

Abstract
The extraordinary developments made in the past decade in proteomic technologies, in particular in mass spectrometry, have enabled investigators to consider designing studies to search for diagnostic and therapeutic biomarkers by scanning complex proteome samples. We developed a method based on extensive fractionation of intact proteins, to comprehensively and quantitatively profile the liver and plasma proteomes in health and disease. We have applied this method to samples collected from patients with early hepatocellular carcinoma (HCC) and from patients with liver cirrhosis as well as to samples collected from three mouse models of HCC. This method allowed for the identification of proteins that differ in expression levels in liver tissue or in plasma with disease progression from liver fibrosis, cirrhosis or steatohepatitis to HCC. The comparative analysis of the liver and plasma proteomes generated from human and mouse specimen, constitutes a novel and powerful strategy for HCC biomarker discovery.
Keywords: Liver cancer, Hepatocellular carcinoma, biomarker, proteomics, mouse model
Introduction to Clinical Proteomics
The significant escalation of interest in the application of proteomics, the study of proteins and their functions, to the study of human diseases is reflected by the explosive growth of a new field named Clinical Proteomics [1]. Clinical proteomics aims at providing clinicians with tools to accurately diagnose and efficiently treat patients in an individualized manner. Such tools could translate into the use of blood biomarkers that may influence daily clinical practice by driving clinical decision-making. Proteomics has been largely used to date for the discovery of new serological biomarkers for early diagnosis of chronic diseases such as cancer and cardiovascular diseases [2-6]. Early diagnosis of cancer improves outcome and survival. It is however particularly difficult to diagnose the early stages of cancer because of the lack of specific symptoms in early disease and the limited understanding of the disease heterogeneity and etiology.
The recent development of mass spectrometry (MS) technology undoubtedly contributed to the explosion of clinical proteomics. MS has improved greatly in sensitivity and accuracy and MS-based proteomics has become the method of choice for the analysis of complex protein samples. Within a decade, the field evolved from having great difficulty in identifying a single protein to being able to assign peaks with high confidence to a large number (sometimes hundreds) of proteins within a single MS run. The inherent analytical advantages of MS, including sensitivity (with routine detection limits currently at the femtomole (10-15) level), resolution, speed and throughput combined with advanced bioinformatics for data interpretation, allow for the rapid and systematic analysis of thousands of proteins. Beyond identification, quantitation is also an important parameter in clinical proteomics, needed to compare two or more samples for discriminating features. Quantitation can be performed by directly looking at peptide counts [7-10]. Therefore, the application of MS to the analysis of tissue and plasma/serum specimens from different stages of disease or different diseases has the potential to provide unique information about disease-associated alterations at the protein level, both qualitatively and quantitatively.
The Challenges of Disease Biomarker Discovery
The process for MS-based biomarker or drug target discovery follows a long and complicated pipeline from collection of clinical samples and associated clinical data, processing and handling of specimens, sample selection for proteomics analysis, fractionation and sample preparation for MS, MS analysis, data interpretation and statistical analysis. It is vital to define precisely a well-framed, relevant clinical problem and focus the experimental design around appropriate study populations and samples. Translation from the laboratory to the clinic is a long process and it will take time to move a new target/biomarker from discovery to regulatory approval. Unreasonable expectations for the possible outcomes of proteomics-based clinical studies are detrimental to the field.
To move the field forward, fundamental biological questions remain to be addressed and experimental designs need to be improved. Most study designs so far have been based on intuitive concepts and assumptions. The initial goal of most clinical proteomics studies is to identify proteins that are differentially abundant between biological samples obtained from healthy and disease-afflicted patients. These experimental designs are based on the assumption that a subset of proteins present in the blood reflects, reproducibly and specifically, a single disease at a particular stage. They are also based on the assumption that we can and know how to identify disease-specific signals over noise in the blood. There is currently no information on the biological variations of the blood proteome (protein changes occurring in healthy individuals of different sex, race, age or with different diet or activity habits). Unless we have knowledge of the composition and dynamic of the blood proteome in health, critical elements of how to design biomarker studies will evade us. A big debate in the proteomics community is which type of sample – blood or diseased tissue – should be used to discover serologic biomarkers. Proteomic studies using blood as a source of proteins still have not resolved the question of whether plasma or serum can serve as a window into the state of a patient's disease and there is currently no scientific evidence that a linear relationship exists between protein changes in diseased tissue and protein changes in blood. In addition, the “one size fits all” rule seems to have been applied to studies targeting different organs and different diseases. A particularly difficult question is how to design a study aimed at discovering biomarkers for early detection of small, localized, pre-invasive cancer lesions which cannot be currently diagnosed with the available tools. Most studies so far have used samples (tissue or plasma/serum) from patients with far more advanced disease, with the hope that some of the identified markers will have utility in early diagnosis. This is based on the oversimplified assumption that the molecular composition of very early tumors and more advanced tumors are very similar.
Novel Strategies for Biomarker Discovery for Hepatocellular Carcinoma
We are currently in a position to make a critical assessment of the lessons learned in order to define future strategies. Efforts to integrate proteomics and basic cellular biology or other growing fields such as imaging and/or mouse models of diseases might greatly accelerate the translation of basic discoveries into daily clinical practice. The ability to understand the underlying molecular dysfunction in human disease (signaling pathways, protein-protein interaction networks) should facilitate the development of diagnostic biomarkers, predictors of disease progression or new therapeutic targets as well as individualized patient management [11,12]. Mouse models of disease can often closely mimic human diseases. The use of relevant animal models in clinical proteomics has the potential to accelerate discovery, as well as the initial validation processes, in part by reducing heterogeneity and sample preparation variability. The past few years have also witnessed increased availability of non-invasive small animal imaging technologies allowing signal detection from deep tissues [13,14]. In general, imaging technologies have undergone explosive growth with increased spatial resolution and already play a central role in clinical oncology [15]. The identification of biomarkers that might be combined with diagnostic imaging techniques will improve sensitivity for detecting disease and have the unique potential to provide a whole-body assessment.
Several proteomics studies of HCC have been reported (reviewed in [16,17]). Most proteomic analyses of HCC tissues, including previous studies by our group, have been performed using 2-D PAGE followed by MS. This method has led to the identification of a large number of proteins modified in HCC compared to adjacent non-tumor tissues. In our analysis, only 30% of the protein changes correlated with changes at the corresponding transcript level, suggesting that post-transcriptional and post-translational alterations account for most protein modifications in HCC [18]. Notably, 33% of the protein spots we identified as modified in HCC corresponded to proteolytic cleavages. Interestingly, some of the proteolytic fragments generated were released in the extracellular compartment and detected in the serum of patients with HCC. For example, fragments of calreticulin and PDIA3 were detected at significantly higher levels in serum of patients with HCC compared to serum from healthy individuals, from patients with chronic hepatitis and patients with liver cirrhosis [18]. We also reported that in the serum of patients with HCC, circulating antibodies against calreticulin can be detected [19]. We can hypothesize that the presence of these autoantibodies is a consequence of the cleavage of calreticulin in HCC. These data are particularly interesting in the light of the recent study published in Nature Medicine, demonstrating that calreticulin cell surface expression dictates the immunogenicity of cancer cell death [20].
Despite increasing sensitivity of the mass spectrometers, protein profiling has not to date reached sufficient depth to allow for the identification of a majority of the low-abundance proteins expressed in a mammalian tissue or biological fluid. The complexity of the peptide mixtures often overwhelms the mass spectrometer and low-abundance peptides can be masked when mixed with peptides of high abundance. Detecting low-abundance proteins and reaching the proteins of interest can be successfully achieved by enhancing the pre-analytical step using multidimensional fractionation strategies or by enrichment of a subproteome (e.g., the glycoproteome) [21-25]. We have recently developed a pipeline based on the use of multidimensional separation of intact proteins with particular attention to the isolation of low-abundance proteins and exhaustive LC-MS/MS analysis of each generated fraction and applied it to the study of liver and plasma proteomes (Fig. 1). The vast dynamic range of protein abundance in serum or plasma, present a formidable challenge for comprehensive protein analysis. High-abundance proteins such as albumin, immunoglobulins, antitrypsin and haptoglobin for example, interfere with the proteomic analysis of low-abundance proteins. Therefore, it is advantageous to specifically remove high-abundance proteins in a sample pre-fractionation step prior to protein analysis. We applied a 3-D separation of intact proteins and selected SDS-PAGE as the last separation step. SDS-PAGE is well-suited for separation of proteins according to their size and protein abundance can be estimated by gel staining. A nano-UPLC system coupled to a tandem mass spectrometer (LTQ-FT or LTQ-Orbitrap) is utilized for peptide mixture analysis. The X!Tandem search engine and the International Protein Index (IPI) database are used for protein identifications and PeptideProphet as well as ProteinProphet scores of ≥ 0.9 are used to filter high-confidence peptide and protein identifications (Fig. 2). We demonstrated that by extensively reducing the complexity of peptide mixtures and by carefully separating proteins of different abundances and sizes, in-depth proteomic profiling can be achieved [26]. In addition, estimates of absolute abundance can be obtained by counting the number of observed peptides. The calculated abundance scores strongly correlated with the abundance of the corresponding transcripts for over 80% of the identified proteins (L. Beretta, personal communication). We further validated this method by demonstrating a strong correlation (R=0.82) obtained between concentration reported in the literature and abundance scores calculated by this method for 50 proteins selected over the complete abundance range [26].
Fig. 1.
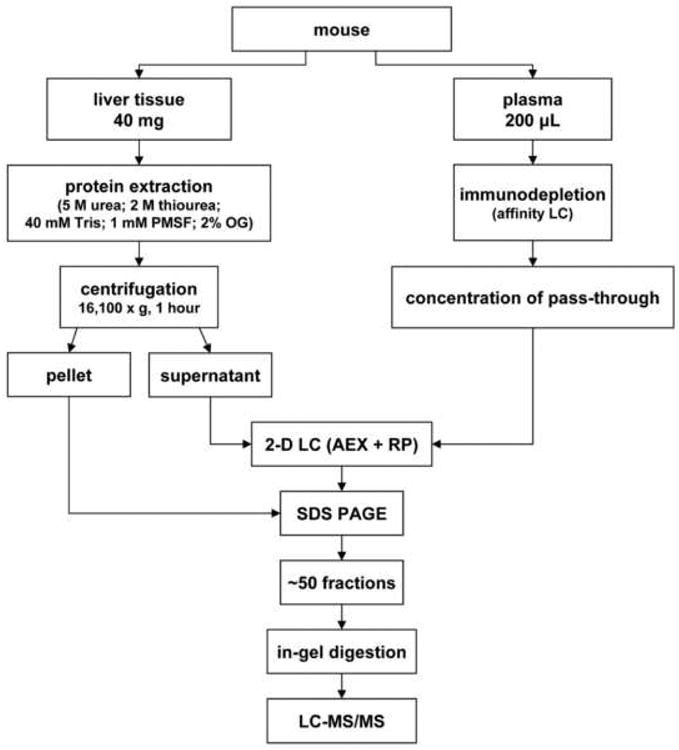
Schematic view of the protein extraction and 3-D separation process. Online 2-D HPLC separation consists of 8 stepwise gradients in the 1st dimension AXC liquid chromatography separation, each of them further separated in the 2nd dimension by RP liquid chromatography separation. Proteins are further separated by SDS-PAGE. The fractionation process generates approximately 50 samples to be analyzed in duplicate by LC-MS/MS.
Fig. 2.
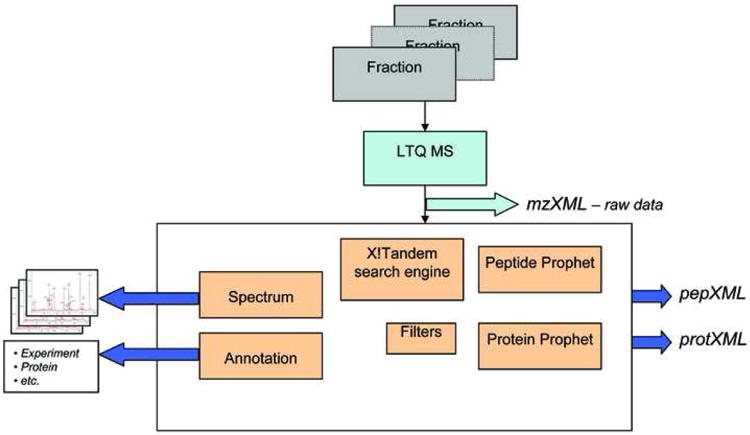
Schematic view of the protein identification process. Approximately 50 fractions generated from 1 sample are analyzed by LC-MS/MS using the highly sensitive LTQ-FT or LTQ-Orbitrap mass spectrometer. The data are searched against the non-redundant human International Protein Index (IPI) protein sequence database with the X!Tandem Search engine. ProteinProphet and PeptideProphet statistical tools are used to filter peptide and proteins identified with less than 1% error rate.
Proteome Profiles Generated in our Studies and their Comparative Analysis
Mouse models of HCC
We are studying in our group three mouse models of HCC: the PTEN knock-out mice, the MAT1A knock-out mice and the PDGF-C transgenic mice. These three mouse models are particularly interesting as PTEN, MAT1A and PDGF-C expression have been show to be frequently impaired in human HCC and because they mimic the disease progression from liver fibrosis and steatohepatitis to HCC as observed in patients. PTEN is a tumor suppressor and a regulator of insulin sensitivity in peripheral tissues. In the mouse liver, PTEN deletion results in massive hepatomegaly and steatohepatitis followed by liver fibrosis and HCC, a phenotype similar to human NASH [27-30]. MAT1A encodes for the essential enzyme methionine adenosyltransferases, which catalyzes the biosynthesis of S-adenosylmethionine (SAMe), the principal methyl donor and, in the liver, a precursor of glutathione. Mice lacking MAT1A spontaneously develop steatohepatitis and HCC [31]. Members of the platelet-derived growth factor (PDGF) ligand family are known to play important roles in wound healing and fibrotic disease. Transgenic expression of PDGF-C in the liver results in the development of liver fibrosis and HCC [32]. We performed proteomic profiling analysis on the liver tissue and the corresponding plasma obtained at different stages of disease on these three mouse models.
Clinical samples
We also performed proteomic profiling analysis on plasma collected from patients with liver cirrhosis without HCC and from patients with early HCC, who had liver transplantation, separately for different etiologies (HCV, HBV, NASH). The rationale to select these groups of patients is the following: 1- the decision to analyze separately HCCs of different etiologies is to reduce the issue of heterogeneity. Indeed, in prior studies, we observed significant differences in the genomic profiles of HCC of different etiologies; 2-because over 90% of the HCC develop on cirrhosis, the control group in this study is cirrhosis; 3- because early HCC can be undetected in patients with liver cirrhosis, we selected patients who had liver transplantation. The histological analysis of the explanted liver ascertains that subjects have not yet developed very early tumors; 4- the goal of this study being to identify biomarkers for early detection, we selected patients with well-differentiated tumor, 1 or 2 tumor nodules of less than 2.5 cm with no vascular invasion and no metastasis confirmed on explant examination. The majority of the specimens have been obtained from patients enrolled by Dr. Jorge Marrero, University of Michigan Medical School and by Dr. Hugo Rosen, University of Colorado Health Sciences Center. Drs. Marrero and Rosen recognize the need for high-quality samples for a biomarker study and have collected a high number of well-characterized samples associated with detailed clinical information.
Comparative analysis
Mining and interpreting each proteome profile obtained either from the liver or from plasma, from human or from mice, is a daunting task. We therefore decided to apply a novel strategy: the integration and comparative analysis of all data sets (Fig. 3). Integration of genomic profiling data between human HCC samples and mouse models of HCC has been previously performed [33,34]. Such comparative analysis allows for further evaluation of the clinical relevance of specific mouse models. Comparative analysis of the proteome profiles obtained from samples collected in different cohorts is also important. A major benefit in analyzing samples from different cohorts is to limit variation and bias due to sample collection. In addition, a careful selection of the samples to be analyzed is needed and multiple sources are often needed to obtain a sufficient sample size. Finally, comparative analysis of the proteomes of liver and corresponding plasma at different stages of disease suggested that protein abundance changes observed in the disease liver don't translate into similar changes in the plasma for the majority of commonly identified proteins (L. Beretta, personal communication). In conclusion, this method allowed for the identification of disease specific protein changes and suggested that tissue-based discovery and plasma-based discovery studies may lead to significantly different results.
Fig. 3.
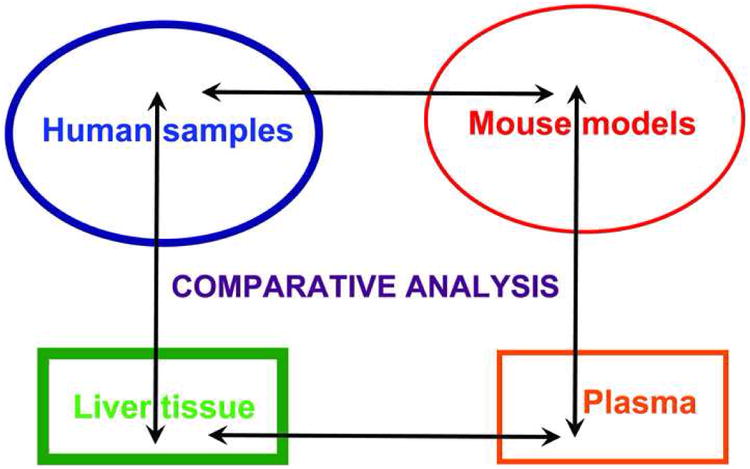
Schematic view of the cross comparative analysis performed between liver and plasma proteomes profiles and between human and mouse samples.
Concluding Remarks
To date, both proteomic and genomic studies have highlighted the heterogeneity of HCC. This heterogeneity emphasizes the complex nature of HCC carcinogenesis and disease progression in which multiple pathogenesis mechanisms seem to be involved. In addition, proteomic studies of HCC to date have highlighted that some information in the proteome cannot be predicted simply from its related nucleic acid sequence. With the rapid development of proteomics and its extensive application in the study of HCC, clinical management of HCC is poised to benefit enormously, with the potential to develop better diagnostic and prognostic tests, to identify new therapeutic agents, and ultimately to allow individualized therapy. Further work is required to enhance the performance and reproducibility of established proteomic tools and issues regarding pre-analytical variables, analytical variability, and biological variation must be addressed if further progress is to be made. To this end, the Human Liver Proteome Project (HLPP) was launched by the Human Proteome Organisation (HUPO) in 2002 and, to date, HLPP members have identified about 5,000 proteins in normal human liver tissue and have generated tools for their analysis in disease [35].
References
- 1.Beretta L. Proteomics from the clinical perspective: many hopes and much debate. Nat Methods. 2007;4:785–786. doi: 10.1038/nmeth1007-785. [DOI] [PubMed] [Google Scholar]
- 2.Conrads TP, Hood BL, Petricoin EF, 3rd, Liotta LA, Veenstra TD. Cancer proteomics: many technologies, one goal. Expert Rev Proteomics. 2005;2:693–703. doi: 10.1586/14789450.2.5.693. [DOI] [PubMed] [Google Scholar]
- 3.Hanash SM. Disease proteomics. Nature. 2003;422:226–232. doi: 10.1038/nature01514. [DOI] [PubMed] [Google Scholar]
- 4.Massion PP, Caprioli RM. Proteomic strategies for the characterization and the early detection of lung cancer. J Thorac Oncol. 2006;1:1027–1039. [PubMed] [Google Scholar]
- 5.Marko-Varga G, Ogiwara A, Nishimura T, Kawamura T, Fujii K, Kawakami T, Kyono Y, Tu HK, Anyoji H, Kanazawa M, Akimoto S, Hirano T, Tsuboi M, Nishio K, Hada S, Jiang H, Fukuoka M, Nakata K, Nishiwaki Y, Kunito H, Peers IS, Harbron CG, South MC, Higenbottam T, Nyberg F, Kudoh S, Kato H. Personalized medicine and proteomics: lessons from non-small cell lung cancer. J Proteome Res. 2007;6:2925–2935. doi: 10.1021/pr070046s. [DOI] [PubMed] [Google Scholar]
- 6.Fu Q, Van Eyk JE. Proteomics and heart disease: identifying biomarkers of clinical utility. Expert Rev Proteomics. 2006;3:237–249. doi: 10.1586/14789450.3.2.237. [DOI] [PubMed] [Google Scholar]
- 7.Bergeron JJ, Hallett M. Peptides you can count on. Nat Biotechnol. 2007;25:61–62. doi: 10.1038/nbt0107-61. [DOI] [PubMed] [Google Scholar]
- 8.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 9.States DJ, Omenn GS, Blackwell TW, Fermin D, Eng J, Speicher DW, Hanash SM. Challenges in deriving high-confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study. Nat Biotechnol. 2006;24:333–338. doi: 10.1038/nbt1183. [DOI] [PubMed] [Google Scholar]
- 10.Gilchrist A, Au CE, Hiding J, Bell AW, Fernandez-Rodriguez J, Lesimple S, Nagaya H, Roy L, Gosline SJ, Hallett M, Paiement J, Kearney RE, Nilsson T, Bergeron JJ. Quantitative proteomics analysis of the secretory pathway. Cell. 2006;127:1265–1281. doi: 10.1016/j.cell.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 11.Ryan DP, Matthews JM. Protein-protein interactions in human disease. Curr Opin Struct Biol. 2005;15:441–446. doi: 10.1016/j.sbi.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Irish JM, Kotecha N, Nolan GP. Mapping normal and cancer cell signalling networks: towards single-cell proteomics. Nat Rev Cancer. 2006;6:146–155. doi: 10.1038/nrc1804. [DOI] [PubMed] [Google Scholar]
- 13.Massoud TF, Paulmurugan R, De A, Ray P, Gambhir SS. Reporter gene imaging of protein-protein interactions in living subjects. Curr Opin Biotechnol. 2007;18:31–37. doi: 10.1016/j.copbio.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villalobos V, Naik S, Piwnica-Worms D. Current state of imaging protein-protein interactions in vivo with genetically encoded reporters. Annu Rev Biomed Eng. 2007;9:321–349. doi: 10.1146/annurev.bioeng.9.060906.152044. [DOI] [PubMed] [Google Scholar]
- 15.Weissleder R. Molecular imaging in cancer. Science. 2006;312:1168–1171. doi: 10.1126/science.1125949. [DOI] [PubMed] [Google Scholar]
- 16.Chignard N, Beretta L. Proteomics for hepatocellular carcinoma marker discovery. Gastroenterology. 2004;127:S120–125. doi: 10.1053/j.gastro.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 17.Feng JT, Shang S, Beretta L. Proteomics for the early detection and treatment of hepatocellular carcinoma. Oncogene. 2006;25:3810–3817. doi: 10.1038/sj.onc.1209551. [DOI] [PubMed] [Google Scholar]
- 18.Chignard N, Shang S, Wang H, Marrero J, Brechot C, Hanash SM, Beretta L. Cleavage of endoplasmic reticulum proteins in hepatocellular carcinoma: Detection of generated fragments in patient sera. Gastroenterology. 2006;130:2010–2022. doi: 10.1053/j.gastro.2006.02.058. [DOI] [PubMed] [Google Scholar]
- 19.Le Naour F, Brichory F, Misek DE, Brechot C, Hanash SM, Beretta L. A distinct repertoire of autoantibodies in hepatocellular carcinoma identified by proteomic analysis. Mol Cell Proteomics. 2002;1:197–203. doi: 10.1074/mcp.m100029-mcp200. [DOI] [PubMed] [Google Scholar]
- 20.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 21.Chen EI, Hewel J, Felding-Habermann B, Yates JR., 3rd Large scale protein profiling by combination of protein fractionation and multidimensional protein identification technology (MudPIT) Mol Cell Proteomics. 2006;5:53–56. doi: 10.1074/mcp.T500013-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Clouthier SG, Galchev V, Misek DE, Duffner U, Min CK, Zhao R, Tra J, Omenn GS, Ferrara JL, Hanash SM. Intact-protein-based high-resolution three-dimensional quantitative analysis system for proteome profiling of biological fluids. Mol Cell Proteomics. 2005;4:618–625. doi: 10.1074/mcp.M400126-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Liu T, Qian WJ, Gritsenko MA, Camp DG, 2nd, Monroe ME, Moore RJ, Smith RD. Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. J Proteome Res. 2005;4:2070–2080. doi: 10.1021/pr0502065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plavina T, Wakshull E, Hancock WS, Hincapie M. Combination of abundant protein depletion and multi-lectin affinity chromatography (M-LAC) for plasma protein biomarker discovery. J Proteome Res. 2007;6:662–671. doi: 10.1021/pr060413k. [DOI] [PubMed] [Google Scholar]
- 25.Stahl-Zeng J, Lange V, Ossola R, Eckhardt K, Krek W, Aebersold R, Domon B. High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol Cell Proteomics. 2007;6:1809–1817. doi: 10.1074/mcp.M700132-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Lai KK, Kolippakkam D, Beretta L. Comprehensive and quantitative proteome profiling of the mouse liver and plasma. Hepatology. 2008;47:1043–1051. doi: 10.1002/hep.22123. [DOI] [PubMed] [Google Scholar]
- 27.Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, Mizuno K, Hasegawa G, Kishimoto H, Iizuka M, Naito M, Enomoto K, Watanabe S, Mak TW, Nakano T. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, Sherwin R, Devaskar S, Lesche R, Magnuson MA, Wu H. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected] Proc Natl Acad Sci USA. 2004;101:2082–2087. doi: 10.1073/pnas.0308617100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe S, Horie Y, Suzuki A. Hepatocyte-specific Pten-deficient mice as a novel model for nonalcoholic steatohepatitis and hepatocellular carcinoma. Hepatol Res. 2005;33:161–166. doi: 10.1016/j.hepres.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 30.Xu X, Kobayashi S, Qiao W, Li C, Xiao C, Radaeva S, Stiles B, Wang RH, Ohara N, Yoshino T, LeRoith D, Torbenson MS, Gores GJ, Wu H, Gao B, Deng CX. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J Clin Invest. 2006;116:1843–1852. doi: 10.1172/JCI27282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez-Chantar ML, Corrales FJ, Martinez-Cruz LA, Garcia-Trevijano ER, Huang ZZ, Chen L, Kanel G, Avila MA, Mato JM, Lu SC. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J. 2002;16:1292–1294. doi: 10.1096/fj.02-0078fje. [DOI] [PubMed] [Google Scholar]
- 32.Campbell JS, Hughes SD, Gilbertson DG, Palmer TE, Holdren MS, Haran AC, Odell MM, Bauer RL, Ren HP, Haugen HS, Yeh MM, Fausto N. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci USA. 2005;102:3389–3394. doi: 10.1073/pnas.0409722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JS, Chu IS, Mikaelyan A, Calvisi DF, Heo J, Reddy JK, Thorgeirsson SS. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet. 2004;36:1306–1311. doi: 10.1038/ng1481. [DOI] [PubMed] [Google Scholar]
- 34.Thorgeirsson SS, Lee JS, Grisham JW. Functional genomics of hepatocellular carcinoma. Hepatology. 2006;43:S145–150. doi: 10.1002/hep.21063. [DOI] [PubMed] [Google Scholar]
- 35.Beretta L. The Human Liver Proteome Project. Mol Cell Proteomics. 2007;6:2043. [Google Scholar]