

Abstract
Collagen VI-related dystrophies and myopathies (COL6-RD) are a group of disorders that form a wide phenotypic spectrum, ranging from severe Ullrich congenital muscular dystrophy (UCMD), intermediate phenotypes, to the milder Bethlem myopathy (BM). Both inter- and intra-familial variable expressivity are commonly observed. We present clinical, immunohistochemical, and genetic data on four COL6-RD families with marked inter-generational phenotypic heterogeneity. This variable expression seemingly masquerades as anticipation is due to parental mosaicism for a dominant mutation, with subsequent full inheritance and penetrance of the mutation in the heterozygous offspring. We also present an additional 5th simplex patient identified as a mosaic carrier. Parental mosaicism was confirmed in the four families through quantitative analysis of the ratio of mutant versus wild-type allele (COL6A1, COL6A2 and COL6A3) in genomic DNA (gDNA) from various tissues; including blood, saliva, and dermal fibroblasts. Consistent with somatic mosaicism, parental samples had lower ratios of mutant versus wild-type allele compared to the fully heterozygote offspring. However, there was notable variability of the mutant allele levels between tissues tested, ranging from 16% (saliva) to 43% (fibroblasts) in one mosaic father. This is the first report demonstrating mosaicism as a cause of intra-familial/inter-generational variability of COL6-RD, and suggests that sporadic and parental mosaicism may be more common than previously suspected.
Keywords: COL6A1, COL6A2 and COL6A3, Collagen VI, Genetic counseling, Bethlem Myopathy, Ullrich Congenital Muscular Dystrophy
Introduction
Collagen type VI-related dystrophies and myopathies (COL6-RD) present with variable expressivity, ranging from severe Ullrich congenital muscular dystrophy (UCMD; MIM# 254090) manifesting with lack of or early loss of ambulation, through phenotypes of intermediate severity, to the milder Bethlem myopathy (BM; MIM# 158810). The clinical manifestations include muscle weakness, joint contractures, joint hyperlaxity, keratosis pilaris, keloid scar formation, scoliosis, and respiratory insufficiency with disease progression, all of which can show significant variability (Bonnemann, 2011). Diagnosis is supported by characteristic patterns on muscle imaging, reduced, absent or mislocalized collagen VI in muscle, and by abnormal matrix deposition in dermal fibroblast culture (Bonnemann, et al., 2003; Jimenez-Mallebrera, et al., 2006; Mercuri, et al., 2005).
COL6-RD can be inherited in an autosomal dominant or autosomal recessive manner. UCMD may be caused by recessive loss of function mutations or severe de-novo dominant-negative mutations. The intermediate phenotype and BM are more commonly caused by dominantly acting mutations of variable severity, and less commonly by recessive mutations (Bonnemann, 2011). Molecular genetic confirmation is obtained through direct sequencing of the COL6A1, COL6A2 and COL6A3 genes. This method identifies point mutations or deletions that result in reduced or functionally impaired collagen VI, an important component of the muscle extracellular matrix. The most commonly identified mutations are dominant-negative mutations, involving in-frame skipping or glycine substitutions in the collagenous triple helical domain Gly-X-Y motif (Butterfield, et al., 2013). Genetic heterogeneity is evident as approximately 10% of patients with a clinical diagnosis of COL6-RD do not have mutations identified in COL6A1, COL6A2 or COL6A3, indicating that another gene such as COL12A1, or currently unidentified genes account for their symptoms (Hicks, et al., 2014; Zou, et al., 2014; Petrini, et al., 2007).
Intra-familial phenotypic variability as well as disease progression in COL6-RD is common. However, the underlying molecular mechanism for this variability is not clearly understood. Mosaicism is the occurrence of two or more genetically different cell lines in an individual that can be limited to the germline, somatic cells, or a combination of both, depending on the timing of the postzygotic mutation during development (Strachan and Read, 1999). Identifying and understanding the underlying genetic mechanism affecting disease severity is essential for providing accurate prognosis, recurrence risk, genetic counseling, and development of therapeutic strategies. Neither germline nor somatic mosaicism have been previously described in COL6-RD. Here, we present clinical, immunohistochemical, and genetic data on four unrelated families with marked intra-familial phenotypic variability due to inherited mosaicism of dominant negative mutations in collagen VI. Mosaicism for the mutant allele was confirmed through quantitative analysis of mutant allele quantity in various tissues, such as blood, dermal fibroblasts, and saliva, in the affected patient versus mosaic parents. Additionally, we present a fifth simplex patient who was identified as a de novo mosaic carrier of a collagen VI mutation. This is the first report demonstrating inheritance of a parental mosaic mutant allele as a cause of intra-familial/inter-generational variability of collagen VI-related dystrophies.
Patients and Methods
Patient recruitment & sample collection
Four families and a fifth unrelated simplex patient were identified in neuromuscular clinics in three different countries. This study was approved by the Institutional Review Board of the National Institute of Neurological Disorders and Stroke, National Institutes of Health. Written informed consent and appropriate assent were obtained from each evaluated member of the family by a qualified investigator. DNA was obtained from blood, fibroblasts, and saliva based on standard procedures. When available, banked muscle biopsy samples were obtained.
Molecular diagnostic investigations
Sequencing of COL6A1, COL6A2, and COL6A3 in Families 1, 2, and 3 was performed by extracting RNA from the patient and mosaic parent fibroblast cells, followed by reverse transcription polymerase chain reaction to obtain cDNA and amplification of triple helical domain of each gene using polymerase chain reaction (PCR). For Families 4 and 5 PCR sequencing was done on gDNA extracted from blood. Sequencing of PCR products was performed on an ABI 3130x1 capillary sequencer in the forward and reverse directions. All families had confirmatory genetic testing by outside laboratories. Variants were numbered according to RefSeq transcripts NM_001848.2 for COL6A1, NM_001849.3 for COL6A2, and NM_004369.3 for COL6A3. For cDNA numbering, nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1. Mutations were submitted to the Leiden Open Variation Database for COL6A1 (www.LOVD.nl/COL6A1), COL6A2 (www.LOVD.nl/COL6A2) and COL6A3 (www.LOVD.nl/COL6A3)
Immunofluorescence labeling of dermal fibroblasts
Dermal fibroblasts established from a normal control, patients, and mosaic parents were grown in Dulbecco’s modified Eagle medium with 10% FBS and 1% Penicillin/ Streptomycin in an 8-well chamber in 5% CO2 at 37°C until 80% confluency. Cells were continuously cultured in the presence of 50 µg/ml L-ascorbic acid phosphate (Wako, Osaka, Japan) for 4-5 days before being fixed with 4% paraformaldehyde at room temperature, blocked in 10% FBS with or without 0.1% Triton X-100 in PBS, and then incubated with a mouse monoclonal antibody specific for collagen VI (Chemicon, Temecula, CA) at 1:2500 dilution. Cells were washed with PBS prior to incubation with goat anti-mouse Alexa Fluor 568-conjugated secondary antibody (Life Technologies, Grand Island, NY) at 1:500 dilution and nuclei were counterstained with DAPI (4’,6’-diamidino-2-phenylindole hydrochloride). Images were obtained using a Nikon Eclipse Ti epifluorescence microscope.
To quantify the granularity of the images, we developed a morphological operations-based image processing protocol that uses the collagen VI staining channel of the fluorescence image as input. The granularity analysis allows us to define a quantitative measure on the discontinuity (or lack of continuity) of the collagen microfibrils, by counting the number of round regions with smaller areas. For each sample, we imaged five random and independent regions. From these fluorescence images, an adaptive thresholding operation was first performed to generate a binary image. The threshold was determined by intensity mean of the image. Next, a connected component analysis was conducted to identify each connected region and their morphologic features, including area, solidity, and major-minor axis lengths. The roundness of each connected component was measured by the ratio between the length of its long axis and its short axis. In our experiment, we considered a ratio between 1.0 and 1.8 as an acceptable roundness range (mathematically, 1.0 corresponds to an exact circle). Connected components with a long/short axis ratio larger than 1.8 or smaller than 1.0 were automatically removed by the computer. After this selection, the final granularity was quantitatively measured by the remaining objects with proper shapes and sizes, and then normalized by the mean fluorescence intensity of the image. The greatest advantage of the proposed computer-guided granularity analysis is its objectivity, which allows us to apply the same criteria for both control and disease groups. The software has produced the same results after three technicians have performed the automatic operations, in order to demonstrate the reproducibility of the method.
Immunofluorescence labeling of muscle biopsies
Nine µm frozen muscle sections were cut using a Microm HM505E cryostat. The muscle sections were fixed with pre-cold methanol at -20°C, blocking in 10% FBS with 0.1% Triton X-100 in PBS. The muscle sections were dual labeled for co-localization with mouse monoclonal anti-collagen VI antibody 1:2500 (Chemicon, Temecula, CA) and anti-Laminin γ1 rabbit polyclonal antibody 1:800 (Sigma Aldrich, St. Louis, MO), a marker for the basement membrane. Alexa Fluor 568-conjugated goat anti-mouse 1:500 (Life Technologies, Grand Island, NY) and Alexa Fluor 4888-conjugated goat anti-rabbit 1:500 (Life Technologies, Grand Island, NY) were used and nuclei were counterstained with DAPI (4’,6’-diamidino-2-phenylindole hydrochloride). Images were obtained using a Leica Microsystems SP5, Wetzlar, Germany confocal microscope.
Quantification of mutant versus normal allele
Genomic DNA was extracted from blood, dermal fibroblasts and saliva. cDNA was obtained using Reverse Transcription polymerase chain reaction from RNA extracted from cultured dermal fibroblasts. Custom TaqMan SNP Genotyping Assays (Life Technologies, Grand Island, NY), consisting of mutation-specific primers and fluorescent-labeled allele discrimination probes, were designed for each mutation using a custom design tool provided by Applied Biosystems (Supp. Table S1). Fifty-five ng of Genomic DNA was used with total volume of 5µl for each reaction. The master mix was ordered from the manufacturer (Life Technologies, 4371353). The fluorescent readings were recorded after 40 amplification cycles. The amplification was performed using the ABI 7900 Real-time PCR system. Reactions were run in triplicates. GAPDH was used as an endogenous quantity control for cDNA samples. Quantitative analysis of the percentage of mosaicism was performed as previously reported (Hsu, et al., 2013). Mutant allele levels were calculated by using the ΔΔ cycle threshold (ΔΔCt) method: ΔΔCt= ΔCt (mosaic)- ΔCt (het). (ΔCt was defined as the difference between the mean Ct values of mutant and WT)(Hsu, et al., 2013).
Results
Clinical and imaging findings
We identified four unrelated families with a parent mosaic for a dominant collagen VI mutation and a fifth mosaic simplex patient, two from the United States, and one each from the Netherlands, France, and the United Kingdom. Detailed clinical information was available on nine affected individuals (Table 1; Fig. 1). Imaging findings were available on six affected individuals (Fig. 2). All four unrelated non-mosaic (i.e. fully heterozygote) children of a mosaic parent [F1P (family 1; fully heterozygote Patient), F2P, F3P and F4P] presented with typical findings of COL6-RD, including delayed milestones, distal hypermobility with contractures, proximal more than distal weakness, and characteristic skin findings (Fig. 1).
Table 1.
Clinical features of the COLVI-RD patients and their mosaic parents
| Patient/ sex/age (yrs.) |
Mutation | Presentation | Motor Development |
Current motor function & strength (MRC grades) |
Contractures & skin findings |
FVC |
|---|---|---|---|---|---|---|
| F1P/F/13 | Heterozygous COL6A2 Exon 7 c.900+1G>A |
Hypotonia and head lag at birth. |
Gowers’ sign at age 2 yrs. Difficulties with stairs at age 8 yrs., used a scooter at age 9 yrs. Loss of independent ambulation following scoliosis surgery at age 10 yrs. |
Unable to transition from supine to sitting. Weakness proximal > distal. Hip flexion 2/5; neck flexion and extension 2/5; remaining proximal muscles 3–4/5. |
Distal finger hyperlaxity with coexisting long finger flexor contractures. Contractures of neck flexion, shoulder rotators, elbow flexors L>R, wrist extension, hips, knees, toes. Moderate to severe scapular winging. Keratosis pilaris. |
Not done |
| F1M/M/45 | Heterozygous COL6A2 Exon 7 c.900+1G>A |
Toe walking at age 2 yrs. |
Difficulties with stairs at age 4 yrs. Run with awaked gait and heel cord contractures at age 10 yrs. Increased falls in 20s. Unable to rise from floor at age 38 yrs. |
Trendelenburg gait. Minimal acceleration but unable to clear ground, with excessive arm pumping. Weakness proximal > distal. Trunk flexion 2/5; hip flexion 3+/5; neck extension 5/5; remaining distal proximal muscles 4/5. |
Distal finger hyperlaxity with coexisting long finger flexor contractures. Contractures of shoulders external rotation, elbows, wrist extension and flexion. Mild scapular wining, rigid spine. Cigarette paper skin, hyperkeratosis pilaris. |
Not done |
| F2P/M/24 | Heterozygous COL6A1 Exon 10 c.859G>A p.Gly287Arg |
Congenital elbow contractures, hyperlaxity of the fingers. |
Started using a walker at age 10 yrs., a scooter at age 11 yrs. Loss of independent ambulation since early teens. |
Few short and narrow based steps with assistance. Strength 4-5-/5 range. Hip flexion and extension 3-/5. |
Distal finger hyperlaxity, contractures of elbows, knees and ankles. Mild scoliosis, mild keratosis pilaris. |
31% |
| F2M/M/60 | Heterozygous COL6A1 Exon 10 c.859G>A p.Gly287Arg |
Toe walker, not very athletic, slow runner. |
Difficulties with stairs in late 50’s possibly related to back pain from L4-5 herniation |
Mild Trendelenburg gait, mild action tremor. Hip flexion 4. Otherwise 5/5 strength. |
Bilateral long finger flexor contractures, elbow contractures, slight pectoralis contractures. Mild scoliosis. |
71% |
| F3P/M/16 | Heterozygous COL6A1 Exon 11 c.930+2T>A |
Delayed motor development noted at 18 months. |
Never able to bike. Frequent falls in childhood. Able to walk 15 minutes at age 9 yrs. |
Unable to jump. Difficulties rising from chair. Increased fatigue. Proximal muscle weakness in hip flexion 3/5. Truncal weakness. Ascends and descends stairs with two hands on banister. |
Distal finger hyperlaxity with coexisting and long finger flexor contractures. Limited shoulder abduction. Rigid spine, keratosis pilaris, keloid scarring. |
83% |
| F3M/M/56 | Heterozygous COL6A1 Exon 11 c.930+2T>A |
Never very athletic. Unable to run and do push- ups. |
Recurrent pulmonary infections in teens. |
Mild facial and proximal weakness: biceps, triceps, hip flexors & hip extensors 4/5. Fatigue in arms while swimming. |
Mild elbow contractures. Scoliosis, lumbar lordosis, hypertrophic scarring. |
Not done |
| F4P/M/43 | Heterozygous COL6A3 Exon 17 c.6238G>T p.Gly2080Cys |
Congenital arthrogryposis and bilateral hip dislocation |
Achieved walking at 2 yrs. Loss of independent ambulation age 40 yrs. Nocturnal non- invasive ventilation since age 32 yrs. |
Severe proximal muscle weakness: deltoids 3/5; biceps and triceps 3+/5; hip flexion 2/5; knee extension 3/5; knee flexion 3/5; dorsiflexion & plantar flexion 3+/5; distal muscles 3/5. |
Distal finger hyperlaxity with long finger contractures. Contractures of the elbows, shoulder, jaw, hips, knees and Achilles tendons. Mild lumbar lordosis, rigid spine. Limited shoulder abduction. |
30% |
| F4M/F/75 | Heterozygous COL6A3 Exon 17 c.6238G>T p.Gly2080Cys |
Never very athletic. |
Normal gait. No muscle weakness. |
Mild long finger flexor contractures. |
Not done |
|
| F5M/M/10 | Heterozygous COL6A2 Exon 10 c.955-2A>G |
Frequent falls noted at age 14 months. Frequent respiratory infections as toddler. |
Gowers’ at age 14 months. Tendency to W sit at age 23 months. Able to fast walk but not run. |
At age 7 yrs. uses stroller for long distances, rides a bicycle on flat surfaces, independently ambulant, ascends and descends stairs with one hand on banister. Toe walking with waddling-type gait. Neck flexion & trunk flexion 2+/5; elbow flexion & extension 4-5/5 & knee extension 5/5. |
Distal finger and toe hyperlaxity. Contractures of the elbows and Achilles tendons. Mild scoliosis. |
70% at age 7 yrs. |
M= male; F= Female; Yrs = years; CK = Serum Creatine Kinase; FVC=Forced Vital Capacity
Figure 1.
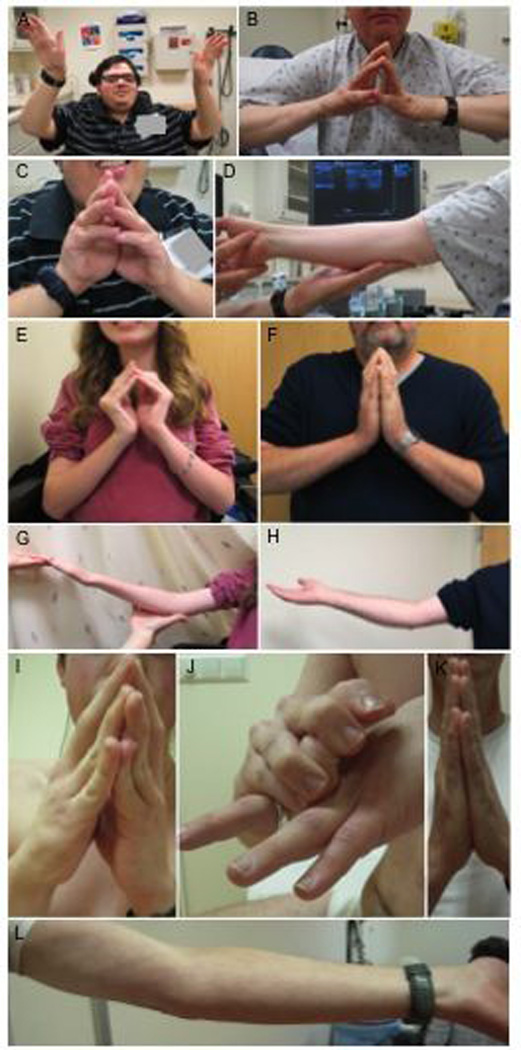
(A, C): 24 year old male non-mosaic proband F2P showing proximal weakness and long finger flexor contractures. (B, D): His 60 year old mosaic father F2M demonstrates mild long finger flexor and elbow contractures with minimal weakness. (E, G): Ten year old female non-mosaic proband F1P with contractures in long finger flexors and elbows and her 40 year old mosaic father F1M with mild long finger flexor and elbow contractures (F, H). (I, J): 17 year old male non-mosaic proband F3P with long finger flexor contractures and distal finger hyperlaxity and his 53 year old mosaic father F3M without long finger flexor contractures and mild elbow contractures (K, L).
Figure 2.
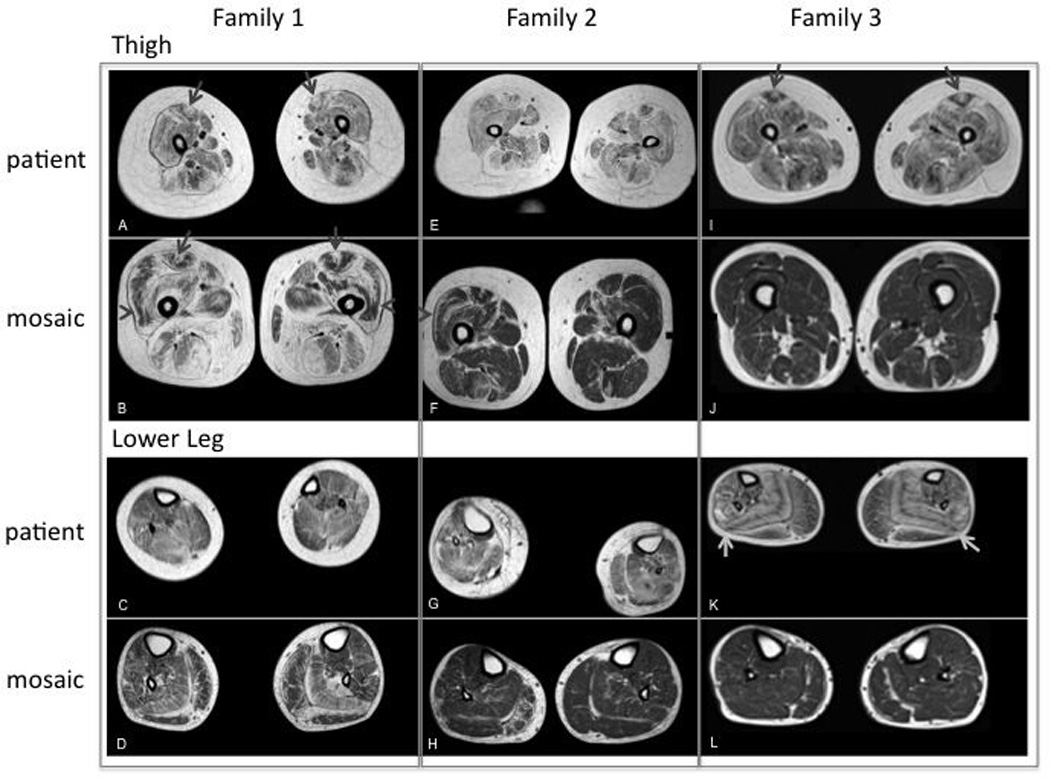
Muscle MRI imaging for patient and mosaic parent from Family 1, 2 and 3. (A): Severe fibrofatty changes and atrophy in all muscles of the thigh. “Central shadow” is visible bilaterally (arrow); (B): Severe fibrofatty changes in the muscles of the posterior thigh (HAM) and adductor magnus. Moderate fibrofatty changes in the muscles of the anterior thigh with evidence of rimming (especially in RF and VL, arrow head) and "central shadow” bilaterally (arrow); (C): Mild fibrofatty changes in the muscles of the distal lower extremity; (D): Mild fibrofatty changes in most muscles of the distal lower extremity. More moderate fibrofatty changes are noted in the SOL and lateral gastrocnemius muscles bilaterally, which also exhibit atrophy; (E): Severe fibrofatty changes and atrophy of all muscles of the thigh. Adductor longus, hamstrings (HAM) and rectus femoris (RF) are most affected. No clear “central shadow” phenomenon; (F): Mild-moderate fibrofatty changes in the muscles of the thigh. On the right, there is a small central shadow in the RF. Rimming of fibrofatty changes in the RF and vastus lateralis (VL) are noted bilaterally; (G): Moderate fibrofatty changes and atrophy in all muscles of the distal lower extremity. (H): Mild fibrofatty changes in the soleus (SOL) bilaterally and peroneus (PER) and medial gastrocnemius (MG) muscle on the right; (I): Severe fibrofatty changes and moderate atrophy in all muscles of the thigh. “Central shadow” in the RF bilaterally; (J): mild fibrofatty changes in the HAM. No rimming or central cloud; (K): Moderate fibrofatty changes are severe in the SOL and PER muscle. Notes striped appearance of the SOL because of rimming with central preservation of muscle. Mild-moderate changes are noted in the remaining anterior and posterior muscles of the distal lower extremity; (L): Mild fibrofatty changes in the MG muscles bilaterally and PER on the right.
Patient F1P and F2P had typical UCMD with hypotonia noted at birth and loss of ambulation at age 10 years and early teens, respectively. F2P also had lower extremity swelling of unclear etiology following blunt force trauma to his right leg. Work-up for deep vein thrombosis was negative. Patient F3P and F4P had an intermediate phenotype, with retained ability to walk short distances at the age of 18 and 40 years, respectively. Patient F4P also had very thin skin, hyperkeratosis, and numerous small ulcers in both legs, requiring continuous dermatological care, facial hyperkeratosis and seborrheic dermatitis.
Three mosaic parents, father F2M (Family 2; Mosaic parent), father F3M and mother F4M, never noticed collagen VI related symptoms and were only clinically evaluated after sequencing studies for carrier status suggested that they were mosaic for the mutant allele. F3M did report abnormal scar tissue prior to collagen VI mutation segregation testing. In hindsight, the mosaic parents reported that they were never very athletic. Imaging studies in mosaic fathers F2M and F3M showed the “central cloud” phenomenon, reflecting degeneration around the central fascia of the rectus femoris, as well as the typical concentric degeneration in other muscles, consistent with a diagnosis of COL6-RD (Fig. 2). On exam, F2M and F4M had contractures and mild weakness. Mosaic mother F4M had a normal clinical examination except for mild finger flexors contractures. However, she had been previously diagnosed with polyarthritis. Lower limb CT scan was normal.
Mosaic father F1M was noted to have difficulty walking and climbing steps at 4 years of age and he was initially diagnosed with Duchenne Muscular Dystrophy (DMD) based on dystrophic changes on muscle biopsy. His diagnosis was amended to spinal muscular atrophy (SMA) based the slow course of the disease and a second muscle biopsy showing “neurogenic changes.” He was counseled on the presumed autosomal recessive inheritance seen in SMA, with a low risk for recurrence. After his daughter was diagnosed with UCMD, his diagnosis of Bethlem myopathy was confirmed by genetic testing.
F5M is a simplex case who was found to be mosaic for an exon skipping c.955-2A>G mutation in COL6A2. He had a COL6-RD phenotype of intermediate severity, as he was still independently ambulant for long distances, able to ride a bicycle, and able to ascend and descend stairs at age 7 years. Given that his phenotype was milder than typical UCMD patients, but more severe than Bethlem myopathy patients, he would best be characterized as having intermediate collagen VI-related dystrophy.
Genetic and histological findings
The family histories and COL6A1-3 sequencing results are shown in Figure 3. Heterozygous mutant alleles were identified in the four patients and in one of their parents, however peak height of the mutant allele in the carrier parents was significantly smaller, suggesting parental somatic mosaicism (Fig. 3). Mosaic patient F5M was found to have a perceived lower-height peak of the COL6A2 mutation compared to typical heterozygous mutations (Fig. 3E2).
Figure 3.
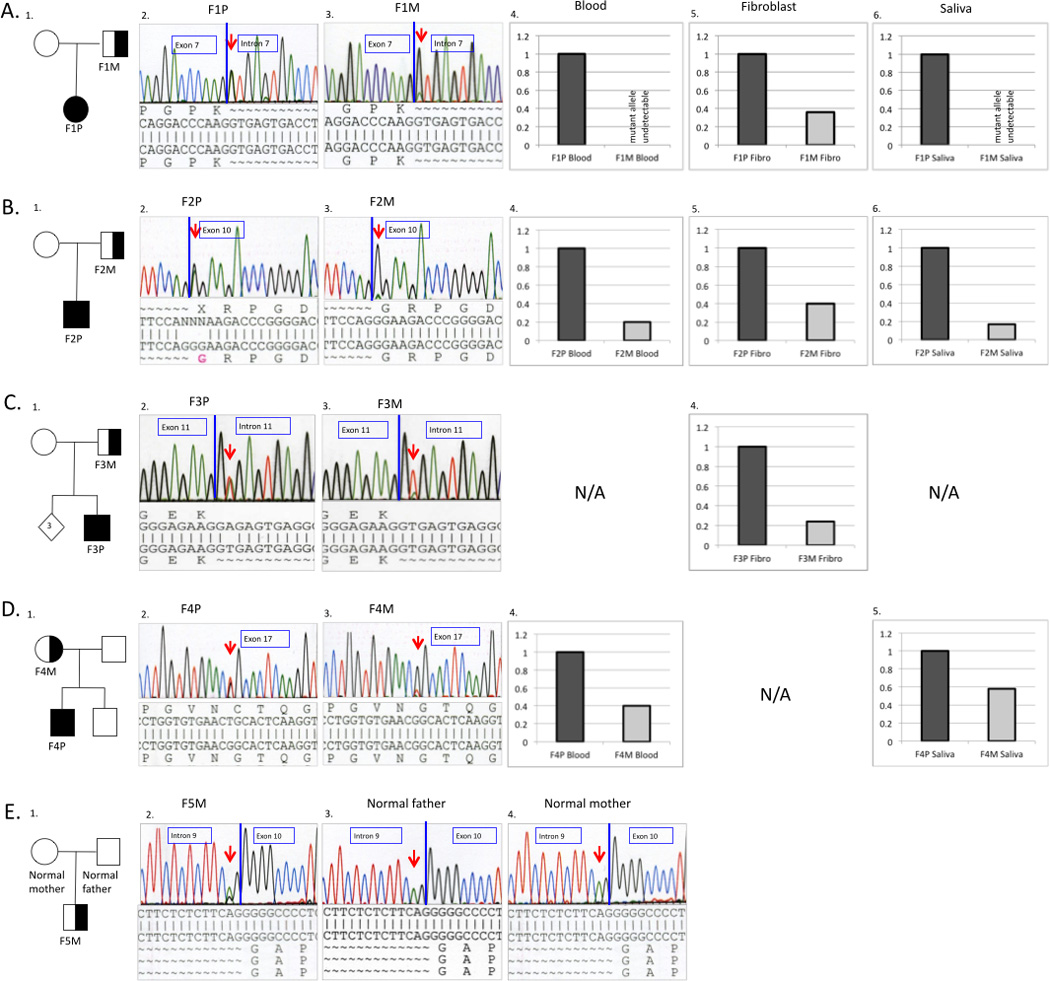
Pedigrees and molecular characterizations of the respective families. Superscript numerals correspond to individuals in Table 1 (A-D). Genomic DNA sequence chromatograms show the somatic mutation in mosaic parent and affected child. Mutations were confirmed on RNA extracted from dermal fibroblasts for Family 1,2 and 3 and gDNA extracted from blood in Family 4 and 5. The mosaic mutation sequence is seen as a lower-height peak in the parent compared to the non-mosaic mutation in the child (A-D). (A): Sequencing results of Family 1 showing heterozygous COL6A2 c.900+1G>A mutation in the patient F1P (A2. left) and mosaic father F1M (A3. right). (B): Sequencing results of Family 2 showing heterozygous COL6A1 c.859G>A (p.Gly287Arg ) mutation in the affected son F2P (B2. left) and mosaic father F2P (B3. right). (C): Sequencing results of Family 3 showing heterozygous COL6A1 c.930+2T>A mutation in the affected child F3P (C2. left) and mosaic father F3M (C3. right) (D): Sequencing results of Family 4 showing heterozygous COL6A3 c.6238G>T (p.Gly2080Cys) mutation in affected child F4P (D2. left) and mosaic mother F4M (D3. right). (E): Sequencing results of family F5 showing a lower-height peak of COL6A2 c.955-2A>G mutation in the mosaic patient F5M (E2. left) compared to the normal parents (E3.; E4.). Quantitative analysis of mutant allele expression from different tissues for the respective families. The graphic represents the percentage of the mutant allele expression in the probands (dark grey) and somatic mosaic parent (light grey).
Collagen VI matrix deposition expressed by immunofluorescence of cultured dermal fibroblasts was variable and showed a range from normal to complete absence in families with different mutations. The matrix formation in F1P (Fig. 4A1) appeared slightly reduced and less organized with a speckled and knotted pattern when compared to the more normal matrix formation shown in the mosaic parent F1M (Fig. 4A2). Since dominant-negative mutations, such as exon-skipping mutations, interfere with normal collagen VI assembly and deposition, which in turns appear disseminated and speckled, we quantified the granularity (number of speckles) of the collagen VI matrix deposited by fibroblasts in culture (Fig. 4B7, 4B8). In F1P the granularity was greater than the mosaic parent (Fig. 4B1, 4B2), supporting the idea that the matrix was less organized when the mutation was fully expressed. Similarly, for patient F3P (Fig. 4B4), collagen VI matrix formation was significantly reduced and the granularity increased when compared to normal extracellular matrix deposition in the mosaic father F3M (Fig. 4B5). In contrast, collagen VI was not detectable in the extracellular matrix of fibroblasts derived from F2P but strong intracellular retention was visible (Fig. 4A3). However, the matrix deposition revealed virtually normal staining in the mosaic father F2M (Fig. 4A4). F4P had no collagen VI secretion in cultured fibroblasts but intracellular retention (data not shown). Fibroblasts from the mosaic parent were not available.
Figure 4.
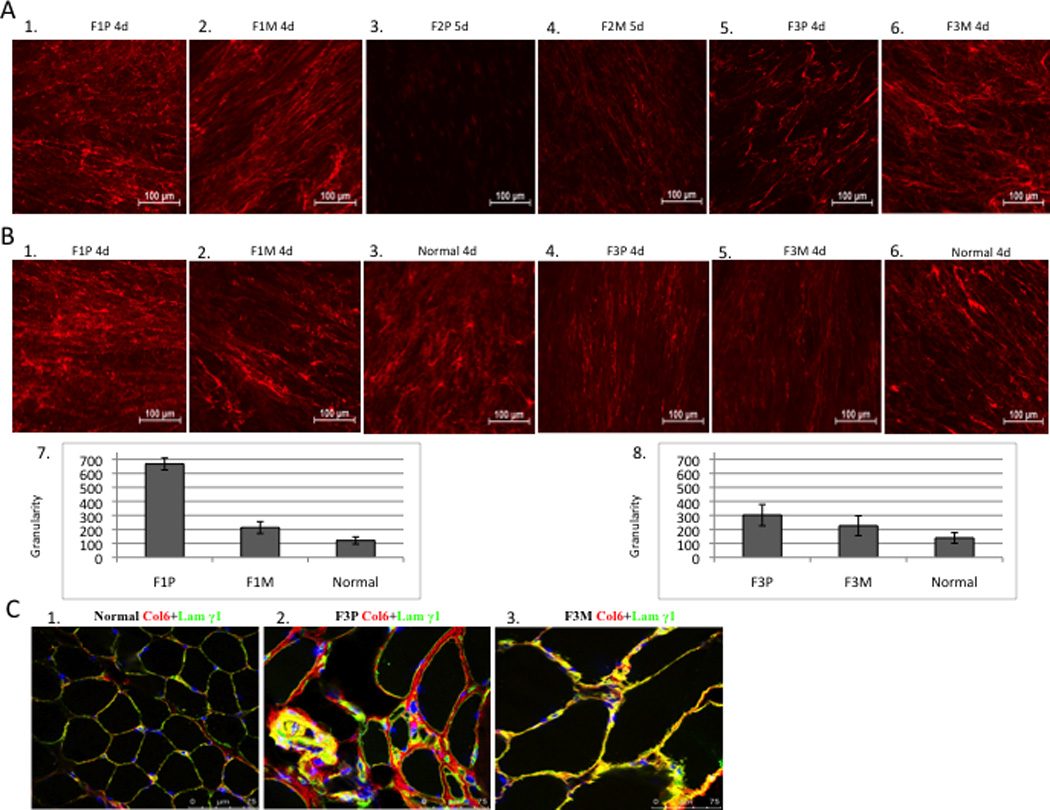
(A) Comparison of collagen VI immunoactivity in dermal fibroblast. (20x). Dermal fibroblasts were grown in the presence of 50ug/ml of L-ascorbic acid for 4-5 days after 80% confluency. Collagen VI specific monoclonal antibody was used (red). Collagen VI was detected in the extracellular matrix of mosaic parent and has a “knotted” and “speckled” pattern in the patient F1P (A1, A2), immunoreactivity is absent in patient F2P (A3, A4) and is reduced in patient F3P (A5, A6). (B) Granularity (quantitation of speckles) of the matrix deposited by fibroblasts was measured and normalized to the mean fluorescence intensity for Family 1 and 3, and normal controls processed concomitantly. Each bar graph represents the average of five images from independent fields of the same experiment. (C) Immunofluorescent staining of collagen VI in skeletal muscle (63x). Normal control muscle showed normal amount of collagen VI (red) with co-localization of basement membrane protein, laminin γ1 (green) resulting in yellow signal (C1); considerable amount of collagen VI in the matrix with partial co-localization of basement membrane protein in the patient’s muscle (see yellow), significant increased connective tissue (C2); mild increased connective tissue with normal amount of collagen VI and co-localization of basement membrane in the mosaic parent (C3).
Immunohistochemical analysis of a muscle biopsy from patient F3P and the mosaic father F3M, compared with a normal control, showed a relatively normal amount of collagen VI protein in the patient (Fig. 4C2), but partial loss of co-localization with the basement membrane labeled with Laminin © 1 antibody compared to the normal control (Fig. 4C1). The muscle of the mosaic father F3M showed a normal amount of collagen VI protein and complete overlay with the basement membrane protein Laminin © 1(Fig. 4C3).
Quantitative analysis of mutant allele ratio in mosaic versus non-mosaic
To determine the degree of mosaicism, the relative ratio of mutant allele quantity in the mosaic parents was analyzed in different somatic tissues, with five pairs of real-time PCR probes specific to either mutant or WT alleles used for corresponding patients (Supp. Fig. S1). In family 1, genomic DNA obtained from peripheral blood, dermal fibroblasts, and saliva was tested. The fibroblast sample showed a lower proportion approximately 36% of the mutant allele in the mosaic father (Fig. 3A5). The signal from the mutant allele was too weak to be detected in blood and in saliva samples from the mosaic father (Fig. 3A4&6). The quantitative data from blood, dermal fibroblast, and saliva from family 2 showed 20%, 42% and 16%, of mutant allele ratio in the father, respectively (Fig. 3B4-6). The percentage of mosaicism on the cDNA level for family 2 was also tested to indicate relative expression from the mutant allele. The mutant allele ratio from the cDNA from dermal fibroblast revealed 35% in the father (data not shown), which is slightly lower than in genomic DNA extracted from fibroblast. The data from family 3 showed approximately 24% of mutant ratio in the fibroblast (Fig 3C4). Family 4 showed 40% of mutant ratio in the blood sample, and 58% in the saliva sample of the mosaic mother (Fig. 3D4,5).
Discussion
Mosaicism is a result of a postzygotic mutation and may be confined to somatic cells, the germline, or both, depending on the developmental stage at which the mutation occurred. The phenotypic expression of the mosaic mutation depends on the tissue distribution and mutation ratio expressed. Mosaicism can complicate clinical diagnosis and genetic counseling. Within the realm of dominant neuromuscular diseases, inherited mosaicism has been reported for several genes including ACTA1 (Nowak, et al., 1999), PMP22 (Sorour, et al., 1995), MPZ (Fabrizi, et al., 2001), LMNA (Makri, et al., 2009) and BAG3 (Odgerel, et al., 2010). Additionally, germline and somatic mosaicism are well established in X-linked disorders such as Duchenne muscular dystrophy (DMD) (Kesari, et al., 2009; van Essen, et al., 2003). Parentally inherited mosaicism has been reported in the collagen gene family, including point mutations in COL1A1 and COL1A2 resulting in osteogenesis imperfecta (Cohn, et al., 1990; Pyott, et al., 2011; Wallis, et al., 1990) and deletions in COL3A1 resulting in Ehlers-Danlos syndrome (EDS) type IV. However, this is the first report of inherited parental mosaicism of dominantly-acting collagen VI mutations.
Phenotypic intra-familial variability in particular for disease severity is a well-established phenomenon in COL6-RD and may be related to epigenetics, modifier genes and/or variable “leaky” splice site mutations. Here, we present an additional mechanism in which the heterogeneous ratio of mutant and normal collagen VI may result in the mosaic parent having a less severe phenotype compared to their children who have a full mutation load. This phenomenon mimics genetic anticipation with disease severity increasing in subsequent generations. Interestingly, the mutation load in blood and saliva does not necessarily correlate with the severity of disease, as illustrated by F4M who has the highest mutation load tested but shows the mildest phenotype, or vice versa the mosaic mutation may be virtually undetectable in certain tissues as illustrated by F1M in blood and saliva in a clearly affected patient who tested positive in fibroblasts. Higher levels of somatic mosaicism may be present in tissues derived from other cell lineages, but these were not analyzed, due to unavailability of other tissues.
Mosaic collagen VI mutations may be difficult to suspect in a single patient on clinical grounds. Since not all mosaic parents reported here complained of COL6-RD symptoms, it may be advisable to evaluate parents of apparent simplex cases for subtle clinical findings as well as to proceed with molecular genetic testing to accurately establish recurrence risk. Additionally, germline mosaicism for a dominant mutation may appear as recessive inheritance when two or more affected children are born to apparently unaffected parents. If the germline is involved in a mosaic affected proband, subsequent generations are at increased risk to be affected by the full mutation, possibly presenting at the severe end of the COL6-RD spectrum. Genetic counseling is complicated as the risk of parents having an affected child is unknown, and depends on the ratio of mutated germline progenitor cells. The level of germline mosaicism, and therefore recurrence risk, cannot be predicted from analysis of other somatic tissues. In reviewing families with parental mosaicism for dominant mutations causing osteogenesis imperfecta and retinoblasma, the recurrence rate was estimated to be 27% and 10% respectively (Pyott, et al., 2011; Sippel, et al., 1998). Caution is necessary, however, as the individual recurrence risk may be as high as 50%.
Our clinical and fibroblast data indicate the importance of the dose load in dominant collagen VI mutation as a pathogenic mechanism. In particular, relative lesser dose of the mutant allele, as seen in the mosaic parent, shows histochemical restoration of collagen VI localization in the fibroblast cell culture in family 2. Clinically, this notion translates into a milder phenotypic expression of the disease in mosaic parents who present with a lower mutation load, compared to their non-mosaic carrier children. Relative dose of dominant collagen VI mutation is capable of modulation disease mechanism, a variable therapeutic approach since known haploinsufficiency does not cause disease (Baker, et al., 2007; Baker, et al., 2005). This data validates the promising development of a therapeutic approach for dominant-negative collagen VI mutations in which knock-down of the mutant allele expression may be achieved through antisense-mediated or RNAi based therapy (Bolduc, et al., 2014; Gualandi, et al., 2012).
This report of somatic mosaicism for dominant collagen VI mutations suggests that parental mosaicism may be more common than previously suspected in COL6-RD. Thorough diagnostic workup, including molecular genetic testing, of family members is needed for patients with COL6-RD in order to better assess the incidence of mosaicism in collagen VI mutations and to ultimately provide accurate recurrence risk information for genetic counseling. Caution is required, as low levels of somatic mosaicism may not be detectable by standard genetic sequencing and pure germline mosaicism will not be detectable by testing of specimen types routinely available to diagnostic laboratories. The phenomenon of parental mosaicism in apparently simplex cases of COL6-RD has important implications for molecular testing, clinical diagnosis, management, and genetic counseling for this patient population. Advancements in understanding the genetics of COL6-RD will aid future clinical trials where modulation of the disease phenotype may be achieved by directly targeting the mutant allele and its expression.
Supplementary Material
Acknowledgments
The authors thank the families for their generous participation in this study. We thank Elizabeth Hartnett for coordinating the patient visits. We are grateful to Corine Gartioux (UMRS974, Paris, France) for technical help with collagen VI immunostaining. Work in CGB’s laboratory is supported by intramural funds by the National Institute for Neurological Disorders and Stroke/NIH. The National Specialist Commissioned Service support of the Dubowitz Neuromuscular Centre is also gratefully acknowledged.
References
- Baker NL, Morgelin M, Pace RA, Peat RA, Adams NE, Gardner RJ, Rowland LP, Miller G, De Jonghe P, Ceulemans B, et al. Molecular consequences of dominant Bethlem myopathy collagen VI mutations. Ann Neurol. 2007;62:390–405. doi: 10.1002/ana.21213. [DOI] [PubMed] [Google Scholar]
- Baker NL, Morgelin M, Peat R, Goemans N, North KN, Bateman JF, Lamande SR. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet. 2005;14:279–293. doi: 10.1093/hmg/ddi025. [DOI] [PubMed] [Google Scholar]
- Bolduc V, Zou Y, Ko D, Bonnemann CG. siRNA-mediated Allele-specific Silencing of a COL6A3 Mutation in a Cellular Model of Dominant Ullrich Muscular Dystrophy. Mol Ther Nucleic Acids. 2014;3:e147. doi: 10.1038/mtna.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nature reviews. Neurology. 2011;7:379–390. doi: 10.1038/nrneurol.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnemann CG, Brockmann K, Hanefeld F. Muscle ultrasound in Bethlem myopathy. Neuropediatrics. 2003;34:335–336. doi: 10.1055/s-2003-44665. [DOI] [PubMed] [Google Scholar]
- Butterfield RJ, Foley AR, Dastgir J, Asman S, Dunn DM, Zou Y, Hu Y, Donkervoort S, Flanigan KM, Swoboda KJ, et al. Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Human mutation. 2013;34:1558–1567. doi: 10.1002/humu.22429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn DH, Starman BJ, Blumberg B, Byers PH. Recurrence of lethal osteogenesis imperfecta due to parental mosaicism for a dominant mutation in a human type I collagen gene (COL1A1) American journal of human genetics. 1990;46:591–601. [PMC free article] [PubMed] [Google Scholar]
- Fabrizi GM, Ferrarini M, Cavallaro T, Jarre L, Polo A, Rizzuto N. A somatic and germline mosaic mutation in MPZ/P(0) mimics recessive inheritance of CMT1B. Neurology. 2001;57:101–105. doi: 10.1212/wnl.57.1.101. [DOI] [PubMed] [Google Scholar]
- Gualandi F, Manzati E, Sabatelli P, Passarelli C, Bovolenta M, Pellegrini C, Perrone D, Squarzoni S, Pegoraro E, Bonaldo P, et al. Antisense-induced messenger depletion corrects a COL6A2 dominant mutation in Ullrich myopathy. Human gene therapy. 2012;23:1313–1318. doi: 10.1089/hum.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks D, Farsani GT, Laval S, Collins J, Sarkozy A, Martoni E, Shah A, Zou Y, Koch M, Bonnemann CG, et al. Mutations in the collagen XII gene define a new form of extracellular matrix-related myopathy. Human molecular genetics. 2014 doi: 10.1093/hmg/ddt637. [DOI] [PubMed] [Google Scholar]
- Hsu AP, Sowerwine KJ, Lawrence MG, Davis J, Henderson CJ, Zarember KA, Garofalo M, Gallin JI, Kuhns DB, Heller T, et al. Intermediate phenotypes in patients with autosomal dominant hyper-IgE syndrome caused by somatic mosaicism. The Journal of allergy and clinical immunology. 2013;131:1586–1593. doi: 10.1016/j.jaci.2013.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Mallebrera C, Maioli MA, Kim J, Brown SC, Feng L, Lampe AK, Bushby K, Hicks D, Flanigan KM, Bonnemann C, et al. A comparative analysis of collagen VI production in muscle, skin and fibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations. Neuromuscular disorders : NMD. 2006;16:571–582. doi: 10.1016/j.nmd.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Kesari A, Neel R, Wagoner L, Harmon B, Spurney C, Hoffman EP. Somatic mosaicism for Duchenne dystrophy: evidence for genetic normalization mitigating muscle symptoms. American journal of medical genetics. Part A. 2009;149A:1499–1503. doi: 10.1002/ajmg.a.32891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makri S, Clarke NF, Richard P, Maugenre S, Demay L, Bonne G, Guicheney P. Germinal mosaicism for LMNA mimics autosomal recessive congenital muscular dystrophy. Neuromuscular disorders : NMD. 2009;19:26–28. doi: 10.1016/j.nmd.2008.09.016. [DOI] [PubMed] [Google Scholar]
- Mercuri E, Lampe A, Allsop J, Knight R, Pane M, Kinali M, Bonnemann C, Flanigan K, Lapini I, Bushby K, et al. Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscular disorders : NMD. 2005;15:303–310. doi: 10.1016/j.nmd.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Nowak KJ, Wattanasirichaigoon D, Goebel HH, Wilce M, Pelin K, Donner K, Jacob RL, Hubner C, Oexle K, Anderson JR, et al. Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet. 1999;23:208–212. doi: 10.1038/13837. [DOI] [PubMed] [Google Scholar]
- Odgerel Z, Sarkozy A, Lee HS, McKenna C, Rankin J, Straub V, Lochmuller H, Paola F, D'Amico A, Bertini E, et al. Inheritance patterns and phenotypic features of myofibrillar myopathy associated with a BAG3 mutation. Neuromuscular disorders : NMD. 2010;20:438–442. doi: 10.1016/j.nmd.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrini S, D'Amico A, Sale P, Lucarini L, Sabatelli P, Tessa A, Giusti B, Verardo M, Carrozzo R, Mattioli E, et al. Ullrich myopathy phenotype with secondary ColVI defect identified by confocal imaging and electron microscopy analysis. Neuromuscular disorders : NMD. 2007;17:587–596. doi: 10.1016/j.nmd.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Pyott SM, Pepin MG, Schwarze U, Yang K, Smith G, Byers PH. Recurrence of perinatal lethal osteogenesis imperfecta in sibships: parsing the risk between parental mosaicism for dominant mutations and autosomal recessive inheritance. Genetics in medicine : official journal of the American College of Medical Genetics. 2011;13:125–130. doi: 10.1097/GIM.0b013e318202e0f6. [DOI] [PubMed] [Google Scholar]
- Sippel KC, Fraioli RE, Smith GD, Schalkoff ME, Sutherland J, Gallie BL, Dryja TP. Frequency of somatic and germ-line mosaicism in retinoblastoma: implications for genetic counseling. American journal of human genetics. 1998;62:610–619. doi: 10.1086/301766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorour E, Thompson P, MacMillan J, Upadhyaya M. Inheritance of CMT1A duplication from a mosaic father. Journal of medical genetics. 1995;32:483–485. doi: 10.1136/jmg.32.6.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan T, Read AP. Human molecular genetics. New York: 1999. [Google Scholar]
- van Essen AJ, Mulder IM, van der Vlies P, van der Hout AH, Buys CH, Hofstra RM, den Dunnen JT. Detection of point mutation in dystrophin gene reveals somatic and germline mosaicism in the mother of a patient with Duchenne muscular dystrophy. American journal of medical genetics. Part A. 2003;118A:296–298. doi: 10.1002/ajmg.a.10056. [DOI] [PubMed] [Google Scholar]
- Wallis GA, Starman BJ, Zinn AB, Byers PH. Variable expression of osteogenesis imperfecta in a nuclear family is explained by somatic mosaicism for a lethal point mutation in the alpha 1(I) gene (COL1A1) of type I collagen in a parent. American journal of human genetics. 1990;46:1034–1040. [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Zwolanek D, Izu Y, Gandhy S, Schreiber G, Brockmann K, Devoto M, Tian Z, Hu Y, Veit G, et al. Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Human molecular genetics. 2014 doi: 10.1093/hmg/ddt627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.