EDTA (as positive control) in an in vitro Rac-GEF activity assay that was done essentially as described.41 All recombinant proteins were purified from Sf9 insect cells. Data were generated by Sarah Donald and are representative of 3 independent experiments.
Regulation of Activity
As is typical for Rac-GEFs, full length P-Rex proteins have low basal activity and are stimulated by the release of intramolecular inhibition, as shown by the analysis of deletion and truncation mutants. However, the combination of signals that activate P-Rex proteins is unique to this Rac-GEF family.
Activation by PIP3 and Gβγ
Native P-Rex1 was originally purified from neutrophils on the basis of being activated by PIP3,7 and the Rac-GEF activity of purified recombinant human P-Rex1 can be directly stimulated more than 30-fold by PIP3, with an EC50 of 1.5 μM.7,42 The fact that neutrophils exhibit significant Gβγ-dependent Rac activity as well as PIP3-dependent Rac activity led to the discovery that purified native or recombinant Gβγ subunits (but not Gα) can directly activate purified P-Rex1, to a similar degree as PIP3.7,42 Furthermore, PIP3 and Gβγ can stimulate P-Rex1 activity synergistically 7,43 (Fig. 2). Similarly, Pak-CRIB pull down assays in Sf9 cells expressing P-Rex1, PI3K and Gβγ showed that P-Rex1 is also synergistically stimulated by PI3K activity and Gβγ in vivo, and suggested that it might act as a coincidence detector for the concomitant stimulation of PI3K-coupled receptors and GPCRs.7 While other Rac-GEFs can also be directly activated by PIP3 (e.g. Swap70 44) or can directly bind Gβγ (Dbl 45), the synergistic activation by PIP3 and Gβγ is unique to the P-Rex family. Several other Rac-GEFs can be activated by Gβγ in vivo, including PLEKHG2 46 and DOCK180/Elmo1,47 but as yet without evidence for direct activation in vitro. The most promising example for direct Gβγ-dependent activation of GEFs other than P-Rex is for DOCK/Elmo complexes in Dictyostelium, where direct Gβγ binding to ElmoE was suggested by FRET,48 although direct Gβγ-dependent activation of DOCK/Elmo remains to be confirmed with recombinant proteins.
A panel of purified deletion and truncation mutant P-Rex1 proteins showed that iDH is sufficient for its basal and Gβγ stimulated Rac-GEF activity,37,42 and iDHPH for its PIP3-stimulated Rac-GEF activity, whereas deletion of the DEP, PDZ of IP4P domains did not fundamentally affect its ability to be stimulated by PIP3 or Gβγ.42 Yet, deletion of the DEP or PDZ domains resulted in constitutively active protein, suggesting that interactions between these domains and the DHPH tandem keep the basal activity of the full-length protein low.42 Phosphoinositide binding assays showed that both PIP3 and PI(34)P2 can bind to purified P-Rex1, although only PIP3 can stimulate its Rac-GEF activity,7 and that iPH is sufficient for PIP3 binding.42 However, a mutant lacking the PH domain (ΔPH) retained some PIP3 binding ability, so there remains some possibility of further PIP3 binding sites.42
Analysis of a range of Gβ and Gγ protein dimers showed that many different, but not all, combinations can activate P-Rex1. Among the different Gβ proteins (tested in complex with Gγ2), Gβ1, Gβ2, Gβ3 and Gβ4 all activated P-Rex1 whereas Gβ5 did not.49 Similarly, among Gγ proteins (in complex with Gβ1), Gγ2, Gγ3, Gγ7, and Gγ13 activated P-Rex1 equally well, Gγ11 and Gγ12 less so, and Gγ1 least.49 Furthermore, P-Rex1 was not activated by any Gα subunits tested (Gi, Gs, Gq, G12 or G13).49 This study suggested that only Gβ5γ2-coupled GPCRs would be unable to signal through P-Rex1 (such GPCRs are unlikely to exist), and indeed all GPCRs examined to date in P-Rex expressing cell types can signal through P-Rex. However, the profile of Gβγs able to activate P-Rex1 mirrors that which activates the Gβγ-dependent haematopoietic PI3K isoform PI3Kγ,49 and PI3Kγ is known not to be activated by all GPCRs.50 This is usually attributed to limited cellular availability of certain Gβγs, and similar limitations are likely to apply during activation of P-Rex.
The fact that the catalytic iDH domain is sufficient for Gβγ-dependent stimulation of GEF activity in vitro 37,42 was surprising, as it implied that other Dbl-type Rho-GEFs may also be activated by Gβγ subunits. This possibility remains to be explored. Convincing signal-to-noise ratios are difficult to obtain in Gβγ binding assays, and while Gβγ binding to full-length P-Rex1 was successfully determined to occur at 1:1 stoichiometry, with a KD of 0.3 μM,43 the affinity for iDH remains to be determined and may well be too low to be relevant in vivo. Expression of P-Rex1 deletion and truncation mutants showed that the iDH is insufficient for robust Gβγ binding in vivo and that an interaction between the second DEP, first PDZ and IP4P domains favors Gβγ association.43 P-Rex1 mutants isolated from this overexpression system retained only part of their Gβγ sensitivity, and endogenous Rac activity remains to be determined. However, both the N- and C-terminal halves of P-Rex1 were clearly required for downstream responses such as cell spreading to be Gβγ sensitive.43 One argument against an essential role of the C-terminal half for the Gβγ sensitivity of P-Rex proteins in vivo is the fact that the Rac-GEF activity of P-Rex2b, which lacks the C-terminal half, is stimulated by the expression of Gβγ, although indirect effects of Gβγ in vivo cannot be excluded.23
Like P-Rex1, P-Rex2 is directly and synergistically activated by PIP3 and Gβγ in vitro (P-Rex2b remains to be assessed), and both P-Rex2 and P-Rex2b are also synergistically stimulated by PI3K activity and Gβγ expression in vivo.8 9,23 Interestingly, inhibition of PI3K in HEK-293 cells abolishes P-Rex2b and Gβγ dependent serum response transcription factor activity, which led to the suggestion that PI3K activity may be a pre-requisite for Gβγ stimulation of P-Rex2b in vivo.23 However, while serum response factor activity does require active Rac, it can also be stimulated through other pathways, some of which mediated through Gβγ or PI3K, independently of P-Rex2b,23 so this hypothesis needs further exploration. Finally, it was suggested that iPH of P-Rex2 may suffice for Gβγ binding, but as mentioned above these assays are difficult, and this particular interaction was indeed not observed at stoichiometrically relevant concentrations of Gβγ.23 Furthermore, iDH is sufficient for Gβγ-dependent Rac1-GEF activity of P-Rex2 in vitro (Fig. 2).
A seminal study by the Kazanietz lab illustrates best how intricate the regulation of P-Rex1 by PI3K-coupled receptors and GPCRs can be in vivo.51 shRNA knockdown of P-Rex1 showed that P-Rex1 mediates Rac1 activity, chemotaxis and transforming capacity in breast cancer cell lines stimulated by heregulin (HRG), a ligand of the receptor tyrosine kinase (RTK) ErbB3. PI3Kγ inhibitors or PI3Kγ shRNA showed furthermore that this typically haematopoietic Gβγ-dependent isoform of PI3K is required. The sensitivity of HRG-stimulated Rac activity to pertussis toxin (PTX) first suggested an involvement of GPCRs in this RTK-dependent response, and the SDF1 receptor CXCR4 was identified as the GPCR mediating it, through knockdown by CXCR4 siRNA. The fact that the CXCR4 antagonist AMD-3100 did not inhibit HRG-stimulated Rac1 activity or migration, while efficiently blocking SDF1 responses, showed that HRG responses do not involve ligand-dependent activation of the CXCR4. Bioluminescence resonance energy transfer (BRET) showed that, instead, stimulation of ErbB3 is sufficient to transactivate the CXCR4 and induce its interaction with downstream effectors. Even more intricately, this transactivation of CXCR4 by the HRG receptor was shown to occur via the EGFR, ErbB1, as the EGFR inhibitor AG1478 or EGFR siRNA inhibited HRG-stimulated CXCR4 phosphorylation and Rac activity.51 Therefore, in breast cancer cells, stimulation of ErbB3 activates Rac through Gβγ-dependent PI3Kγ and P-Rex1, via the GPCR CXCR4, but independently of the natural CXCR4 ligand SDF-1α, through a transactivation mechanism which involves the EGFR.
Activation by PP1α
P-Rex2 was identified as a putative binding partner of the serine phosphatase PP1α in a screen for proteins containing motifs which typically confer PP1 binding.52 Characterization of the interaction showed that purified P-Rex1 or P-Rex2 can both bind directly to purified PP1α (Fig. 3A), that binding is conferred through the RVxF motif in their IP4P domains, and that endogenous P-Rex1 constitutively interacts with endogenous PP1α in cells.41 The Rac-GEF activity of P-Rex1 was directly stimulated by PP1α in vitro, around two-fold, and this was largely dependent on the phosphatase activity of PP1α. Co-stimulation with PP1α and either PIP3 or Gβγ had additive effects. In HEK-293 cells, P-Rex1 dependent Rac activity was stimulated four-to-five fold by coexpression of PP1α, required an intact RVxF motif, and could also be elicited by the closely related PP1α isoform PP1β, although to a lesser extent. Mass spectrometric analysis and mutagenesis showed that PP1α dephosphorylates at least 3 serine residues, S834, S1001 and S1165 in P-Rex1, of which S1165 is the major target site and conserved in P-Rex2 and throughout vertebrate evolution 41 (Fig. 3A). As P-Rex2b does not contain the RVxF motif, nor S1165, this splice variant must therefore escape the regulation through PP1α. Questions that remain are: is the interaction between P-Rex proteins and PP1α regulated by signaling events, does it affect their subcellular localization, and which kinases counteract the effects of PP1α?
Figure 3.
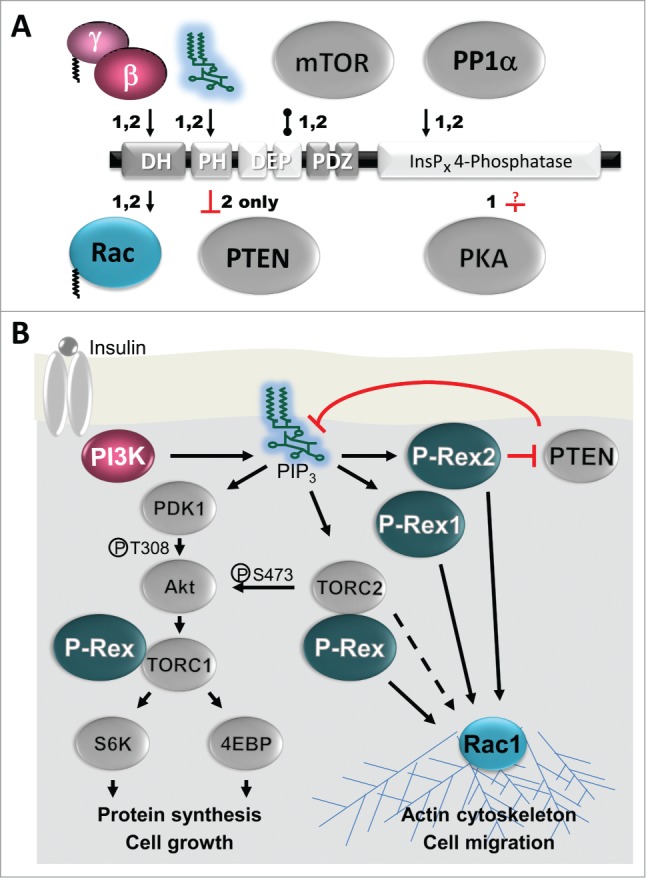
Binding proteins. (A) Direct binding partners of P-Rex1 (1) and P-Rex2 (2). Considering their size and the abundance of protein interaction domains in P-Rex family Rac-GEFs, remarkably few proteins have been identified that bind directly. The substrate Rac and the activator Gβγ bind directly to the DH domain and the phosphoinositide activator PIP3 to the PH domain. PP1α-binds directly to the RVXF motif and dephosphorylates S1165 of P-Rex1, another direct mechanism of activation.41 PKA directly inactivates P-Rex1 (P-Rex2 was not tested) but the binding and targets sites are unknown (as denoted by the question mark).53 Apart from these regulators, mTOR and PTEN are known to bind directly. P-Rex1 and P-Rex2 bind to mTOR through their DEP domains and form part of TORC1 and TORC2. P-Rex1 affects mTOR responses, but it is unclear whether up- or downstream.60 P-Rex2, but not P-Rex1, binds to the tumor suppressor PTEN through its PH domain, thus inhibiting PTEN phosphatase activity,58 although the IP4P domain also contributes to PTEN binding.11 Additional interactions, which may be indirect, are seen in vivo. Unidentified serine kinases regulate P-Rex1 activity upon RTK stimulation of breast cancer cells,10,54 and PKCδ or Akt regulate its activity upon overexpression in COSphox cells.55 P-Rex2 interacts with IGF1 receptor in HEK-293 cells.11 P-Rex1 interacts with Akt1 in overexpressing HEK-293 cells, likely through TORC,56 with overexpressed PDGFRβ in immortalized human fibroblasts,38 with ephrin-B1 in mouse embryonic cortex,62 with EHBP1 in prostate cancer cells,63 with the Rap1-GTP binding domain of RalGDS in HUVECs,65 and its isolated PDZ domains interact with Edg1 in overexpressing HEK-293 cells.61 Arrows denote activation, blunt ends show inhibition, and bobble-ends show direct binding. (B) Roles of P-Rex1 and P-Rex2 in Insulin signaling. Insulin or IGF-1 binding stimulate the Insulin receptor which leads to activation of tyrosine-kinase sensitive class 1A PI3Ks.50 These PI3Ks produce PIP3 which directly activates both P-Rex1 54,56,63,74,75 and P-Rex2 11,58 to stimulate Rac GTP-loading and Rac-dependent responses such as cell migration. P-Rex2, but not P-Rex1, inhibits PTEN, thereby blocking the removal of PIP3 by PTEN and enhancing PI3K signaling.11,58 PIP3 also directly activates PDK1 which activates Akt by phosphorylating T308 and the downstream signaling cascade that stimulates cell growth.50 In addition, PIP3 activates the TORC2 complex (=PDK2), leading to further activation of Akt (phospho-S473), and activation of Rac through an unknown pathway that may involve P-Rex.50,60 P-Rex1 and P-Rex2 also both directly bind to the TORC1 and TORC2 complexes, although it is still unclear how this affects both mTOR signaling and P-Rex activity.60
Inhibition by PKA
Treatment of purified recombinant P-Rex1 with purified cAMP-dependent kinase (PKA) completely blocks its activation by Gβγ/PIP3 or PIP3 alone (and presumably also its basal activity but that was not tested) (Fig. 3A), whereas treatment with λ-phosphatase increases it.43,53 Interestingly, surface plasmon resonance showed that PKA-treated purified P-Rex1 cannot bind Gβγ.43 Overexpressed P-Rex1 is also inhibited by endogenous PKA in vivo, as shown by stimulation of HEK-293 cells with isoproterenol, an agonist of Gs-coupled β-adrenergic GPCRs which lead to the activation of PKA, or with the direct PKA activator Sp-cAMPS.53 Expression of the catalytic subunit of PKA in HEK-293 cells blocks the interaction between overexpressed P-Rex1 and Gβγ as well as between the isolated N- and C-terminal halves of P-Rex1, suggesting that PKA regulates both Gβγ and interdomain interactions.43 However, the effects of PKA on endogenous P-Rex1 remain to be tested. Furthermore, P-Rex1 contains 28 putative PKA phosphorylation sites,53 and the residues that are phosphorylated by PKA remain to be identified. In any event, it is unlikely that PKA is the kinase which counteracts the activation of P-Rex proteins by PP1α, as the main PP1α target residue S1165 does not lie within a PKA consensus sequence.41
Other regulation through kinases
Further research into P-Rex phosphorylation has revealed important indirect mechanisms that regulate P-Rex1 activity. Mass spectrometric analysis of a ∼180 kDa phosphoprotein in MCF7 breast cancer revealed P-Rex1 phosphorylation on S315, S319, S605/606 and S1169.10 Phosphospecific ABs showed that stimulation of breast cancer cells with neuregulin (NRG), a ligand of Erb-family RTKs, decreases phosphorylation of S313 and S319 whereas phosphorylation of S605 and S1169 is increased, with S1169 showing the biggest response. Phosphorylation of S1169 was also observed in NRG-stimulated breast cancer cells from patient ascites fluid and in breast cancer cell lines stimulated with other RTK ligands, including IGF-I (but not EGF) in T47D cells or PDGF in HS578T cells. Expression of P-Rex1 mutant S313A increased basal Rac activity in MCF7 cells whereas S1169A decreased basal or NRG-stimulated Rac activity. Importantly, proliferation of MCF7 cells, when inhibited by downregulation of P-Rex1, could be rescued by WT P-Rex1 but not by the S1169A mutant, suggesting that S1169 phosphorylation is required. Furthermore, proliferation of HCC3153 cells was stimulated by the expression of S313A but not WT P-Rex1, suggesting that relief of inhibitory S313 phosphorylation may be sufficient. Use of mTOR inhibitors or mTOR shRNA in MCF7 cells has ruled out the possibility of IGF-1 stimulated P-Rex1 S1169 phosphorylation being mediated by mTOR activity.54 Therefore, the serine kinases and phosphatases that mediate P-Rex1 serine phosphorylation of upon cell stimulation through RTKs remain to be identified.
P-Rex1 expression mediates fMLP stimulated ROS formation in COSphox cells (COS cells which express essential components of the NADPH oxidase and the fMLP receptor for reconstitution of the phagocyte oxidative burst).55 In this system, P-Rex1 dependent ROS formation is potentiated by coexpression of PKCδ or Akt, and sensitive to the Akt inhibitor SHX, suggesting that PKCδ and Akt may be further regulators of P-Rex1 activity.55 However, expression of kinase-dead Akt reduced P-Rex1 dependent ROS, and Akt phosphorylation was affected by P-Rex1 expression, suggesting that Akt is also at least partially downstream of P-Rex1. The effects of PKCδ or Akt on P-Rex activity have not yet been investigated in vitro, so may be indirect, nor on the endogenous level. However, Akt1 was also separately described as a P-Rex1 interacting protein, and in that study too, there was evidence for Akt1 being both upstream and downstream of P-Rex1 (see Section “mTOR and Akt1”).56
Regulation of Subcellular Localization
Rac proteins are activated at cellular membranes, and membrane localization of Rac-GEFs is required for the activation of Rac. However, like most Rac-GEFs, P-Rex1 and P-Rex2 are largely cytosolic in basal cells, and their membrane translocation requires cell stimulation.7,8,19,37,57,58
In some situations, stimulation of PI3K-coupled receptors is sufficient for P-Rex membrane translocation. For example, TNFα-stimulation induces membrane translocation of endogenous P-Rex1 endothelial cells, in a manner sensitive to the PI3K inhibitor LY294002,36 and SDF1- or HRG-stimulation induce translocation in MCF-7 cells.51 However, PDGF is not sufficient for significant translocation of overexpressed P-Rex1 or P-Rex2 in endothelial cells.7,8,37 Similarly, the presence of PIP3 alone is insufficient to stimulate a robust association of the purified iDHPH domains of P-Rex1 with liposomes.59
In contrast, cell stimulation that leads to the concomitant production of PIP3 and Gβγ does induce robust membrane translocation. For example, stimulation of monocytic THP1 cells with the GPCR ligand MCP-1, which leads to concomitant Gβγ release and PIP3 production (the latter through Gβγ-dependent haematopoietic PI3Kγ), induces robust plasma membrane translocation of P-Rex1.37 Similarly, subcellular fractionation of human neutrophils showed that fMLP stimulates translocation of endogenous P-Rex1 from the cytosol to a plasma-membrane enriched fraction.57 In addition, immunofluorescence microscopy suggested possible polarized localization of P-Rex1 at the leading edge of neutrophils upon GPCR stimulation,57 but as expression levels in primary neutrophils are difficult to manipulate, this requires confirmation, for example through the use of P-Rex1−/− mouse neutrophils. Similar to the studies in myeloid cells, subcellular fractionation of Sf9 cells expressing P-Rex1 in combination with PI3K or Gβγ also showed that both PI3K activity and Gβγ are required for robust membrane localization, and the use of P-Rex1 mutants demonstrated furthermore that iDHPH is sufficient for synergistic stimulation.37 Hence, PIP3 and Gβγ not only synergistically stimulate the Rac-GEF activity of P-Rex1, but also its membrane recruitment (Fig. 2).
Membrane-derived P-Rex1 migrates on gels similarly to P-Rex1 activated by PP1α whereas cytosolic P-Rex1 migrates more like the phosphorylated inactive protein.37,53 Furthermore, P-Rex1 purified from Sf9 cell membrane has higher Rac-GEF activity than cytosol-derived P-Rex1, although both can be further stimulated by PIP3 and Gβγ 37. These observations led to the suggestion that dephosphorylation of P-Rex1 by PP1α in the cytosol may facilitate its access to PIP3 and Gβγ in the membrane, a possibility which remains to be explored. Finally, use of the GEF-dead P-Rex1 mutant E56A/N238A showed that Rac-GEF activity is not required for the synergistic stimulation of P-Rex1 membrane localization by PIP3 and Gβγ 37.
Binding Proteins
Considering their size and the abundance of protein interaction domains in P-Rex family Rac-GEFs, remarkably few P-Rex binding proteins have been identified to date, and of these, only mTOR and PTEN have been shown to bind P-Rex proteins directly (Fig. 3). However, a number of new interactions in cells have been identified over the past year.
mTOR and Akt1
A yeast 2-hybrid screen using the tandem DEP domains of P-Rex1 as bait identified mTOR as a potential interactor.60 Characterization of the interaction showed that iDEP/DEP or full-length P-Rex1 interact with full-length mTOR or its isolated C-terminus when overexpressed in HEK-293 cells, and that endogenous mTOR interacts with endogenous P-Rex1 in HeLa cells. The in vitro-translated C-terminus of mTOR interacted with iDEP/DEP purified from HEK-293 cells, which strongly suggested that the interaction is direct, although direct binding is difficult to ascertain when proteins are isolated from mammalian cells with undefined purity. The interaction with mTOR is not restricted to P-Rex1 but also occurs with P-Rex2 and P-Rex2b (Fig. 3). Unsurprisingly, considering its direct interaction with mTOR, P-Rex1 interacted in HEK-293 cells with both mTOR-containing protein complexes, TORC1 which is central in cell growth and TORC2 which controls morphology and migration 56,60 (Fig. 3B). Furthermore, shRNA knockdown of endogenous P-Rex1, or expression of a dominant negative ΔDH mutant of P-Rex1 in HeLa cells inhibits leucine-stimulated endogenous Rac activity and migration, suggesting that P-Rex1 is involved in TORC2 dependent processes. ΔDH did not inhibit leucine-stimulated p70S6K activity which led to the suggestion that P-Rex1 may preferentially signal through TORC2 rather than TORC1.60 However, shRNA knockdown of P-Rex1 did inhibit p70S6K activity,60 suggesting that P-Rex1 might contribute to TORC1 signaling as well as TORC2. These intriguing differences revealed by the comparison of the ΔDH mutant with shRNA imply that the effects of P-Rex1 on TORC1 would be Rac-GEF activity dependent whereas those on TORC2 might be Rac-GEF activity independent. This possibility should be elucidated further, for example through the use of GEF-dead P-Rex mutants.
Furthermore, while the original identification of the P-Rex/mTOR interaction favored a model which situated P-Rex1 downstream of mTOR, further research suggested that P-Rex1 can likely act both upstream and downstream of mTOR.56 More work is required, for example through in vitro assays for mTOR kinase and P-Rex1 GEF activity or the reconstitution of lost TORC1 or TORC2 function by active P-Rex1, to ascertain under which conditions P-Rex1 signals in sequence with or parallel to mTOR and if it affects one of the mTOR complexes preferentially.
TORC2 is the kinase that phosphorylates Akt on S473, thereby fully activating Akt (whereas the PIP3-dependent PDK1 phosphorylates T308) (Fig. 3B). P-Rex1 was shown to interact with Akt1 as well as with TORC1 and TORC2 upon overexpression in HEK-293 cells, and this constitutively increased Akt1 phosphorylation on S473.56 In SKOV3 ovarian carcinoma cells, where Akt1 mediates migration downstream of TORC2, shRNA knockdown of P-Rex1 inhibited both Akt1 activity and cell migration, suggesting that P-Rex1 functions upstream of both TORC2 and Akt1.56 However, constitutively active Akt1 was unable to overcome the inhibition of SKOV-3 cell migration induced by shRNA knockdown of P-Rex1, leading to the proposal that P-Rex1 may also signal downstream of Akt1, with a positive feedback loop between Akt1 and P-Rex1-dependent Rac activity.56 It remains to be seen if the interaction of P-Rex1 with Akt1 is entirely mediated through mTOR, or indeed if it might be direct and if Akt1 can phosphorylate P-Rex1. Finally, P-Rex1 dependent activation of Akt was also observed in platelets stimulated through the GPCR for thromboxane 2.17 However, P-Rex1-dependent regulation of Akt has not been observed in all situations. For example, Akt activity is normal in fMLP-stimulated P-Rex1−/− neutrophils,21 or in HRG-stimulated T47D breast cancer cells treated with P-Rex1 siRNA.51
PTEN
The tumor suppressor PTEN was identified by mass spectrometry to be a binding protein of P-Rex2, in a study by the Parsons lab which propelled the P-Rex family from the interest of a select few to superstardom in cancer research (see section “Cancer”).58 The interaction between P-Rex2 and PTEN occurs on the endogenous level in HEK-293 cells, mouse embryonic fibroblasts (MEFs) and mouse liver; it requires the C2 domain and C-terminal tail of PTEN and the PH domain of P-Rex2; and it is direct, as shown by the binding of bacterially derived purified GST-PTEN to in vitro-translated iPH11,58 (Fig. 3). CoIP experiments with P-Rex2 and PTEN mutants showed complex interactions between the 2 proteins in vivo. The IP4P domain of P-Rex2 interacts with the C-terminal PDZ-binding domain of PTEN, and the PH domain binds the catalytic and C2 domains of PTEN.11
Crucially, full-length P-Rex2 or iDHPH inhibit the phosphatase activity of PTEN at equimolar concentrations when purified from HEK-293 cells and tested with soluble di-C8-PIP3 as substrate.58 The effects on PTEN are specific to P-Rex2, as P-Rex1 does not coIP with PTEN from overexpressing HEK-293 cells, nor does its PH domain inhibit PTEN activity 11,58 (Fig. 3). In contrast, the effects of P-Rex2 on PTEN do require the lipid phosphatase activity of PTEN as shown by the inability of P-Rex2 to restore Akt phosphorylation in U87 glioma cells expressing lipid phosphatase-dead PTEN. Furthermore, while phosphorylation of the PTEN tail on regulatory residues is not required for P-Rex2 interaction, mutation of these residues renders PTEN phosphatase activity insensitive to P-Rex2 in U87 cells. Similarly, purified recombinant P-REX2 is unable to inhibit unphosphorylated purified PTEN in vitro, but does inhibit the phosphorylation mimic PTEN-3E at equimolar concentrations.11
Importantly, inhibition of PTEN by P-Rex2 is Rac-GEF activity independent. This was initially concluded from the fact that full-length P-Rex2 or a GEF-dead P-Rex2 N212A mutant in U87 cells were similarly able to inhibit PTEN when coIPed from HEK-293 cells, or to restore Akt phosphorylation (on T308 and S473) lost upon PTEN expression.58 While P-Rex2 N212A may still have residual GEF activity, as extrapolated from P-Rex1 which requires double-mutation of E56A and N238A to be fully GEF-dead, iPH of P-Rex2 was later shown to be sufficient for PTEN inhibition, proving unequivocally that the Rac-GEF activity of P-Rex2 is not required.11
The authors proposed the following model: The P-Rex2 DH domain blocks access of the PH domain to the PTEN tail as long as the tail is unphosphorylated, with IP4P providing structural support. Phosphorylation of the tail is somehow “read” by the P-Rex2 DH domain, which frees the PH domain to inhibit PTEN.11 The charge change in the phosphorylated PTEN tail was proposed to be the message “read” by the DH domain, but another possibility could be that PTEN phosphorylation induces a conformational change robust enough to affect P-Rex2 interdomain interactions. If the proposed freeing of the DH domain occurs, one would predict – in analogy with P-Rex1 – that PTEN tail phosphorylation affects P-Rex2 Rac-GEF activity as well as P-Rex2-dependent PTEN inhibition. However, the effects of the PTEN interaction on P-Rex2 Rac-GEF activity remain to be investigated.
Other interactions (possibly indirect)
Receptors
In serum-starved U87 cells, P-Rex2 restores Akt phosphorylation lost upon PTEN expression when the cells are stimulated with Insulin or PDGF (but not EGF)58 (Fig. 3B). Similarly, P-Rex2−/− MEFs show reduced Akt phosphorylation when stimulated with IGF1 (but not PDGF or EGF).11 This prompted investigations into interactions between P-Rex2 and RTKs. Indeed, overexpressed P-Rex2 coIPs with the phosphorylated IGF1 receptor from HEK-293 cells upon insulin-stimulation, but not with EGF receptor upon EGF stimulation,11 suggesting that P-Rex2 can be recruited selectively into some RTK containing complexes. Similarly, P-Rex1 coIPs with overexpressed PDGFRβ but not PDGFRα from immortalized human fibroblasts, demonstrating that RTK interaction is not restricted to P-Rex2.38 Furthermore, PDGFRβ coIPs both with WT or GEF-dead P-Rex1, showing that Rac-GEF activity is not required.38 The possibility of direct association of P-Rex family GEFs with RTKs remains to be investigated. In addition to interacting with RTKs, they may also bind to GPCRs, as suggested by coIP of the S1P receptor Edg1 with the iPDZ/PDZ domains of P-Rex1 from overexpressing HEK-293 cells. Unsurprisingly, this interaction is mediated by the intracellular C-terminus of the GPCR,61 but it remains unknown whether full-length P-Rex1 can also interact with the GPCR and whether binding might be direct.
Ephrin-B1
P-Rex1 has recently been shown to bind to the transmembrane protein ephrin-B1, a ligand of the RTK EphB which controls neuronal migration and axonal guidance. This was shown through coIP from mouse embryonic cortex transiently overexpressing P-Rex1 by in utero electroporation.62 This required the PDZ domains of P-Rex1 and PDZ-binding domain of ephrin-B1, as P-Rex1 ΔPDZ or an ephrin-B1 PDZ-binding domain mutant did not interact.62 It has been proposed that P-Rex1 might mediate the effects of ephrin-B1 during the development of the cerebral cortex in mice (see section “Neurons and Behavior”).62
EHBP1
Statins have recently been reported to inhibit nuclear Akt phosphorylation and matrigel invasion in insulin-stimulated prostate cancer cells at concentrations too low to affect small G prenylation, by acting through the purinergic GPCR P2×7.63 EHBP1, an adaptor protein involved in GLUT4 trafficking,64 was shown to mediate these effects in some but not all prostate cancer cell lines, and variations of P-Rex1 expression levels in these cell lines led the authors to investigate a possible interaction. Proximity ligation assays suggested that P-Rex1 and EHBP1 may indeed interact in the cytosol of insulin-stimulated DU145 cells and in the nucleus of cells treated additionally with statins,63 but this remains yet to be confirmed by other methods. Confocal microscopy furthermore suggested the possibility of an insulin-dependent interaction between PTEN and P-Rex1, from similarities in their subcellular localizations, but this also requires confirmation through other approaches.
Rap1-GTP/RalGDS
P-Rex1 has recently been proposed as a potential target of Rap1-GTP in human umbilical vein endothelial cells (HUVEC; see section “Endothelial Cells and Vascular Biology”), based on siRNA knockdown of the Rap1-GEF Epac1 or of P-Rex1 having similar effects on Weibel Pallade body secretion and on the fact that P-Rex1 could be isolated from HUVEC lysates with the Rap1-GTP binding domain of RalGDS.65 It is unknown whether P-Rex1 is able to associate with RalGDS independently of Rap1-GTP, or if the association is with Rap1, perhaps independently of RalGDS or Rap1-GTP loading, and if binding might be direct. Interestingly, both Rap1-GTP and P-Rex1 have also recently been shown to mediate E-selectin dependent slow rolling of neutrophils along the vessel wall during inflammation.31
Physiological Function
The functional roles of P-Rex1 and P-Rex2 have largely been studied through siRNA or shRNA-mediated knockdown, dominant-negative mutants and mouse knockouts. Two strains of P-Rex1−/− mice 21,30 and 2 of P-Rex2−/− mice 11,12 have been generated, and one P-Rex1−/− P-Rex2−/− strain.12 All are viable, fertile and apparently healthy, except for slightly reduced body size, liver size, mild neutrophilia 21 and a characteristic skin pigmentation phenotype 66 in P-Rex1−/− mice, and some reduction in body weight in aging female P-Rex2−/− mice.12 However, all P-Rex deficient strains show distinct phenotypes when taxed, as detailed in this section on the physiological P-Rex functions and in sections “Insulin Resistance and Type 2 Diabetes” and “Cancer” on pathophysiological P-Rex functions.
Leukocytes and inflammation
ROS formation through the NADPH oxidase complex, of which active Rac is an essential component, was the first physiological role identified for P-Rex family GEFs, through DNA antisense knockdown of P-Rex1 in myeloid NB4 cells.7 Neutrophils isolated from P-Rex1−/− mice display impaired GPCR (fMLP or C5a receptor)-dependent Rac2 and RhoG activity, ROS production, F-actin formation and polarity, as well as a small reduction in cell motility 21,22,30 (Fig. 4). The motility defect is seen in chemokinesis as well as chemotaxis, and comes from a reduction in cell speed rather than directionality, consistent with an impairment in Rac2 activity.21,30 The largest defect in fMLP- or C5a-dependent ROS formation is seen upon LPS priming, implying a role for P-Rex1 in TLR4 signaling that remains to be explored.21 P-Rex1−/− mice show impaired neutrophil recruitment during inflammation, including thioglycollate-induced sterile peritonitis 21 and ischemia reperfusion of the kidney 31 (Fig. 4). Analysis of P-Rex1−/− neutrophils under flow conditions and through intravital microscopy of the inflamed cremaster muscle, together with the analysis of myeloid HL60 cells with shRNA knockdown of P-Rex1, showed that P-Rex1 controls E-selectin dependent activation of the integrins LFA1 and Mac1, affecting the slow rolling and crawling of neutrophils along the vessel wall prior to firm adhesion and transmigration into the inflamed tissue.31
Figure 4.
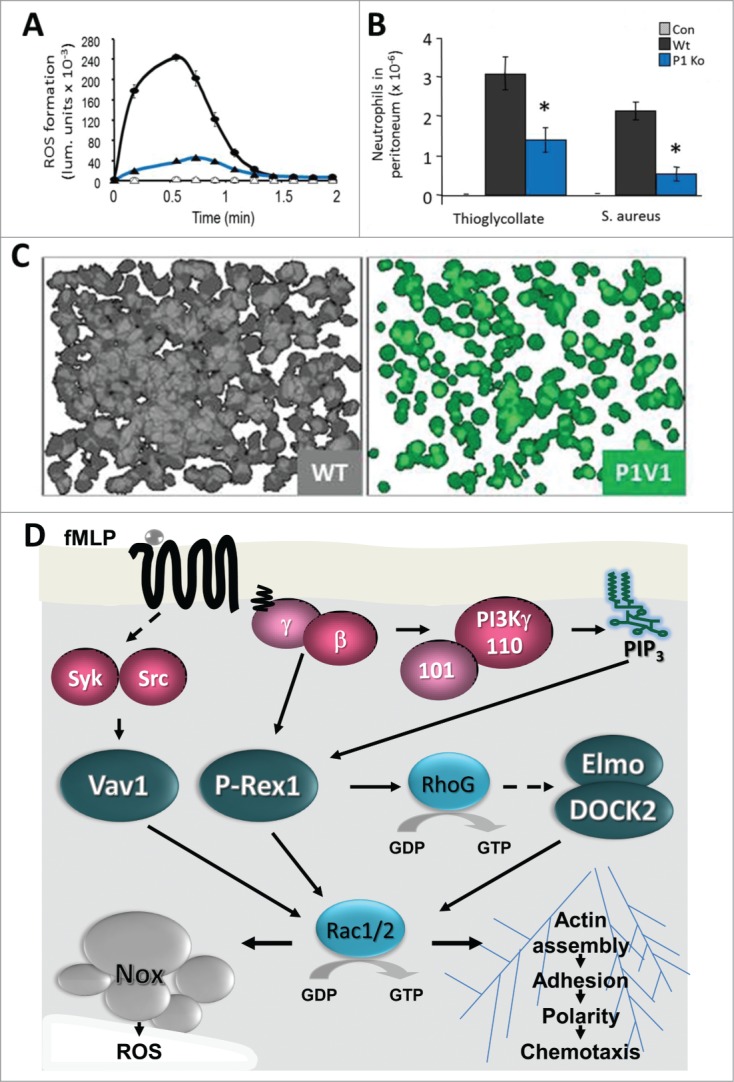
P-Rex1 in neutrophil responses and inflammatory recruitment. (A) Neutrophils isolated from P-Rex1−/− mice (Ko, blue) show impaired GPCR-dependent production of ROS stimulated with fMLP, here shown upon LPS priming.21,30 In addition, P-Rex1−/− neutrophils show defects in GPCR-dependent Rac2 activity, F-actin formation and polarity, and migration.21,30 (B) In P-Rex1−/− mice (Ko, blue), neutrophil recruitment to sites of inflammation is impaired, including as shown here in thioglycollate-induced sterile peritonitis and in septic peritonitis induced by IP injection of Staphylococcus aureus.21,30,31,36 (C) P-Rex1−/− Vav1−/− neutrophils show more profound defects in GPCR-dependent responses than cells lacking the entire P-Rex family or the entire Vav family, showing that P-Rex1 and Vav1 cooperate.20 These pseudocoloured images show 24 superimposed fields of view of WT (gray) and P-Rex1−/− Vav1−/− neutrophils (green) adhering to pRGD upon stimulation with fMLP. Data in (A) and (B) were adapted from ref.21 and data in (C) from ref.20 (D) Neutrophil Rac-GEF signaling stimulated through the fMLP receptor. Stimulation of the GPCR for fMLP releases Gβγ and activates PI3K and protein tyrosine kinases. Gβγ and PIP3 synergistically activate P-Rex1, and Src or Syk activate Vav1. P-Rex1 activates RhoG which leads to activation of Rac1/2, most likely via DOCK2/Elmo22. Much of the P-Rex1 dependent Rac1/2 activity is through RhoG, but it seems likely that at least some is direct, as P-Rex1−/− neutrophils have defects in actin polymerization and P-Rex1−/− mice in peritoneal recruitment during inflammation that are not seen with RhoG−/− cells or mice, respectively.21,30,32 P-Rex1 and Vav1 cooperate in fMLP-dependent activation of Rac1/2 and Rac-dependent responses, including ROS formation through the NADPH oxidase complex (Nox) of which active Rac2 is an integral part, and through the Rac1/2 dependent actin polymerization which is required for adhesion, polarization and migration.20
The fMLP-dependent responses of P-Rex1−/− neutrophils were at best partially inhibited by the P-Rex1 deficiency. However, Rac activity is absolutely required for fMLP-stimulated ROS formation and chemotaxis, suggesting that other Rac-GEF families contribute to these responses. As neutrophils lacking the Vav-family Rac-GEF Vav1, which is regulated by tyrosine phosphorylation, showed similar GPCR-dependent defects as P-Rex1−/− cells, double-deficient mice were generated. Indeed, P-Rex1−/− Vav1−/− neutrophils showed profound defects in fMLP-receptor-dependent Rac1 and Rac2 activity, ROS formation, adhesion and migration (Fig. 4). These defects were stronger than in cells lacking the entire P-Rex or the entire Vav family, suggesting that, despite being activated through different mechanisms, P-Rex1 and Vav1 cooperate to generate thresholds of Rac activity required for GPCR-dependent neutrophil responses. It remains to be shown what effect such cooperation of different Rac-GEF families has on the inflammatory response.
In addition to neutrophil responses, P-Rex1 is also important in other myeloid cells. It regulates GPCR-dependent responses, including C5a-stimulated Rac1 activity or ROS formation and C5a- or MCP1-stimulated chemotaxis in mouse macrophages,15 and P-Rex1−/− mice show impaired recruitment of monocytes/macrophages to the thioglycollate-inflamed peritoneum.21
Platelets and haemostasis
P-Rex1 is expressed in human and mouse platelets, and it binds to Rac1 upon thrombin stimulation.16 Despite normal platelet development, P-Rex1−/− mice show mild defects in haemostasis, as incisions in the tail of P-Rex1−/− mice bleed longer and the incidence of re-bleeding of fresh wounds is higher than in control mice.17 Isolated P-Rex1−/− mouse platelets show reduced endogenous Rac1, Akt and Jnk activities upon stimulation of the thromboxane A2 receptor.17 They adhere and spread normally on fibrinogen in response to thrombin or ADP stimulation, or on fibrillar collagen,16,17 but show defects in aggregation and dense granule secretion (ATP release) upon stimulation of the GPCRs for thromboxane A2 or thrombin.17 However, these defects were only observed at low doses of stimuli, suggesting that P-Rex1 is not absolutely required.
Endothelial cells and vascular biology
Overexpression of P-Rex family Rac-GEFs in endothelial cell lines induces Rac1 activity, cell spreading, lamellipodia formation, membrane ruffling, chemotaxis and matrigel invasion in response to a range of stimuli, as would be expected for a typical Rac-GEF.7,8,18,61 Interestingly, overexpression of the isolated PDZ domains of P-Rex1 was sufficient to promote S1P-dependent chemotaxis of pig aortic endothelial cells, which implies a potential Rac-GEF activity independent function and merits further investigation.61 Yet, endogenous P-Rex proteins also play important roles in various types of endothelial cells. P-Rex1 is expressed in a range of human endothelial cell lines to a similar level as in macrophages,36 and shRNA knockdown of endogenous P-Rex1 in human microvascular endothelial cells (HMEC) inhibited SDF1-stimulated Rac1 activity, as well as chemotaxis and in vitro angiogenesis (formation of capillary-like structures in matrigel) stimulated by SDF1 but not VEGF or FBS.18 Similarly, P-Rex2b siRNA inhibited S1P-stimulated Rac1 activity and chemotaxis in human umbilical vein endothelial cells (HUVECs).23 As HUVECs express both P-Rex1 and P-Rex2b,23,65 they might be a useful system for exploring the possibility of differential physiological roles, as the only other non-cancer cell type known to endogenously express both P-Rex1 and P-Rex2 isoforms are cerebellar Purkinje neurons which are very difficult to manipulate.
Rac1 activity is required for the secretion of Weibel Pallade bodies (which contain von Willebrand factor) in HUVECs induced by costimulation with the adrenergic GPCR ligand epinephrine and with IBMX (a phosphodiesterase inhibitor that blocks cAMP metabolism, thereby stabilizing PKA activity and other cAMP-dependent responses).65 This Rac1-dependent response is mediated by the Rap1-GEF Epac1, and in a search for possible Rap1 targets, P-Rex1 was recently found to interact with the Rap1-GTP binding domain of RalGDS.65 P-Rex1 siRNA showed that P-Rex1 mediates epinephrine/IBMX-stimulated but not thrombin-stimulated Rac1 activity and Weibel Pallade body secretion.65 However, this sensitivity to P-Rex1 siRNA was not observed at high doses of epinephrine, suggesting that other Rac-GEFs are able to compensate. Furthermore, epinephrine/IBMX-stimulated Rac1 activity requires PKA as well as P-Rex1, as shown by use of PKA inhibitors, but it is currently unknown how this apparent cooperation of P-Rex1 and PKA relates to the fact that PKA inhibits the Rac-GEF activity of P-Rex1.53,65
Stimulation of monolayers of human lung microvascular endothelial cells (HLMVEC) with TNFα decreases their barrier function, as assessed by transendothelial electrical resistance (TER), through a weakening of VE-cadherin dependent cell/cell contacts and the induction of gaps between cells in a Rac1-dependent manner.36 P-Rex1 siRNA showed that this response, as well as TNFα-stimulated Rac1 activity, VE-cadherin phosphorylation, apoptosis and ROS formation, are mediated by P-Rex1.36 In addition, TNFα-stimulated migration of WT or P-Rex1−/− neutrophils across monolayers of HLMVECs treated with P-Rex1 or control siRNA showed that P-Rex1 expression in endothelial cells is more important for “transmigration” than P-Rex1 expression in neutrophils.36
Intratracheal stimulation of mice with TNFα increases the vascular permeability of the lung, as measured by the leakage of Evans blue albumin from the circulation, resulting in edema. Interestingly, P-Rex1−/− mice were completely protected from these effects in one study,36 whereas another study equally convincingly showed normal vascular permeability in kidney vessels during ischemia-reperfusion injury.31 Different P-Rex1−/− strains were used in these studies, but both strains have comparable defects in neutrophil function and in the inflammatory recruitment of neutrophils or macrophages,21,30,31,36 so the reason for the discrepancy in vascular permeability remains unclear.
Neurons and behavior
P-Rex1 is widely expressed throughout the nervous system whereas P-Rex2 is in quite selectively expressed in the cerebellar Purkinje neuron which controls motor coordination.12,19
NGF stimulation of P-Rex1 overexpressing neuronal PC12 cells induces Rac1 activity, lamellipodia formation, membrane ruffling, cell spreading and migration, whereas expression of a dominant-negative ΔDH mutant of P-Rex1 or siRNA knockdown of endogenous P-Rex1 inhibit these responses.19 Interestingly, a separate study using stable shRNA knockdown of P-Rex1 in the same cell line showed that P-Rex1 activates overexpressed Rac3 and inhibits neurite outgrowth and maturation, consistent with reports suggesting that Rac3 activity can have opposite effects to Rac1 activity in neurons, but as mentioned in section “Substrate Specificity”, the conditions which may alternatively promote P-Rex1 dependent Rac1 or Rac3 activity require further study.34
Transient expression of the dominant-negative ΔDH mutant of P-Rex1 in mouse embryos by in utero electroporation was shown to block neuronal migration within the cerebral cortex.19 In a recent study which demonstrates that ephrin-B1 controls pyramidal neuron migration and interacts with P-Rex1 during the development of the cerebral cortex,62 overexpression of the P-Rex1 ΔDH mutant was used to suggest that P-Rex1 mediates the effects of ephrin-B1.62 However, as discussed in section “Tools for Assessing P-Rex Function”, overexpression of ΔDH is a difficult tool for assessing P-Rex function in isolation, so the role of P-Rex1 in this process should be confirmed through use of P-Rex1 knockdown, deficiency or GEF-dead point mutant. To emphasize this point, P-Rex1−/− mice show no obvious defects in cerebral development.12
P-Rex2−/− mice have impaired Purkinje cell morphology, with shorter and narrower principal dendrites, and exhibit a mild motor coordination defect which worsens during aging specifically in females 12 (Fig. 5). The cause of the gender selectivity of this phenotype remains unexplored. It would be interesting to generate conditional P-Rex2 deficiency that could be induced specifically in Purkinje neurons of mature animals to dissect developmental defects from acute impairment of signaling pathways. Interestingly, Vav3−/− mice show Purkinje neuronal morphology defects early during development that are compensated for in mature cells, and they show motor coordination defects in postnatal but not older animals.67 It would be useful to assess the combined roles of P-Rex2 and Vav3 in Purkinje neuron function.
Figure 5.
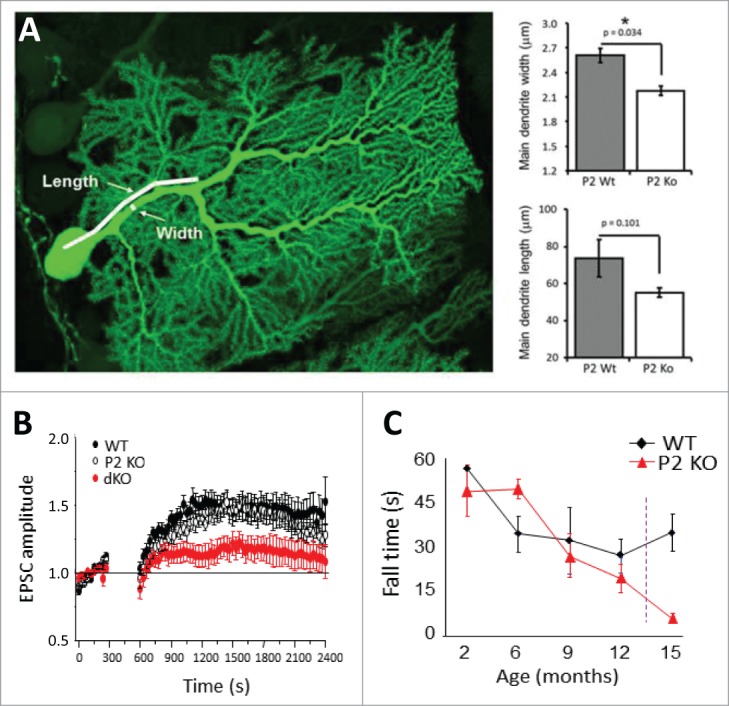
P-Rex1 and P-Rex2 in neuronal morphology, synaptic plasticity and behavior. (A) P-Rex2−/− but not P-Rex1−/− mice have impaired Purkinje cell morphology, with shorter and narrower principal dendrites.12 The image shows a single Purkinje neuron in a cerebellar slice from a P-Rex2−/− Pcp2-GFP mouse brain which expresses GFP to enable detailed morphological analysis. (B) P-Rex1−/− P-Rex2−/− Purkinje neurons can largely compensate for the dendrite morphology defect, but patch clamp electrophysiology showed that they have impaired maintenance of long-term potentiation.68 (C) P-Rex2−/− mice exhibit a mild motor coordination defect which worsens during aging specifically in females.12 In P-Rex1−/− P-Rex2−/− mice, this phenotype is exacerbated.12 Data in (A) and (C) were adapted from ref.12 (B) from ref.68
Like P-Rex2, P-Rex1 is also expressed in the cerebellum, but P-Rex1−/− mice show normal Purkinje neuronal morphology and motor behavior.12 In contrast, double-deficiency in P-Rex1−/− P-Rex2−/− mice exacerbates the phenotype seen in P-Rex2−/− mice, with similar morphological defects but a more severe impairment of motor activity, posture and gait that is consistent with cerebellar dysfunction and evident in both males and females from a young age.12 Patch clamp electrophysiology showed that P-Rex1−/− P-Rex2−/− Purkinje neurons largely compensate for the dendrite morphology defect, as they show normal passive membrane properties and basal synaptic transmission. However, P-Rex expression is required for the maintenance of cerebellar long-term potentiation, whether evoked through electric stimulation or through nitric oxide donors 68 (Fig. 5). Finally, a recent report suggested that P-Rex2 may also be expressed in mouse retinal neurons,69 but this requires confirmation through knockdown or P-Rex2−/− controls. For a detailed recent review on the role P-Rex1 and P-Rex2 in neuronal synaptic plasticity and behavior, see reference number.70
Melanocytes and pigmentation
When crossed onto a C57Bl6 genetic background, P-Rex1−/− mice have black fur except for white bellies, feet and tail tips.66 This pattern of pigmentation is caused by impaired melanoblast migration from the neural crest around the body of the developing embryo, rather than by impaired proliferation, and it cannot be overcome by genetically induced hyperproliferation of melanoblasts66 (Fig. 6). This role of P-Rex1 in melanoblast migration is biologically relevant as it translates into a critical role in metastasis upon transformation of mature melanocytes into melanoma cells (see section “Melanoma”).66 To investigate whether this role in melanoblast migration is entirely due to P-Rex1-dependent activation of Rac1, P-Rex1−/− mice were crossed to a mouse strain with a melanocyte-specific deletion of Rac1 that shows a similar white-belly phenotype as P-Rex1−/− mice, but from impaired melanoblast proliferation as well as impaired migration,71 The expectation was that melanocyte P-Rex1 would signal largely through Rac1 in melanocytes and that, therefore, the double-mutant strain should have a similar coat color. Surprisingly though, P-Rex1−/− Tyr::Cre Rac1fl/fl double-mutant mice were almost entirely white, due to a substantial reduction in melanocyte numbers caused by Rac1-independent decrease in melanoblast proliferation as well as through Rac1-dependent impairment in melanoblast migration.72 Therefore, P-Rex1 clearly has Rac1-independent as well as Rac1-dependent functions in melanocyte development. It has been suggested that the Rac1-independent function of P-Rex1 in melanoblast proliferation might be mediated through its recently described role as a RhoG-GEF,22 but this hypothesis remains to be tested experimentally.
Figure 6.
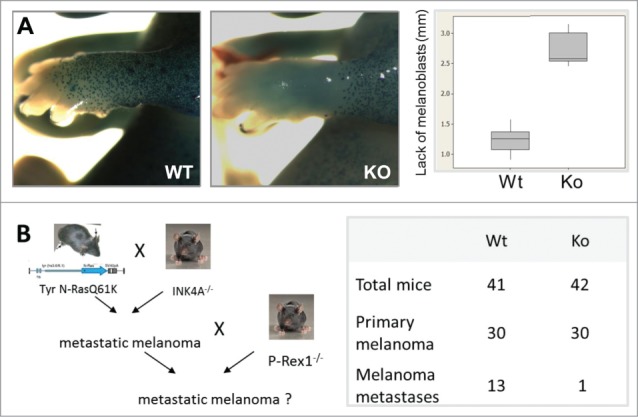
P-Rex1 in melanocyte migration and melanoma metastasis. (A) P-Rex1−/− mice on C57Bl6 genetic background have white bellies, feet and tail tips, caused by impaired melanoblast migration from the neural crest in the developing embryo.66 This role of P-Rex1 translates into a critical function in metastasis upon transformation of mature melanocytes into melanoma cells. (B) P-Rex1−/− mice show normal formation and growth of primary tumors when crossed to the metastatic melanoma model Tyr::NrasQ61K Ink4a−/−, but a drastic reduction in the formation of metastases in the lung, liver and brain, which results in a significant improvement in survival.66 Data were adapted from reference.66
Zebrafish development
Only one study has so far investigated P-Rex family Rac-GEFs in non-mammalian vertebrates. In zebrafish, the TGF-β like cytokine Nodal is important in endoderm and mesoderm formation, by stimulating transcription factor expression and cell migration during gastrulation, and at least the latter occurs in a Rac1 dependent manner.73 Microarray analysis identified P-Rex1 as a potential target of Nodal in zebrafish endoderm.73 Morpholino knockdown in endodermal cells of zebrafish embryos showed that P-Rex1 mediates Rac1 activity and affects the persistence and speed of cell migration in a manner similar to Nodal, and P-Rex1 overexpression partially rescued endodermal cell migration lost upon inhibition of Nodal.73 However, it is unknown whether P-Rex1 is sufficient to affect development into mature fish. Furthermore, P-Rex1 may affect embryonic development specifically in fish. In mammals, its role in embryonic development seems minor, as P-Rex1−/− mice develop into healthy animals.
Insulin Resistance and Type 2 Diabetes
A role of P-Rex family Rac-GEFs in metabolic syndrome is currently emerging. P-Rex2 controls insulin signaling at least partially through its inhibition of PTEN and the resulting increase in PIP3 11 (Fig. 3B). Insulin-stimulated P-Rex2−/− MEFs show reduced phosphorylation of Akt, Foxo1/3 and GSK3-β, and loss of PTEN inhibition.11 Similarly, Akt, Foxo1/3 and Gsk3-β phosphorylation are reduced in adipose tissue and liver of P-Rex2−/− mice injected with insulin. PTEN activity is elevated and PIP3 levels are reduced in P-Rex2−/− livers despite normal PI3K activity. Importantly, P-Rex2 deficiency is sufficient to affect glucose homeostasis and insulin sensitivity. Despite normal baseline blood glucose levels, P-Rex2−/− mice show increased peak and sustained blood glucose levels upon glucose challenge, suggesting defects in glucose uptake.11 Furthermore, while blood glucose levels drop initially upon insulin injection of P-Rex2−/− mice, as expected, the response is unsustained, suggesting that P-Rex2 deficiency might cause insulin resistance.11 This seems to be relevant in humans, as adipose tissue from insulin-resistant people shows reduced P-Rex2 protein levels and higher PTEN activity than that of healthy subjects.11 As insulin resistance can lead to type 2 diabetes, it is likely that reduced P-Rex2 expression in highly insulin-responsive tissues may contribute to the development of this disease, a possibility that requires urgent further research. It would be valuable to generate a GEF-dead P-Rex2 knock-in mouse (not trivial seen that 2 residues in different exons need to be mutated) to evaluate the relative contributions of GEF-activity independent effects (on PTEN and perhaps other targets) and of the Rac-GEF activity of P-Rex2 in insulin signaling and glucose homeostasis.
Like P-Rex2, P-Rex1 also mediates insulin signa-ling 54,56,63,74,75 (Fig. 3B) and has been tentatively linked to type 2 diabetes,76,77 although in contrast to P-Rex2, P-Rex1 dependent insulin signaling seems to be entirely mediated through its Rac-GEF activity.54,74,75 P-Rex1 is endogenously expressed in 3T3-L1 adipocytes, and P-Rex1 siRNA reduces insulin-stimulated glucose uptake by these cells.74 This could be due to effects of P-Rex1 on the trafficking of the transmembrane glucose transporter Glut4. Coexpression of P-Rex1 with an exofacially-tagged form of the transporter in 3T3-L1 adipocytes showed that P-Rex1 promotes the insulin-stimulated plasma membrane localization of Glut4.74 While the effect of P-Rex1 on total cellular Glut4 levels remains to be determined, a dominant-negative Rac1 mutant was shown to block P-Rex1 dependent Glut4 membrane localization, suggesting that P-Rex1 mediates Glut4 trafficking via Rac.74
SNP analysis provided a potential link of P-Rex1 with type 2 diabetes. The human PREX1 locus is near a region on chromosome 20 associated with type 2 diabetes (20q12–13.1). Sequencing of this region in type 2 diabetes patients with end-stage renal disease identified 3 SNPs in the perigenic 3′ region of PREX1 that are significantly disease-associated, and one of these SNPs was also found in a separate cohort of patients without renal disease.76 A follow-up study with further patient cohorts and additional analysis by body mass index showed no association with obesity alone but identified further SNPs significantly associated with type 2 diabetes, and 6 of these were seen specifically in obese type 2 diabetes patients.77 It was concluded that SNPs in the 3’ perigenic region of the PREX1 locus, while not predisposing people to obesity, may predispose obese people to developing type 2 diabetes.77 However, it remains to be seen if any of these SNPs actually affect the PREX1 gene rather than other genes in the vicinity, and in which way.
Cancer
P-Rex1 and P-Rex2 are important for tumor growth and/or metastasis in several types of cancer, particularly in breast and prostate cancer and in melanoma. The roles in of P-Rex family Rac-GEFs in cancer generally4,78,79, in breast cancer 80,81 and in melanoma82-84 have recently been reviewed in detail elsewhere, so they will only be briefly summarised and updated here. It should be noted that there are no reports yet demonstrating spontaneous tumor formation in P-Rex2−/− mice, as might have been expected from the P-Rex2-dependent inhibition of the tumor suppressor PTEN.
Overexpression and mutation
P-Rex1 is overexpressed in thyroid, breast, kidney, prostate and colon cancer as well as in melanoma,51,66,85 and P-Rex2 is overexpressed in breast, prostate, pancreatic and ovarian cancer and glioma.58 The PREX1 locus is amplified in some but not all breast cancer cell lines that show overexpression, suggesting that amplification is an important mechanism in the deregulation of P-Rex1, but not the only one.51 In metastatic prostate cancer cells, upregulation of P-Rex1 is a consequence of lost repression through HDACs.24 However, as discussed in section “Tissue Distribution and Regulation of Expression,” it is currently unknown why this repression is specifically lost in metastatic rather than non-metastatic cells, or whether the same mechanism applies in other types of cancer. The PREX2 locus is in a genomic region linked to aggressive cancers and metastasis and is frequently amplified in melanoma, breast, prostate and colorectal cancers.58,86 In melanoma, the locus also undergoes also chromosomal translocations and rearrangements, suggesting that multiple mechanisms contribute to the deregulation of P-Rex2.86
Stringent analysis of combinations of databases revealed somatic mutations in P-Rex2 in an average of 3% of samples from different types of human cancers, including pancreatic, colorectal and lung cancers, whereas somatic mutations in P-Rex1 are around 10-fold rarer.58 Mutation rates vary between cancer types and are particularly high for P-Rex2 in melanoma 86 and potentially for P-Rex1 and P-Rex2 in lung cancer.87 Large-scale whole genome sequencing revealed that 14% of human melanomas show mutations in P-Rex2 (detailed in section “Melanoma,”), making P-Rex2 the most mutated Rac-GEF in cancer and the third most mutated protein in melanoma after B-Raf and N-Ras.86 Furthermore, exome sequencing of lung cancer samples from 97 Japanese patients recently suggested that P-Rex1 and P-Rex2 may be among the 10 most frequently mutated genes in lung cancer. Five non-synonymous mutations were found in P-Rex1 (4 of which in the DH domain) and seven in P-Rex2 (6 of these in the PH and DEP domains).87 However, the mutations described in this study have not yet been confirmed by other methods or evaluated experimentally.
Breast cancer
P-Rex1 is highly overexpressed in human breast tumors, and P-Rex2 expression is increased in breast tumors with wild-type PTEN status and correlated with activating mutations of PI3K.58 Both Rac-GEFs promote the growth of breast cancer cells, and at least P-Rex1 also promotes tumor growth in mice.51,58 Analysis of breast cancer cell lines suggested that P-Rex1 affects both growth and motility through its Rac-GEF activity, whereas P-Rex2 promotes growth at least in part through its GEF-activity independent inhibition of PTEN.
P-Rex1 is not expressed in normal breast tissue but is found in 58% of human breast tumor samples, with higher expression in metastasising tumors and highest in lymph node metastases.10,51,81 Comparing breast cancer subtypes, P-Rex1 expression is highest in the luminal B subtype and lowest in basal-like tumors and correlates with estrogen receptor and ErbB2 expression.51 Disease-free survival is significantly reduced in breast cancer patients with tumors that express high levels of P-Rex1 compared to patients with tumors expressing low levels.10
Like in the patient samples, P-Rex1 is overexpressed in human breast cancer cell lines that are derived from luminal but not from basal-type breast tumors.51 Knockdown of P-Rex1 in T47D, BT474 or MCF7 breast cancer cells inhibits Rac1 activity stimulated through various RTKs.10,51,54 shRNA knockdown of P-Rex1 impairs the HRG-stimulated chemotaxis and soft agar colony formation of T47D or BT474 cells51 and the NRG- or IGF1-stimulated adhesion to fibronectin, motility in scratch wound assays, matrigel invasion, and proliferation of MCF7 cells.10,54 Furthermore, shRNA knockdown of P-Rex1 in BT474 or MCF7 cells impairs tumor growth in nude mice when these cells are injected into the flanks or orthotopically into the mammary fat pad.10,51
As discussed in section “Activation by PIP3 and Gβγ,” ErbB-dependent activation of P-Rex1 Rac-GEF activity in breast cancer cells is at least in part mediated through transactivation of CXCR4, Gβγs and GPCR-dependent PI3K activity, but CXCR4 also signals through Gβγ- and PI3K-independent routes, and ErbB through tyrosine kinase-dependent PI3Ks, with the relative contributions of these pathways remaining to be investigated.51 Furthermore, as detailed in section “Regulation through Kinases,” some but not all RTKs regulate P-Rex1 in breast cancer cells through unidentified serine kinases, and phosphosite mutants suggested that these phosphorylation events are sufficient to affect the P-Rex-dependent proliferation of MCF7 and HCC3153 breast cancer cells.10,54 A recent investigation into the Erk pathway in breast cancer cell lines with activating PI3K mutations and/or ErbB2 amplifications revealed that PI3K activates the Raf/Mek/Erk pathway in these but not in other types of breast cancer cells, and that this activation occurs in a Rac1/Pak dependent manner.88 P-Rex1 was identified as the PI3K-sensitive Rac-GEF in this pathway, through knockdown of P-Rex1 in T47D or MCF7 cells which reduced the phosphorylation of c-Raf, Mek and Erk but not Akt.88 Moreover, P-Rex1 expression levels were shown in a range of breast cancer cell lines to correlate with the sensitivity of Rac1 and Erk activity in these cells to PI3K inhibitors, although not with total Rac1 activity. It was proposed that global Rac1 activity is irrelevant for Erk signaling in these cells, but that a specific PI3K/P-Rex1 sensitive pool of Rac1 is required.88 It remains to be seen if such specialized pools of Rac1 activity exist and how the cell would be able to identify and interpret them, and also it remains to be shown if the P-Rex1-dependent Rac/Pak/Raf/Mek/Erk pathway is in any way linked to specific upstream receptors.
Unlike P-Rex1,51 P-Rex2 expression correlates with activating PI3K mutations in human breast tumors.58 shRNA knockdown of P-Rex2 inhibits AKT phosphorylation and cell proliferation in PTEN expressing MCF7 cells but not in PTEN-deficient BT549 breast cancer cells.58 Overexpression of P-Rex2 or iDHPH promotes proliferation of the immortalized human mammary cell line MCF10A under low-serum conditions, and coexpression with constitutively active PI3K induces serum-independent growth, the formation of “multilobulated mammospheres” in matrigel with signs of invasion, and soft agar colony formation.58 Rescue experiments with active PTEN in P-Rex2 knockdown cells and comparison of WT and GEF-dead P-Rex2 in PTEN expressing cells remain to be done to evaluate the extent of breast cancer cell growth being mediated through P-Rex2 dependent inhibition of PTEN. Finally, the effects on P-Rex2 expression in breast cancer cells on tumor growth in mice also remain to be assessed.
Melanoma
P-Rex1 is not detectable in adult human skin, but is strongly expressed in 80% of primary and metastatic melanoma, with highest expression in lymph node metastases.66 P-Rex1 is also upregulated in human melanoma cell lines, and its expression level correlates with invasiveness, regardless of N-Ras or B-Raf status.66 P-Rex1 siRNA reduces matrigel invasion of the melanoma cell lines CHL1 and WM852.38,66 P-Rex1−/− mice show normal formation and growth of primary tumors when crossed to the metastatic melanoma model Tyr::NrasQ61K Ink4a−/−, but a drastic reduction in metastases of the lung, liver and brain that results in a significant improvement in survival.66 Stable overexpression of WT but not of GEF-dead P-Rex1, nor of the related Rac-GEF Tiam1, restores matrigel or organotypic invasion of melanocyte lines derived from the skin of Tyr::NrasQ61K Ink4a−/−/P-Rex1−/− pups, which shows that P-Rex1 mediates these responses specifically through its Rac-GEF activity.66
As mentioned above, P-Rex2 is frequently mutated in human melanoma.82,84,86,89 Whole-genome sequencing of 25 human metastatic melanomas showed that 11 genes are significantly mutated, led by B-Raf and N-Ras as expected, but closely followed by P-Rex2. In these 25 metastatic melanomas, 13 non-synonymous mutations were found in P-Rex2, which were distributed throughout the length of the protein. Four of these mutations were truncations, the others missense. Capillary sequencing of a further 107 melanoma samples revealed 15 more non-synonymous mutations, of which one was a truncation, the rest missense. From these numbers, it was concluded that 14% of human melanomas carry P-Rex2 mutations. At least some of these mutations are pathobiologically relevant, as tumor incidence was increased and tumor-free survival reduced in nude mice injected with immortalized human melanocytes that have an N-RasG12D-activating mutation and express the P-Rex2 mutants K278*, E824*, G844D or Q1430*, whereas coexpression of P948S or G106E had little effect and WT P-Rex2 none.86
Future analysis of these P-Rex2 mutations on the molecular level for their effects on Rac-GEF activity, subcellular localization and PTEN interaction is imperative. Studies with purified mutated recombinant P-Rex1 or P-Rex2 proteins would predict that only one of these P-Rex2 mutants, K278* – which results in an iDH protein – should have considerable constitutive Rac-GEF activity 37,42 and be unable to interact with and inhibit PTEN 11 or translocate robustly to the plasma membrane.37 In contrast, E730* or E824* which result in ΔIP4P proteins not unlike P-Rex2b should not harbor substantial constitutive activities and be responsive to PIP3 and Gβγ, although likely not with the same sensitivity as the full-length protein.42 Similarly, the Q1430* mutant which lacks the very C-terminal tail of P-Rex2 might have reduced affinity for Gβγ.43 Due to the lack of structural information on P-Rex family Rac-GEFs, the effects of the point mutants are even harder to predict. Yet, even prior to the elucidation of the underlying mechanisms, the substantial impacts of the deregulation of both P-Rex-family Rac-GEFs in melanoma, together with the frequent occurrence of Rac1 mutations in this disease,90,91 are regarded as the most compelling evidence for driver functions of Rho-family small GTPases and their GEFs in cancer.4
Prostate cancer
P-Rex1 expression is low in normal human prostate and primary prostate tumors but high in lymph node metastases.85 P-Rex1 is also highly overexpressed in human metastatic prostate cancer cell lines.24,63,85 siRNA knockdown of P-Rex1 in PC3 metastatic prostate cancer cells reduces Rac1 activity, transwell migration and matrigel invasion stimulated by EGF or SDF1.63,85 Similarly, overexpression of P-Rex1 in non-metastatic prostate cancer cell lines promotes transwell migration and matrigel invasion.85 However, it is insufficient to promote tumor formation upon subcutaneous injection into nude mice, suggesting that P-Rex1 does not affect tumor growth in this type of cancer.85 In contrast, it is sufficient to induce the formation of lymph node metastases upon orthotopic implantation into NOD/SCID mice. Moreover, this pro-metastatic effect of P-Rex1 in prostate cancer seems to require the Rac-GEF activity of P-Rex1, as overexpression of a P-Rex1 ΔDH mutant (see section “Tools for Assessing P-Rex Function”) does not promote lymph node metastasis.85
Other cancers
In addition to melanoma, breast and prostate cancer, P-Rex family Rac-GEFs have been functionally implicated in neuroblastoma and in ovarian, lung and gastric cancers, mainly through studies in cell lines but also through the recently identified mutations in tumors from lung cancer patients which remain to be evaluated (see section “Overexpression and Mutation”).87
P-Rex1 is expressed in the human ovarian carcinoma cell line SKOV-3, and shRNA knockdown inhibited IGF-1 stimulated Akt phosphorylation and migration in these cells.56 Similarly, shRNA knockdown of endogenous P-Rex1 in A549 lung epithelial cancer cells inhibited their IGF-1 stimulated migration and matrigel invasion.56 In the lung cancer cells, Rac1 activity, chemotaxis and matrigel invasion, as well as the secretion of MMP-2 gelatinase could also be synergistically stimulated through GPCRs and RTKs, for example with combinations of LPA and EGF, and shRNA knockdown of P-Rex1 abolished these synergistically stimulated responses.75
P-Rex2 was recently shown to be overexpressed in gastric cancer samples from 2 patients compared to their paired normal tissue.26 This is obviously a small sample size, but P-Rex2 siRNA also suppressed the growth of the gastric cancer cell line BGC-823, possibly through mild effects on cell cycle progression and rate of apoptosis.26 Finally, P-Rex2 is also expressed in several human neuroblastoma cell lines, and P-Rex2 siRNA in SH-SY5Y or GI-Li-N cells reduced Akt phosphorylation, migration and matrigel invasion in the presence of serum, as well as soft agar colony formation.25
Targeting P-Rex in cancer
In summary, overexpression of P-Rex1 promotes the growth of breast tumors and the metastasis of melanoma and prostate cancer, overexpression of P-Rex2 promotes the growth of breast cancer cells, and at least some of the P-Rex2 mutations found in melanoma can increase Ras-dependent tumor formation. Roles in the growth or migration of cells from other types of cancers are also emerging. These important roles in a wide range of cancers make P-Rex family Rac-GEFs attractive targets for cancer therapy.
Targeting of P-Rex Rac-GEF activity in cancer is preferable to direct targeting of Rac, which would likely have considerable cytotoxic effects, considering past endeavors to target other small G proteins.92 Targeting P-Rex Rac-GEF activity seems more promising, as these proteins are expressed in a limited number of tissues but strongly overexpressed in tumors, and because they are coupled to specific signaling pathways. Yet, targeting of Rac-GEFs is not a trivial task. To block P-Rex activity efficiently, the catalytic domain would need to be targeted, and this implies affecting the interaction with Rac. The targeting of protein/protein interactions carries inherent problems, as they occur with relatively low affinities and usually over large stretches of surface that are difficult to affect specifically. In addition, the fact that the catalytic DH domains of P-Rex family Rac-GEFs are homologous to those of 70 other Dbl-type GEFs must further complicate the development of P-Rex specific inhibitors. However, the task cannot be impossible, as small molecule inhibitors for other Rac-GEFs are slowly beginning to emerge, the best example being NSC23766 which inhibited Tiam1 and Trio, but not Vav1, at micromolar concentrations.93 Therefore, despite not expected to be straightforward, the development of small molecule inhibitors of P-Rex Rac-GEF activity seems both extremely worthwhile and potentially feasible.
In addition to Rac-GEF activity, another targetable aspect of P-Rex biology is the GEF-activity independent inhibition of PTEN by the P-Rex2 PH domain in breast cancer cells.11,58 Blocking this protein/protein interaction would affect P-Rex2 specifically, as P-Rex1 does not interact with PTEN. Furthermore, although PH domains are conserved in a multitude of signaling proteins, it seems likely that the PH domain of P-Rex2 is sufficiently different from others to be specifically targetable. However, as discussed in section “PTEN,” despite experiments in cell lines that suggest an important role of this interaction in cell growth, it is not yet clear how much the P-Rex2 dependent inhibition of PTEN contributes to tumor growth in breast cancer.
As well as targeting P-Rex proteins directly, other promising avenues of targeting these GEFs in cancer include blocking their activation indirectly through their upstream signaling pathways. This could be useful for targeting P-Rex, despite the inherent danger of affecting P-Rex-independent as well as P-Rex-dependent pathways. Another possible approach could be the blockade of the interaction of P-Rex with mTOR.79 Experiments in breast cancer cell lines have already begun to show that the former of these avenues holds promise, as P-Rex1 expression levels correlate with the sensitivity of cell growth to the PI3K inhibitor GDC-0941 in cells that harbor ErbB2 amplifications or PI3K activating mutations, whereas growth inhibition of xenografts from breast cancer cells with low P-Rex1 expression in xenografts required the concomitant treatment with GDC-0941 and the MEK inhibitor AZD6244.88 These experiments led to the proposal that P-Rex1 expression levels could be used as a biomarker for the suitability of breast cancers for treatment with single-agent inhibitors.
Future Directions
What is the next big thing in P-Rex research? Conceptually, the most exciting emerging role is that of P-Rex1 in protein and vesicle trafficking. Coexpression of P-Rex1 with the GPCR for S1P in endothelial cells attenuates the S1P-stimulated internalization of the receptor,61 and coexpression with the glucose transporter Glut4 in adipocytes promotes the insulin-stimulated plasma membrane localization of the transporter,74 although whether these protein trafficking events occur on the endogenous protein level is yet to be investigated. Yet, effects of P-Rex1 on vesicle trafficking have been shown to occur on the endogenous level and are sufficient to alter cell responses. Platelets from P-Rex1−/− mice show defects in dense granule secretion upon stimulation of the thromboxane or thrombin receptors,17 and P-Rex1 siRNA impairs Weibel Pallade body secretion in HUVECs upon stimulation with epinephrine and IBMX.65 Rac activity, although not required for constitutive trafficking such as ER to Golgi transport, is required for regulated endocytic, phagocytic and secretory processes.94 Few Rac-GEFs are known to mediate these responses, and the mechanisms through which they exert this regulation are even less understood.
In addition to this exciting new functional role of P-Rex1, many fundamental questions remain in the P-Rex field which have been alluded to throughout. Among the most urgent issues are structural information for both P-Rex1 and P-Rex2, the molecular characterization of the P-Rex2 mutations observed in melanoma, the possibility of P-Rex2 regulation through PTEN, the paucity of known direct binding partners for both P-Rex1 and P-Rex2, work with human patients on the emerging role of P-Rex family Rac-GEFs in insulin resistance and possibly type 2 diabetes, and an investigation whether the impairments in leukocyte recruitment and function in P-Rex1−/− mice translate into roles in immunodeficiencies or inflammatory conditions in humans.
APPENDIX: Assays and Tools
Many different useful tools have been generated for P-Rex research, including DNA constructs, purified recombinant proteins, various probes for expression, and knockdown tools. These are summarized in Table 1. P-Rex ABs which have been validated by publication and through the use of specificity controls are listed in Table 2.
Table 1.
P-Rex Research Tools. For each new resource, up-to 3 of the earliest publications are listed, regardless of epitope tags, vector backgrounds etc., therefore the table is not exhaustive. Proteins purified from mammalian cells were not included. P-Rex deficient mouse strains were omitted as they are described in detail in the text. P-Rex ABs and their applications are listed in Table 2
| P-Rex1 | P-Rex2 | P-Rex2b | Applications, Comments | ||
|---|---|---|---|---|---|
| DNA | Full length | 7, 19 | 8 | 9 | |
| Deletions and truncations | 42 | ΔPH, ΔDEP, ΔPDZ, ΔIP4P, iIP4P, iDHPH, iPH | |||
| 19 | ΔDH (tricky DN), ΔPH, ΔDEP, ΔPDZ, ΔIP4P | ||||
| 37 | Sf9 cell-derived iDH | ||||
| 43 | Lots of mutants, incl. iDEP2/PDZ1, ΔC34 | ||||
| 61 | iDEP, iPDZ | ||||
| 40 | iDHPH, iDH, iPH | ||||
| 58 | ΔDHPH | ||||
| 23 | iPH | ||||
| GEF-dead | 42 | 58 | E56A/N238A (dominant negative), N212A (may have residual activity) | ||
| PP1α-binding deficient | 41 | RvXF motif VAFA mutant | |||
| Phosphosite | 41 10, 54 | S1165A and “serine cluster”, S315A, S319A, S605A and S1169A | |||
| Expression | 24 | 25 | Promoter region, miR-338–3p binding | ||
| Melanoma | 86 | K278*, E824* G844D, Q1430*, P948S, G106E | |||
| Recombinant proteins | Full-length | 7, 14 | 8, 11 | Sf9 cell-derived bacterial | |
| Deletion, truncation | 14, 42 | Sf9 cell: ΔPH, ΔDEP, ΔPDZ, ΔIP4P, iIP4P, iDHPH, iPH | |||
| 37 | Sf9 cell-derived iDH | ||||
| 40 | Bacterial: iDHPH, iDH, iPH | ||||
| 41 | Bacterial: aa983–1187 | ||||
| 58 | In vitro translated iDHPH, iDH, iPH | ||||
| 23 | Bacterial: iPH | ||||
| GEF-dead | 14, 42 | Sf9 cell-derived E56A/N238A | |||
| Probes | Northern | 7 | 8 | 9 | Multiple tissues |
| 21 | Neutrophils | ||||
| 12, 19 | Neurons | ||||
| In situ hyb | 12, 19 | 12 | |||
| rtPCR primers | 10 | 58 | |||
| Knockdown tools | siRNA | 19, 51, 74 | in PC12, breast cancer cells and adipocytes, respectively | ||
| shRNA | 60, 34, 18 | in HeLa, PC12 and endothelial cells, respectively | |||
| 10, 51 | 58, 25 | in breast cancer and neuroblastoma cells, respectively | |||
| DNA antisense | 7 | in myeloid cells | |||
| Morpholino | 73 | zebrafish |
Table 2.
P-Rex Antibodies. Included are all antibodies for P-Rex family Rac-GEFs that have been validated through publication and shown to recognize the endogenous protein in at least one application. Studies that used ABs in new cell or tissue types or applications without specificity controls were omitted. “No” means an AB was tested for an application and found not to work. Empty fields mean “not known."
| AB | Source | WB | IP | IF | IHC | Specificity controls | Comments | Original Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| P-Rex1 | rat mAB 3A11 | Hoshino lab | 19 | 19 | WB after siRNA | WB: clean in neurons; | 19 | ||
| no control shown for IF; | |||||||||
| 60 | WB after siRNA | clean in HeLa cells; | 19 | ||||||
| 18 | WB after shRNA | clean in endothelial cells | 19 | ||||||
| rat mAB 4A3 | Hoshino lab | 85 | 85 | P-Rex1 siRNA | Excellent staining | 19 | |||
| mouse mAB 6F12 | Thelen lab | 21 | No | No | No | P-Rex1-/- cells | clean in neutrophils | 21 | |
| 85 | P-Rex1 siRNA | clean in prostate cells | 21 | ||||||
| rabbit pAB | Mitchell lab | 34 | 34 | WB after siRNA peptide- adsorbed for IF | Not very clean for WB in PC12 cells | 34 | |||
| 74 | P-Rex1 siRNA | Not very clean for WB in adipocytes | 34 | ||||||
| rabbit pABs | Pandiella lab | 10 | 10 | Reciprocal IP and WB | 1 good Ab for IP & WB, 1 good for WB | 10 | |||
| Rabbit pAB | Sigma | 38 | 38 | P-Rex1 siRNA | HPA001927, OK in melanoma cells | 38 | |||
| ? | Kazanietz lab | 51 | P-Rex1 siRNA and shRNA | clean in breast cancer cells | 51 | ||||
| ? | Sigma | 51 | P-Rex1 siRNA and shRNA | clean in breast cancer cells | 51 | ||||
| goat pAB | Sta Cruz D-20 | 16 | Rac1-IP | 85805; OK in platelets | 16 | ||||
| goat pAB | Sta Cruz K-19 | 31 | 31 | P-Rex1 shRNA | 85806; good for WB and IF in neutrophils | 31 | |||
| ? | Abcam | 65 | P-Rex1 siRNA | clean in endothelial cells | 65 | ||||
| ? | Sigma | 63 | P-Rex1 siRNA | OK in prostate cancer cells | 63 | ||||
| Phospho-specific rabbit pAbs | Pandiella lab | 10 54 |
10 54 |
Phosphosite mutants | WB ABs for phospho- S313, S319, S605, S1169 | 10 | |||
| P-Rex2 | sheep pAB | Welch lab | 12 | 12 | No | No | P-Rex2-/- tissue | OK for WB, good for IP | 12 |
| rabbit pAB | Welch lab | 12 | P-Rex2-/- tissue | Good for WB | 12 | ||||
| rabbit pAB | Parsons lab | 11 | P-Rex2-/- tissue | OK in MEFs, liver and adipose cells | 11 | ||||
| Mouse pAB | Cell Signaling Technologies | 26 | 26 | P-Rex2 siRNA | WB not very clean in gastric cancer cells. No control shown for IHC | 26 | |||
| P-Rex2b | No isoform specific ABs published yet |
Assays for P-Rex activity
Two types of in vitro assays have been employed for measuring P-Rex GEF activity in vitro, a liposome-based assay that measures the loading of 35S-labeled GTPγS onto the recombinant small G protein and provides excellent signal-to-noise ratio,7,14 and a mant fluorescence based assay that measures the loading of mant-labeled GTP onto the small G protein in real time.35 Similarly, in vivo, 2 types of assays have been used for measuring the effects of P-Rex on cellular Rac activity, the Pak-CRIB pull down assay 95 which is used most often (see for example references 7,20,21) and GLISA.34 The Pak-CRIB assay is more labor-intensive but yields better signal-to-noise ratio. Another assay that has proved very useful for assessing P-Rex dependent activation of Rac in vivo which, although indirect, is very sensitive, is the assessment of cell morphology for Rac activity-sensitive features such as lamellipodia formation, membrane ruffling and spreading by imaging (see for example references 7,19). For measuring the RhoG-GEF activity of P-Rex1 in neutrophils, a GST-ELMO based pull down assay has been successfully employed.35
As mentioned in section “Regulation of Subcellular Localization,” membrane translocation has been used to monitor P-Rex activity. While membrane localization can indeed be a good indicator of activity in many instances, translocation should not be used as a stand-alone readout for P-Rex activity, both because GEF-dead P-Rex1 can translocate to the plasma membrane and because cytosolic P-Rex1 has detectable Rac-GEF activity.7,37 Also, as with many other Rac-GEFs, it can be tricky to assess P-Rex membrane translocation by immuno-fluorescence microscopy, because P-Rex-dependent Rac activity results in lamellipodia formation and membrane ruffling. This induces the cell to have a very different morphology from control cells and can look deceptively like membrane translocation of the Rac-GEF. To avoid confusion of the accumulation of cytosolic P-Rex in lamellipodia and ruffles with membrane translocation, ideally the plasma membrane should be labeled in parallel for comparison, e.g. with lipophilic dye.
As serine phosphorylation events can modulate P-Rex activity and phosphospecific ABs are becoming available10,41,54 it might be tempting to use these ABs to monitor P-Rex activity by Western blot. However, phosphorylation is only one of several mechanisms that regulate P-Rex activity, and too little is currently known to correlate phosphorylation of specific sites with total activity. Finally, with the recent development of a Rac-FRET reporter mouse,96 it should soon be possible to monitor P-Rex Rac-GEF activity in living primary cells, once the reporter mouse strain has been crossed to P-Rex-deficient strains.
Tools for assessing P-Rex function
RNAi has been used successfully by many labs to elucidate P-Rex function in cell lines, for example in the papers listed in Table 1. In addition, 2 different types of dominant-negative P-Rex mutants have been developed: a GEF-dead mutant with 2 point mutations in the DH domain and a ΔDH domain mutant. As always with dominant-negative mutants, careful control of expression levels is important, and they should not be used in isolation to elucidate function. Use of the ΔDH mutant is particularly tricky, as the lack of the entire DH domain is likely to unbalance downstream signaling pathways. For example, it is conceivable that the overexpressed ΔDH mutant could act as a PIP3 sponge and inhibit PI3K responses rather than providing a specific readout of P-Rex function. Finally, as mentioned in section “Physiological Function,” the P-Rex-deficient mouse strains P-Rex1−/−,21,30 P-Rex2−/−11,12 and P-Rex1−/− P-Rex2−/−12 have proven invaluable for evaluating function, as have the related P-Rex1−/− Vav1−/− and P-Rex1−/− Vav3−/− mice,20 a P-Rex2−/− eGFP-Pcp2 strain which expresses GFP in Purkinje neurons,12 and the recently described P-Rex1−/− Tyr-Cre Rac1fl/fl strain with a global P-Rex1 deficiency and additional melanocyte-specific Rac1-deficiency.72 The phenotypes of these P-Rex-related strains were described in detail in sections “Physiological Function,” “Insulin Resistance and Type 2 Diabetes,” and “Cancer.”
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
I gratefully acknowledge Dr Simon Andrews, Babraham Bioinformatics Facility, for his help with evaluating database entries, Dr Sarah Donald, who generated the data shown in Figure 2C, and Dr Phill Hawkins and members of the HW lab for critically reading this manuscript.
Funding
This review was funded by Institute Strategic Programme funding from the Biotechnology and Biological Sciences Research Council to the Babraham Institute Signalling Programme.
References
- 1. Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 2008; 9:690-701; PMID:18719708; http://dx.doi.org/ 10.1038/nrm2476 [DOI] [PubMed] [Google Scholar]
- 2. Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci 2005; 118:843-6; PMID:15731001; http://dx.doi.org/ 10.1242/jcs.01660 [DOI] [PubMed] [Google Scholar]
- 3. Hall A. Rho GTPases and the actin cytoskeleton. Science 1998; 279:509-14; PMID:9438836; http://dx.doi.org/ 10.1126/science.279.5350.509 [DOI] [PubMed] [Google Scholar]
- 4. Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene 2013; 33:4021-35; PMID:24037532; http://dx.doi.org/ 10.1038/onc.2013.362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rossman KL, Der CJ, Sondek J. GEF means go: turning on Rho GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 2005; 6:167-80; PMID:15688002; http://dx.doi.org/ 10.1038/nrm1587 [DOI] [PubMed] [Google Scholar]
- 6. Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer 2010; 10:842-57; PMID:21102635; http://dx.doi.org/ 10.1038/nrc2960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Welch HC, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H, Tempst P, Hawkins PT, Stephens LR. P-Rex1, a PtdIns(3,4,5)P3- and Gbg-regulated guanine-nucleotide exchange factor for Rac. Cell 2002; 108:809-21; PMID:11955434; http://dx.doi.org/ 10.1016/S0092-8674(02)00663-3 [DOI] [PubMed] [Google Scholar]
- 8. Donald S, Hill K, Lecureuil C, Barnouin R, Krugmann S, John Coadwell W, Andrews SR, Walker SA, Hawkins PT, Stephens LR, et al. P-Rex2, a new guanine-nucleotide exchange factor for Rac. FEBS Lett 2004; 572:172-6; PMID:15304343; http://dx.doi.org/ 10.1016/j.febslet.2004.06.096 [DOI] [PubMed] [Google Scholar]
- 9. Rosenfeldt H, Vazquez-Prado J, Gutkind JS. P-Rex2, a novel PI-3-kinase sensitive Rac exchange factor. FEBS letters 2004; 572:167-71; PMID:15304342; http://dx.doi.org/ 10.1016/j.febslet.2004.06.097 [DOI] [PubMed] [Google Scholar]
- 10. Montero JC, Seoane S, Ocana A, Pandiella A. P-Rex1 participates in Neuregulin-ErbB signal transduction and its expression correlates with patient outcome in breast cancer. Oncogene 2011; 30:1059-71; PMID:21042280; http://dx.doi.org/ 10.1038/onc.2010.489 [DOI] [PubMed] [Google Scholar]
- 11. Hodakoski C, Hopkins BD, Barrows D, Mense SM, Keniry M, Anderson KE, Kern PA, Hawkins PT, Stephens LR, Parsons R. Regulation of PTEN inhibition by the pleckstrin homology domain of P-REX2 during insulin signaling and glucose homeostasis. Proc Nat Acad Sci U S A 2014; 111:155-60; PMID:24367090; http://dx.doi.org/ 10.1073/pnas.1213773111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donald S, Humby T, Fyfe I, Segonds-Pichon A, Walker SA, Andrews SR, Coadwell WJ, Emson P, Wilkinson LS, Welch HCE. P-Rex2 regulates Purkinje cell dendrite morphology and motor coordination. Proc Nat Acad Sci U S A 2008; 105:4483-8; PMID:18334636; http://dx.doi.org/ 10.1073/pnas.0712324105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rynkiewicz NK, Liu HJ, Balamatsias D, Mitchell CA. INPP4A/INPP4B and P-Rex proteins: related but different? Adv Biol Regul 2012; 52:265-79; PMID:21925199; http://dx.doi.org/ 10.1016/j.advenzreg.2011.09.001 [DOI] [PubMed] [Google Scholar]
- 14. Hill K, Welch HC. Purification of P-Rex1 from neutrophils and nucleotide exchange assay. Meth Enzymol 2006; 406:26-41; PMID:16472647; http://dx.doi.org/ 10.1016/S0076-6879(06)06003-4 [DOI] [PubMed] [Google Scholar]
- 15. Wang Z, Dong X, Li Z, Smith JD, Wu D. Lack of a significant role of P-Rex1, a major regulator of macrophage Rac1 activation and chemotaxis, in atherogenesis. Prostaglandins Other Lipid Mediat. 2008; 87:9-13; PMID:18502673; http://dx.doi.org/ 10.1016/j.prostaglandins.2008.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aslan JE, Spencer AM, Loren CP, Pang J, Welch HC, Greenberg DL, McCarty OJ. Characterization of the Rac guanine nucleotide exchange factor P-Rex1 in platelets. J Mol Signal 2011; 6:11; PMID:21884615; http://dx.doi.org/ 10.1186/1750-2187-6-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Qian F, Le Breton GC, Chen J, Deng J, Christman JW, Wu D, Ye RD. Role for the guanine nucleotide exchange factor phosphatidylinositol-3,4,5-trisphosphate-dependent rac exchanger 1 in platelet secretion and aggregation. Arterioscler Thromb Vasc Biol 2012; 32:768-77; PMID:22207728; http://dx.doi.org/ 10.1161/ATVBAHA.111.243675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carretero-Ortega J, Walsh CT, Hernández-García R, Reyes-Cruz G, Heller Brown J, Vázquez-Prado J. Phosphatidylinositol 3,4,5-triphosphate-dependent Rac exchanger 1 (P-Rex-1), a guanine nucleotide exchange factor for Rac, mediates angiogenic responses to stromal cell-derived factor-1/chemokine stromal cell derived factor-1 (SDF-1/CXCL-12) linked to Rac activation, endothelial cell migration, and in vitro angiogenesis. Mol Pharmacol 2010; 77:435-42; PMID:20018810; http://dx.doi.org/ 10.1124/mol.109.060400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoshizawa M, Kawauchi T, Sone M, Nishimura YV, Terao M, Chihama K, Nabeshima Y, Hoshino M. Involvement of a Rac activator, P-Rex1, in neurotrophin-derived signaling and neuronal migration. J Neurosci 2005; 25:4406-19; PMID:15858067; http://dx.doi.org/ 10.1523/JNEUROSCI.4955-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lawson C, Donald S, Anderson K, Patton D, Welch H. P-Rex1 and Vav1 cooperate in the regulation of fMLF-dependent neutrophil responses. J Immunol 2011; 186:1467-76; PMID:21178006; http://dx.doi.org/ 10.4049/jimmunol.1002738 [DOI] [PubMed] [Google Scholar]
- 21. Welch HC, Condliffe AM, Milne LJ, Ferguson GJ, Hill K, Webb LM, Okkenhaug K, Coadwell WJ, Andrews SR, Thelen M, et al. P-Rex1 regulates neutrophil function. Curr Biol 2005; 15:1867-73; PMID:16243035; http://dx.doi.org/ 10.1016/j.cub.2005.09.050 [DOI] [PubMed] [Google Scholar]
- 22. Damoulakis G, Gambardella L, Rossman KL, Lawson CD, Anderson KE, Fukui Y, Welch HC, Der CJ, Stephens LR, Hawkins PT. P-Rex1 directly activates RhoG to regulate GPCR-driven Rac signalling and actin polarity in neutrophils. J Cell Sci 2014; 127:2589-600; PMID:24659802; http://dx.doi.org/ 10.1242/jcs.153049 [DOI] [PubMed] [Google Scholar]
- 23. Li Z, Paik JH, Wang Z, Hla T, Wu D. Role of guanine nucleotide exchange factor P-Rex2b in sphingosine 1-phosphate-induced Rac1 activation and cell migration in endothelial cells. Prostaglandins Other Lipid Mediat 2005; 76:95-104; PMID:15967165; http://dx.doi.org/ 10.1016/j.prostaglandins.2005.02.002 [DOI] [PubMed] [Google Scholar]
- 24. Wong CY, Wuriyanghan H, Xie Y, Lin MF, Abel PW, Tu Y. Epigenetic regulation of phosphatidylinositol 3,4,5-triphosphate-dependent Rac exchanger 1 gene expression in prostate cancer cells. J Biol Chem 2011; 286:25813-22; PMID:21636851; http://dx.doi.org/ 10.1074/jbc.M110.211292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen X, Pan M, Han L, Lu H, Hao X, Dong Q. miR-338-3p suppresses neuroblastoma proliferation, invasion and migration through targeting PREX2a. FEBS Lett 2013; 587:3729-37; PMID:24140344; http://dx.doi.org/ 10.1016/j.febslet.2013.09.044 [DOI] [PubMed] [Google Scholar]
- 26. Guo B, Liu L, Yao J, Ma R, Chang D, Li Z, Song T, Huang C. miR-338-3p suppresses gastric cancer progression through a PTEN-AKT axis by targeting P-REX2a. Mol Cancer Res 2014; 12:313-21; PMID:24375644; http://dx.doi.org/ 10.1158/1541-7786.MCR-13-0507 [DOI] [PubMed] [Google Scholar]
- 27. Jaiswal M, Dvorsky R, Ahmadian MR. Deciphering the molecular and functional basis of Dbl family proteins: a novel systematic approach toward classification of selective activation of the Rho family proteins. J Biol Chem 2013; 288:4486-500; PMID:23255595; http://dx.doi.org/ 10.1074/jbc.M112.429746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev 2002; 16:1587-609; PMID:12101119; http://dx.doi.org/ 10.1101/gad.1003302 [DOI] [PubMed] [Google Scholar]
- 29. Gu Y, Filippi MD, Cancelas JA, Siefring JE, Williams EP, Jasti AC, Harris CE, Lee AW, Prabhakar R, Atkinson SJ, et al. Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science 2003; 302:445-9; PMID:14564009; http://dx.doi.org/ 10.1126/science.1088485 [DOI] [PubMed] [Google Scholar]
- 30. Dong X, Mo Z, Bokoch G, Guo C, Li Z, Wu D. P-Rex1 is a primary Rac2 guanine nucleotide exchange factor in mouse neutrophils. Curr Biol 2005; 15:1874-9; PMID:16243036; http://dx.doi.org/ 10.1016/j.cub.2005.09.014 [DOI] [PubMed] [Google Scholar]
- 31. Herter JM, Rossaint J, Block H, Welch H, Zarbock A. Integrin activation by P-Rex1 is required for selectin-mediated slow leukocyte rolling and intravascular crawling. Blood 2013; 121:2301-10; PMID:23343834; http://dx.doi.org/ 10.1182/blood-2012-09-457085 [DOI] [PubMed] [Google Scholar]
- 32. Condliffe AM, Webb LMC, Ferguson GJ, Davidson K, Turner M, Vigorito E, Manifava M, Chilvers ER, Stephens LR, Hawkins PT. RhoG regulates the neutrophil NADPH oxidase. J Immunol 2006; 176:5314-20; PMID:16621998; http://dx.doi.org/ 10.4049/jimmunol.176.9.5314 [DOI] [PubMed] [Google Scholar]
- 33. de Curtis I. Functions of Rac GTPases during neuronal development. Dev Neurosci 2008; 30:47-58; PMID:18075254; http://dx.doi.org/ 10.1159/000109851 [DOI] [PubMed] [Google Scholar]
- 34. Waters JE, Astle MV, Ooms LM, Balamatsias D, Gurung R, Mitchell CA. P-Rex1 – a multidomain protein that regulates neurite differentiation. J Cell Sci 2008; 121:2892-903; PMID:18697831; http://dx.doi.org/ 10.1242/jcs.030353 [DOI] [PubMed] [Google Scholar]
- 35. Damulakis G. P-Rex1 directly activates RhoG to regulate GPCR-driven Rac signalling and actin polarity in neutrophils. J Cell Sci 2014; 127:2589-600; PMID:24659802; http://dx.doi.org/ 10.1242/jcs.153049; In Press; [DOI] [PubMed] [Google Scholar]
- 36. Naikawadi RP, Cheng N, Vogel SM, Qian F, Wu D, Malik AB, Ye RD. A critical role for phosphatidylinositol (3,4,5)-trisphosphate-dependent Rac exchanger 1 in endothelial junction disruption and vascular hyperpermeability. Circ Res 2012; 111:1517-27; PMID:22965143; http://dx.doi.org/ 10.1161/CIRCR-ESAHA.112.273078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barber MA, Donald S, Thelen S, Anderson KE, Thelen M, Welch HCE. Membrane translocation of P-Rex1 is mediated by G protein bg subunits and phosphoinositide 3-kinase. J Biol Chem 2007; 282:29967-76; PMID:17698854; http://dx.doi.org/ 10.1074/jbc.M701877200 [DOI] [PubMed] [Google Scholar]
- 38. Campbell AD, Lawn S, McGarry LC, Welch HC, Ozanne BW, Norman JC. P-Rex1 cooperates with PDGFRbeta to drive cellular migration in 3D microenvironments. PloS One 2013; 8:e53982; PMID:23382862; http://dx.doi.org/ 10.1371/journal.pone.0053982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Welch H, Eguinoa A, Stephens LR, Hawkins PT. Protein kinase B and Rac are activated in parallel within a phosphatidylinositide 3OH-kinase-controlled signaling pathway. J Biol Chem 1998; 273:11248-56; PMID:9556616; http://dx.doi.org/ 10.1074/jbc.273.18.11248 [DOI] [PubMed] [Google Scholar]
- 40. Joseph RE, Norris FA. Substrate specificity and recognition is conferred by the pleckstrin homology domain of the Dbl family guanine nucleotide exchange factor P-Rex2. J Biol Chem 2005; 280:27508-12; PMID:15897194; http://dx.doi.org/ 10.1074/jbc.M412495200 [DOI] [PubMed] [Google Scholar]
- 41. Barber MA, Hendrickx A, Beullens M, Ceulemans H, Oxley D, Thelen S, Thelen M, Bollen M, Welch HC. The guanine-nucleotide-exchange factor P-Rex1 is activated by protein phosphatase 1alpha. Biochem J 2012; 443:173-83; PMID:22242915; http://dx.doi.org/ 10.1042/BJ20112078 [DOI] [PubMed] [Google Scholar]
- 42. Hill K, Krugmann S, Andrews SR, Coadwell WJ, Finan P, Welch HC, Hawkins PT, Stephens LR. Regulation of P-Rex1 by phosphatidylinositol (3,4,5)-trisphosphate and Gbg subunits. J Biol Chem 2005; 280:4166-73; PMID:15545267; http://dx.doi.org/ 10.1074/jbc.M411262200 [DOI] [PubMed] [Google Scholar]
- 43. Urano D, Nakata A, Mizuno N, Tago K, Itoh H. Domain-domain interaction of P-Rex1 is essential for the activation and inhibition by G protein bg subunits and PKA. Cell Signal 2008; 20:1545-54; PMID:18514484; http://dx.doi.org/ 10.1016/j.cellsig.2008.04.009 [DOI] [PubMed] [Google Scholar]
- 44. Shinohara M, Terada Y, Iwamatsu A, Shinohara A, Mochizuki N, Higuchi M, Gotoh Y, Ihara S, Nagata S, Itoh H, et al. SWAP-70 is a guanine-nucleotide-exchange factor that mediates signalling of membrane ruffling. Nature 2002; 416:759-63; PMID:11961559; http://dx.doi.org/ 10.1038/416759a [DOI] [PubMed] [Google Scholar]
- 45. Nishida K, Kaziro Y, Satoh T. Association of the proto-oncogene product dbl with G protein betagamma subunits. FEBS Lett 1999; 459:186-90; PMID:10518015; http://dx.doi.org/ 10.1016/S0014-5793(99)01244-2 [DOI] [PubMed] [Google Scholar]
- 46. Runne C, Chen S. PLEKHG2 promotes heterotrimeric G protein betagamma-stimulated lymphocyte migration via Rac and Cdc42 activation and actin polymerization. Mol Cell Biol 2013; 33:4294-307; PMID:24001768; http://dx.doi.org/ 10.1128/MCB.00879-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fritsch R, de Krijger I, Fritsch K, George R, Reason B, Kumar MS, Diefenbacher M, Stamp G, Downward J. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 2013; 153:1050-63; PMID:23706742; http://dx.doi.org/ 10.1016/j.cell.2013.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yan J, Mihaylov V, Xu X, Brzostowski JA, Li H, Liu L, Veenstra TD, Parent CA, Jin T. A Gbetagamma effector, ElmoE, transduces GPCR signaling to the actin network during chemotaxis. Dev Cell 2012; 22:92-103; PMID:22264729; http://dx.doi.org/ 10.1016/j.devcel.2011.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mayeenuddin LH, McIntire WE, Garrison JC. Differential sensitivity of P-Rex1 to isoforms of G protein bg dimers. J Biol Chem 2006; 281:1913-20; PMID:16301321; http://dx.doi.org/ 10.1074/jbc.M506034200 [DOI] [PubMed] [Google Scholar]
- 50. Stephens L, Hawkins P. Signalling via class IA PI3Ks. Adv Enzyme Regul 2011; 51:27-36; PMID:21035483; http://dx.doi.org/ 10.1016/j.advenzreg.2010.09.007 [DOI] [PubMed] [Google Scholar]
- 51. Sosa MS, Lopez-Haber C, Yang C, Wang H, Lemmon MA, Busillo JM, Luo J, Benovic JL, Klein-Szanto A, Yagi H, et al. Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol Cell 2010; 40:877-92; PMID:21172654; http://dx.doi.org/ 10.1016/j.molcel.2010.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hendrickx A, Beullens M, Ceulemans H, Den Abt T, Van Eynde A, Nicolaescu E, Lesage B, Bollen M. Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem Biol 2009; 16:365-71; PMID:19389623; http://dx.doi.org/ 10.1016/j.chembiol.2009.02.012 [DOI] [PubMed] [Google Scholar]
- 53. Mayeenuddin LH, Garrison JC. Phosphorylation of P-Rex1 by the cyclic AMP-dependent protein kinase inhibited the phosphatidylinositiol (3,4,5)-trisphosphate and Gbetagamma-mediated regulation of its activity. J Biol Chem 2006; 281:1921-8; PMID:16301320; http://dx.doi.org/ 10.1074/jbc.M506035200 [DOI] [PubMed] [Google Scholar]
- 54. Montero JC, Seoane S, Pandiella A. Phosphorylation of P-Rex1 at serine 1169 participates in IGF-1R signaling in breast cancer cells. Cell Signal 2013; 25:2281-9; PMID:23899556; http://dx.doi.org/ 10.1016/j.cellsig.2013.07.018 [DOI] [PubMed] [Google Scholar]
- 55. Nie B, Cheng N, Dinauer MC, Ye RD. Characterization of P-Rex1 for its role in fMet-Leu-Phe-induced superoxide production in reconstituted COSphox cells. Cell Signal 2010; 22:770-82; PMID:20074642; http://dx.doi.org/ 10.1016/j.cellsig.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim EK, Yun SJ, Ha JM, Kim YW, Jin IH, Yun J, Shin HK, Song SH, Kim JH, Lee JS, et al. Selective activation of Akt1 by mammalian target of rapamycin complex 2 regulates cancer cell migration, invasion, and metastasis. Oncogene 2011; 30:2954-63; PMID:21339740; http://dx.doi.org/ 10.1038/onc.2011.22 [DOI] [PubMed] [Google Scholar]
- 57. Zhao T, Nalbant P, Hoshino M, Dong X, Wu D, Bokoch GM. Signaling requirements for translocation of P-Rex1, a key Rac2 exchange factor involved in chemoattractant-stimulated human neutrophil function. J Leukoc Biol 2007; 81:1127-36; PMID:17227822 [DOI] [PubMed] [Google Scholar]
- 58. Fine B, Hodakoski C, Koujak S, Su T, Saal LH, Maurer M, Hopkins B, Keniry M, Sulis ML, Mense S, et al. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-Rex2a. Science 2009; 325:1261-5; PMID:19729658; http://dx.doi.org/ 10.1126/science.1173569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang SC, Gremer L, Heise H, Janning P, Shymanets A, Cirstea IC, Krause E, Nurnberg B, Ahmadian MR. Liposome reconstitution and modulation of recombinant prenylated human Rac1 by GEFs, GDI1 and Pak1. PloS One 2014; 9:e102425; PMID:25014207; http://dx.doi.org/ 10.1371/journal.pone.0102425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hernandez-Negrete I, Carretero-Ortega J, Rosenfeldt H, Hernandez-Garcia R, Calderon-Salinas JV, Reyes-Cruz G, Gutkind JS, Vazquez-Prado J. P-Rex1 links mammalian target of rapamycin signaling to Rac activation and cell migration. J Biol Chem 2007; 282:23708-15; PMID:17565979; http://dx.doi.org/ 10.1074/jbc.M703771200 [DOI] [PubMed] [Google Scholar]
- 61. Ledezma-Sánchez BA, García-Regalado A, Guzmán-Hernández ML, Vázquez-Prado J. Sphingosine-1-phosphate receptor S1P1 is regulated by direct interactions with P-Rex1, a Rac guanine nucleotide exchange factor. Biochem Biophys Res Commun 2010; 391:1647-52; PMID:20036214; http://dx.doi.org/ 10.1016/j.bbrc.2009.12.108 [DOI] [PubMed] [Google Scholar]
- 62. Dimidschstein J, Passante L, Dufour A, van den Ameele J, Tiberi L, Hrechdakian T, Adams R, Klein R, Lie DC, Jossin Y, et al. Ephrin-B1 controls the columnar distribution of cortical pyramidal neurons by restricting their tangential migration. Neuron 2013; 79:1123-35; PMID:24050402; http://dx.doi.org/ 10.1016/j.neuron.2013.07.015 [DOI] [PubMed] [Google Scholar]
- 63. Ghalali A, Wiklund F, Zheng H, Stenius U, Hogberg J. Atorvastatin prevents ATP-driven invasiveness via P2´7 and EHBP1 signaling in PTEN-expressing prostate cancer cells. Carcinogenesis 2014; 35:1547-55; PMID:24451147; http://dx.doi.org/ 10.1093/carcin/bgu019 [DOI] [PubMed] [Google Scholar]
- 64. Guilherme A, Soriano NA, Bose S, Holik J, Bose A, Pomerleau DP, Furcinitti P, Leszyk J, Corvera S, Czech MP. EHD2 and the novel EH domain binding protein EHBP1 couple endocytosis to the actin cytoskeleton. J Biol Chem 2004; 279:10593-605; PMID:14676205; http://dx.doi.org/ 10.1074/jbc.M307702200 [DOI] [PubMed] [Google Scholar]
- 65. van Hooren KW, van Breevoort D, Fernandez-Borja M, Meijer AB, Eikenboom J, Bierings R, Voorberg J. Phosphatidylinositol-3,4,5-triphosphate-dependent Rac exchange factor 1 regulates epinephrine-induced exocytosis of Weibel-Palade bodies. J Thromb Haemostasis: JTH 2014; 12:273-81; PMID:24283667; http://dx.doi.org/ 10.1111/jth.12460 [DOI] [PubMed] [Google Scholar]
- 66. Lindsay CR, Lawn S, Campbell AD, Faller WJ, Rambow F, Mort RL, Timpson P, Li A, Cammareri P, Ridgway RA, et al. P-Rex1 is required for efficient melanoblast migration and melanoma metastasis. Nat Commun 2011; 2:555; PMID:22109529; http://dx.doi.org/ 10.1038/ncomms1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Quevedo C, Sauzeau V, Menacho-Marquez M, Castro-Castro A, Bustelo XR. Vav3-deficient mice exhibit a transient delay in cerebellar development. Mol Biol Cell 2010; 21:1125-39; PMID:20089829; http://dx.doi.org/ 10.1091/mbc.E09-04-0292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jackson C, Welch HC, Bellamy TC. Control of cerebellar long-term potentiation by P-Rex-family guanine-nucleotide exchange factors and phosphoinositide 3-kinase. PloS one 2010; 5:e11962; PMID:20694145; http://dx.doi.org/ 10.1371/journal.pone.0011962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sherry DM, Blackburn BA. P-Rex2, a Rac-guanine nucleotide exchange factor, is expressed selectively in ribbon synaptic terminals of the mouse retina. BMC Neurosci 2013; 14:70; PMID:23844743; http://dx.doi.org/ 10.1186/1471-2202-14-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Miller MB, Yan Y, Eipper BA, Mains RE. Neuronal Rho GEFs in synaptic physiology and behavior. Neuroscientist 2013; 19:255-73; PMID:23401188; http://dx.doi.org/ 10.1177/1073858413475486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li A, Ma Y, Yu X, Mort RL, Lindsay CR, Stevenson D, Strathdee D, Insall RH, Chernoff J, Snapper SB, et al. Rac1 drives melanoblast organization during mouse development by orchestrating pseudopod- driven motility and cell-cycle progression. Dev Cell 2011; 21:722-34; PMID:21924960; http://dx.doi.org/ 10.1016/j.devcel.2011.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lindsay CR, Li A, Faller W, Ozanne B, Welch H, Machesky LM, Sansom OJ. A Rac1-independent role for P-Rex1 in melanoblasts. J Invest Dermatol. 2015 Jan; 135(1):314-8. doi: 10.1038/jid.2014.323. Epub 2014 Jul 30. PMID: 25075639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Woo S, Housley MP, Weiner OD, Stainier DY. Nodal signaling regulates endodermal cell motility and actin dynamics via Rac1 and Prex1. J Cell Biol 2012; 198:941-52; PMID:22945937; http://dx.doi.org/ 10.1083/jcb.201203012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Balamatsias D, Kong AM, Waters JE, Sriratana A, Gurung R, Bailey CG, Rasko JE, Tiganis T, Macaulay SL, Mitchell CA. Identification of P-Rex1 as a novel Rac1-guanine nucleotide exchange factor (GEF) that promotes actin remodeling and GLUT4 protein trafficking in adipocytes. J Biol Chem 2011; 286:43229-40; PMID:22002247; http://dx.doi.org/ 10.1074/jbc.M111.306621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kim EK, Yun SJ, Ha JM, Kim YW, Jin IH, Woo DH, Lee HS, Ha HK, Bae SS. Synergistic induction of cancer cell migration regulated by Gbetagamma and phosphatidylinositol 3-kinase. Exp Mol Med 2012; 44:483-91; PMID:22627809; http://dx.doi.org/ 10.3858/emm.2012.44.8.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bento JL, Palmer ND, Zhong M, Roh B, Lewis JP, Wing MR, Pandya H, Freedman BI, Langefeld CD, Rich SS, et al. Heterogeneity in gene loci associated with type 2 diabetes on human chromosome 20q13.1. Genomics 2008; 92:226-34; PMID:18602983; http://dx.doi.org/ 10.1016/j.ygeno.2008.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lewis JP, Palmer ND, Ellington JB, Divers J, Ng MC, Lu L, Langefeld CD, Freedman BI, Bowden DW. Analysis of candidate genes on chromosome 20q12-13.1 reveals evidence for BMI mediated association of PREX1 with type 2 diabetes in European Americans. Genomics 2010; 96:211-9; PMID:20650312; http://dx.doi.org/ 10.1016/j.ygeno.2010.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Barrio-Real L, Kazanietz MG. Rho GEFs and cancer: linking gene expression and metastatic dissemination. Sci Signal 2012; 5:pe43; PMID:23033535; http://dx.doi.org/ 10.1126/scisignal.2003543 [DOI] [PubMed] [Google Scholar]
- 79. Pandiella A, Montero JC. Molecular pathways: P-Rex in cancer. Clin Cancer Res: Off J Am Assoc Cancer Res 2013; 19:4564-9; PMID:23753921; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-1662 [DOI] [PubMed] [Google Scholar]
- 80. McCarthy N. Signalling: REX rules. Nat Rev Cancer 2011; 11:83; PMID:21322294; http://dx.doi.org/ 10.1038/nrc3013 [DOI] [PubMed] [Google Scholar]
- 81. Wertheimer E, Gutierrez-Uzquiza A, Rosemblit C, Lopez-Haber C, Sosa MS, Kazanietz MG. Rac signaling in breast cancer: a tale of GEFs and GAPs. Cell Signalling 2012; 24:353-62; PMID:21893191; http://dx.doi.org/ 10.1016/j.cellsig.2011.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mense S, Hodakoski C, Parsons R. PREX2, a new breed of cancer gene with too many spots? Pigment Cell Melanoma Res 2012; 25:409-10; PMID:22578146; http://dx.doi.org/ 10.1111/j.1755-148X.2012.01006.x [DOI] [PubMed] [Google Scholar]
- 83. Walker G. P-REX1, a Rac guanine exchange factor, links melanocyte development and melanoma progression. Pigment Cell Melanoma Res 2011; 24:1086-7; PMID:22216451; http://dx.doi.org/ 10.1111/j.1755-148X.2011.00934.x [DOI] [PubMed] [Google Scholar]
- 84. Wangari-Talbot J, Chen S. Genetics of melanoma. Front Genetics 2012; 3:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Qin J, Xie Y, Wang B, Hoshino M, Wolff DW, Zhao J, Scofield MA, Dowd FJ, Lin MF, Tu Y. Upregulation of PIP3-dependent Rac exchanger 1 (P-Rex1) promotes prostate cancer metastasis. Oncogene 2009; 28:1853-63; PMID:19305425; http://dx.doi.org/ 10.1038/onc.2009.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012; 485:502-6; PMID:22622578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Suzuki A, Mimaki S, Yamane Y, Kawase A, Matsushima K, Suzuki M, Goto K, Sugano S, Esumi H, Suzuki Y, et al. Identification and characterization of cancer mutations in Japanese lung adenocarcinoma without sequencing of normal tissue counterparts. PloS One 2013; 8:e73484; PMID:24069199; http://dx.doi.org/ 10.1371/journal.pone.0073484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ebi H, Costa C, Faber AC, Nishtala M, Kotani H, Juric D, Della Pelle P, Song Y, Yano S, Mino-Kenudson M, et al. PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. Proc Nat Acad Sci U S A 2013; 110:21124-9; PMID:24327733; http://dx.doi.org/ 10.1073/pnas.1314124110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Turajlic S, Furney SJ, Lambros MB, Mitsopoulos C, Kozarewa I, Geyer FC, Mackay A, Hakas J, Zvelebil M, Lord CJ, et al. Whole genome sequencing of matched primary and metastatic acral melanomas. Genome Res 2012; 22:196-207; PMID:22183965; http://dx.doi.org/ 10.1101/gr.125591.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell 2012; 150:251-63; PMID:22817889; http://dx.doi.org/ 10.1016/j.cell.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012; 44:1006-14; PMID:22842228; http://dx.doi.org/ 10.1038/ng.2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003; 3:11-22; PMID:12509763; http://dx.doi.org/ 10.1038/nrc969 [DOI] [PubMed] [Google Scholar]
- 93. Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Nat Acad Sci U S A 2004; 101:7618-23; PMID:15128949; http://dx.doi.org/ 10.1073/pnas.0307512101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Symons M, Rusk N. Control of vesicular trafficking by Rho GTPases. Curr Biol: CB 2003; 13:R409-18; PMID:12747855; http://dx.doi.org/ 10.1016/S0960-9822(03)00324-5 [DOI] [PubMed] [Google Scholar]
- 95. Sander EE, van Delft S, ten Klooster JP, Reid T, van der Kammen RA, Michiels F, Collard JG. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by Phosphatidylinositol 3-Kinase. J Cell Biol 1998; 143:1385-98; PMID:9832565; http://dx.doi.org/ 10.1083/jcb.143.5.1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Johnsson AK, Dai Y, Nobis M, Baker MJ, McGhee EJ, Walker S, Schwarz JP, Kadir S, Morton JP, Myant KB, et al. The Rac-FRET mouse reveals tight spatiotemporal control of Rac activity in primary cells and tissues. Cell Rep 2014; 6:1153-64; PMID:24630994; http://dx.doi.org/ 10.1016/j.celrep.2014.02.024] [DOI] [PMC free article] [PubMed] [Google Scholar]