

The X-ray crystal structure of the enteropathogenic E. coli gatekeeper protein SepL contains three X-bundle domains.
Keywords: bacterial pathogenesis, protein secretion
Abstract
During infection, enteropathogenic Escherichia coli assembles a complex multi-protein type III secretion system that traverses the bacterial membranes and targets the host cell membrane to directly deliver virulence or effector proteins to the host cytoplasm. As this secretion system is composed of more than 20 proteins, many of which form oligomeric associations, its assembly must be tightly regulated. A protein called the gatekeeper, or SepL, ensures that the secretion of the translocon component, which inserts into the host membrane, occurs before the secretion of effectors. The crystal structure of the gatekeeper SepL was determined and compared with the structures of SepL homologues from other bacterial pathogens in order to identify SepL residues that may be critical for its role in type III secretion-system assembly.
1. Introduction
Many Gram-negative bacterial pathogens, including enteropathogenic Escherichia coli (EPEC), Salmonella, Chlamydia and Yersinia, use a type III secretion system (T3SS) to deliver effector proteins directly into the cytoplasm of target host cells. The T3SS is composed of a cytoplasmic export apparatus, a membrane-spanning basal body, an extracellular needle filament and a tip complex (also called the translocon) that inserts into the target host cell membrane (Worrall et al., 2011 ▸). A substructure called the inner rod forms a channel through the basal body, which connects to the lumen of the needle, creating a conduit for effector proteins (Marlovits et al., 2004 ▸). Effector proteins secreted into the host cell by the T3SS manipulate a number of host cell processes, such as the ubiquitination pathway, cytoskeleton dynamics and inflammatory signalling, to the benefit of the bacterium (Raymond et al., 2013 ▸).
Owing to the complexity of the T3SS, its assembly must be tightly regulated to ensure that components are secreted with the correct hierarchy. After the basal body and export apparatus assemble, the inner rod, needle, tip and translocon subunits are recognized by the export apparatus and secreted through the nascent basal body in an ATP-dependent manner (Diepold & Wagner, 2014 ▸). The needle translocon proteins bind specialized chaperones in the bacterial cytoplasm, which are presumably required for recognition by the ATPase and subsequent secretion of the translocon protein (Sal-Man et al., 2013 ▸; Chatterjee et al., 2013 ▸). Needle length is tightly regulated and is optimized for contact with the host cell (Mota et al., 2005 ▸). An accessory protein called the type III specificity switch protein is proposed to regulate needle length by acting as a molecular ruler (Journet et al., 2003 ▸). After the needle reaches the correct length, subunits that form the translocon complex are secreted. This step is regulated in part by a component of the export apparatus called the autoprotease, which has a conserved motif that undergoes self-cleavage via an intein-like mechanism (Zarivach et al., 2008 ▸). Mutation of this conserved motif to a noncleaving mutant results in normal secretion of the needle subunits but diminished translocon secretion (Zarivach et al., 2008 ▸). Once the translocon has been correctly assembled at the needle tip, effectors can then be secreted.
The switch from secretion of translocon subunits to effectors is controlled by a protein referred to as the gatekeeper, called SepL in EPEC (Diepold & Wagner, 2014 ▸). The gatekeeper is a key regulatory component of type III secretion, as deletion of the gene encoding the gatekeeper has been shown to attenuate virulence (Deng et al., 2004 ▸). A ΔsepL mutant of Citrobacter rodentium (an EPEC-related mouse pathogen) fails to secrete translocon proteins, form pedestals that are characteristic of attaching/effacing (A/E) pathogens and has attenuated virulence in mice (Deng et al., 2004 ▸). SepL binds a protein called SepD, which is also required for the regulation of type III secretion (Deng et al., 2005 ▸; O’Connell et al., 2004 ▸). Generally, the deletion of sepL or sepD results in decreased secretion of tip and translocon proteins (EspA, EspB and EspD in EPEC) and increased secretion of effector proteins (Deng et al., 2005 ▸), a trend also observed on deletion of the Salmonella homologue InvE and the Shigella homologue MxiC (although there are conflicting reports on whether translocator secretion is diminished in Shigella) (Kubori & Galán, 2002 ▸; Martinez-Argudo & Blocker, 2010 ▸; Botteaux et al., 2009 ▸). In contrast, deletion of the Yersinia homologue YopN results in constitutive secretion of effector and translocon proteins, suggesting some variability in phenotype between species (Day et al., 2003 ▸; Ferracci et al., 2005 ▸). The mechanism by which the gatekeeper regulates secretion of the translocon and effectors is not well understood; however, the recently characterized interaction between the Chlamydia gatekeeper CopN and the translocon chaperone Scc3 have led to a model in which a complex formed by the gatekeeper, translocon chaperone and translocon is required to form prior to the secretion of the translocon (Archuleta & Spiller, 2014 ▸). In other species, such as EPEC, interaction between the gatekeeper and the translocon chaperones has not been observed, which may reflect a weaker affinity or perhaps a variation in mechanism.
Much like effector proteins, the gatekeeper interacts with a specialized dimeric chaperone in the bacterial cytoplasm, and in some species the gatekeeper is secreted into the host cell. A crystal structure of Yersinia YopN in complex with its heterodimeric chaperone SycN/YscB shows how the extended N-terminal region of YopN wraps around the heterodimeric chaperone complex (Schubot et al., 2005 ▸). In addition to SepD, SepL interacts with a small acidic protein called CesL (Younis et al., 2010 ▸). CesL has limited sequence similarity to SycN, and the genes encoding SepD and YscB have a conserved position, indicating that CesL and SepD may be homologous to the heterodimeric chaperone of YopN (Younis et al., 2010 ▸). While secretion of SepL has not been directly observed, there is evidence that homologues are secreted in Shigella (Botteaux et al., 2009 ▸) and Chlamydia (Fields & Hackstadt, 2000 ▸). In fact, the Chlamydia gatekeeper CopN acts as an effector in the host cell cytoplasm and binds directly to αβ-tubulin to prevent microtubule polymerization (Archuleta et al., 2011 ▸). This effector-like activity of CopN has yet to be observed in other gatekeeper proteins and may be a divergent function.
Despite low sequence conservation among the homologues, the overall structure of the gatekeeper is well conserved and may serve as a scaffold for protein–protein interactions. Crystal structures of the gatekeeper from Shigella (MxiC), Yersinia (YopN/TyeA; the gatekeeper is expressed as two polypeptides, with TyeA homologous to gatekeeper domain 3) and Chlamydia (CopN) show a conserved architecture of three helical X-bundle domains (Schubot et al., 2005 ▸; Archuleta & Spiller, 2014 ▸; Nawrotek et al., 2014 ▸; Deane et al., 2008 ▸). The similarity between the domains may indicate a gene-duplication event, ultimately increasing the surface area accessible for mediating protein–protein interactions (Deane et al., 2008 ▸). In the structure of the CopN–Scc3 complex, Scc3 contacts CopN through two sites, which span the second and third domains of CopN (Archuleta & Spiller, 2014 ▸). Some evidence suggests that the inner rod (MxiI in Shigella) interacts with the second domain of MxiC (Cherradi et al., 2013 ▸). The interaction between MxiC and the inner rod may form a physical plug to prevent the secretion of effector proteins, and disruption of the interaction deregulates effector secretion (Cherradi et al., 2013 ▸). Additionally, the third domain of SepL is required for interaction with Tir, a type III secreted receptor required for intimate attachment of A/E pathogens to the host cell (Wang et al., 2008 ▸). The SepL–Tir interaction has been shown to be important to delay the release of effector proteins (Wang et al., 2008 ▸). A complete understanding of how protein–protein interactions mediated by the gatekeeper regulate the secretion hierarchy has yet to be reached.
In this study, we present the crystal structure of a gatekeeper protein from EPEC, SepL. SepL consists of three helical X-bundle domains and structural alignment of SepL with its homologues reveals regions in domains 2 and 3 with conserved surface-exposed residues. Our structure of SepL enables the design of structure-based mutations to test the roles of key residues in the regulation of translocon and effector secretion.
2. Materials and methods
2.1. Macromolecule production
DNA encoding SepL residues 70–351 was cloned into pET-28a vector by restriction-free cloning to create a construct encoding N-terminally 10×histidine-tagged SepL (70–351) (Table 1 ▸). This construct was selected because N-terminal sequencing of full-length SepL protein after limited proteolysis with trypsin revealed cleavage of the SepL N-terminus after residue 69. DNA encoding the same region of SepL was also cloned into pET-21a vector using restriction-free methods to create a construct encoding untagged SepL (70–351) (Chen et al., 2000 ▸). DNA encoding SepD (6–151) was cloned into pET-28a and pET-21a vectors using restriction-free methods to generate constructs expressing 10×histidine-tagged SepD (6–151) and untagged SepD, respectively. This N-terminal truncation of SepD was chosen to eliminate potentially disordered N-terminal residues (as predicted by the XtalPred server; Slabinski et al., 2007 ▸) that could potentially interfere with crystallization.
Table 1. SepL production information.
| Source organism | E. coli |
| DNA source | E. coli K12 |
| Forward primer | CACAGCAGCGGCCTGGTGCCGCGCGGCAGCGCTAATGGTATTGAATTTAATCAAAACC |
| Reverse primer | GGTGGTGGTGGTGGTGCTCGAGTTCATCACATAACATCCTCCTTATAATCTATCAC |
| Cloning vector | pET-28a/pET-21a |
| Expression vector | pET-28a/pET-21a |
| Expression host | E. coli |
| Complete amino-acid sequence of the construct produced | GSARKEEEGTTIEKLLNEMQELLTLTDSDKIKELSLKNSGLLEQHDPTLAMFGNMPKGEIVALISSLLQSKFVKIELKKKYAKLLLDLLGEDDWELALLSWLGVGELNQEGIQKIKKLYEKAKDEDSENGASLLDWFMEIKDLPEREKHLKVIIRALSFDLSYMSSFEDKVRTSSIISDLCRIIIFLSLNNYTDIIAISIKKDKDVILNEMLSIIEHVWLTEDWLLESPSRVSIVEDKHVYYFHLLKEFFASLPDACFIDNEQRSNTLLMIGKVIDYKEDVM |
To overexpress the native SepL (70–351)–SepD (6–151) complex, pET-28a-SepL (70–351) and pET-21a-SepD (6–151) were co-transformed in E. coli BL21 cells by electroporation (Yang et al., 2001 ▸). The resulting colonies were inoculated into LB medium with 50 µg ml−1 kanamycin and 100 µg ml−1 ampicillin and were grown at 37°C with shaking at 200 rev min−1 until an OD600 of 0.6 was reached. Expression was induced with 1 mM IPTG at 20°C for 18 h. The cells were harvested by centrifugation and resuspended in lysis buffer (20 mM HEPES pH 7.5, 500 mM NaCl, 50 mM imidazole). The cells were lysed using a French press and the lysates were clarified by centrifugation at 45 000 rev min−1 for 45 min. The lysate was injected onto a 1 ml HisTrap column, washed with lysis buffer and eluted with a gradient of elution buffer (20 mM HEPES pH 7.5, 500 mM NaCl, 500 mM imidazole). During this step, untagged SepD (6–151) co-purified with 10×histidine-tagged SepL (70–351). Protein-containing fractions were pooled and dialyzed overnight at 4°C into buffer (20 mM HEPES pH 7.5, 350 mM NaCl) with added thrombin to remove the polyhistidine tag. As a second purification step, the protein was injected onto a Superdex 200 10/30 column pre-equilibrated in buffer (20 mM HEPES pH 7.5, 350 mM NaCl). The SepL (70–351)–SepD (6–151) complex eluted in a single peak on gel filtration. Fractions containing SepL (70–351)–SepD (6–151) were pooled and concentrated to 20 mg ml−1.
To prepare selenomethionine-derivatized (SeMet) protein, M9 medium (supplemented with 1 mM MgSO4, 0.1 mM CaCl2, 0.01 mM FeCl3, 1 mg thiamine and 1% glucose) with 50 µg ml−1 kanamycin was inoculated with E. coli BL21 (DE3) cells harbouring pET-28-SepL (70–351), grown to an OD600 of 0.6, and 0.05 g selenomethionine was added per litre of culture. After 30 min the cells were induced with 1 mM IPTG and harvested as described above. N-terminally histidine-tagged SepD (6–151) was expressed as described for the unlabelled protein (no selenomethionine) and the cells were combined with SeMet SepL (70–351)-expressing cells. Both proteins were co-purified and concentrated as described above, including 10 mM TCEP throughout the purification.
2.2. Crystallization
Initially, crystallization conditions were screened by the vapour-diffusion method with 5 mg ml−1 SepL (70–351)–SepD (6–151) complex, and tiny needle-like crystals grew after 1 d in 3 M NaCl with 0.1 M Tris pH 8.5 buffer (Table 2 ▸). Optimization of this condition failed to produce diffraction-quality crystals. Instead, this condition was used as an additive for further screening by microbatch crystallization under oil, using paraffin oil to cover the drop and Al’s oil to cover the plate. Using 0.3 µl 20 mg ml−1 protein solution, 0.1 µl initial crystal condition (3 M NaCl, 0.1 M Tris pH 8.5) and 0.2 µl PACT condition produced cube-shaped or thick plate-like crystals in over 30% of PACT conditions after 9–25 d at room temperature. The PACT condition used in this study was 0.1 M SPG buffer pH 6, 25% PEG 1500. Crystals were cryoprotected in perfluoropolyether oil and flash-cooled in liquid nitrogen. Crystals were also grown using 3 M NaBr, 0.1 M Tris pH 8.5 as an additive for an additional phasing strategy.
Table 2. Crystallization.
| Method | Microbatch |
| Temperature (K) | 300 |
| Protein concentration (mgml1) | 20 |
| Buffer composition of protein solution | 20mM HEPES pH 7.5, 500mM NaCl |
| Composition of screen solution | 0.1M SPG buffer pH 6, 25% PEG 1500 + 0.1M Tris pH 8.5 and 3M NaCl additive |
| Volume of drop (l) | 0.6 |
2.3. Data collection and processing
Multi-wavelength anomalous diffraction data from a selenomethionine-derivative crystal and single-wavelength anomalous diffraction data from a crystal grown in the presence of NaBr were collected on beamline 5.0.2 at the Advanced Light Source at Lawrence Berkeley National Laboratory. Data sets for the NaBr-derivative and selenomethionine-derivative crystals were processed with xia2, using XDS to index all frames, XSCALE for scaling and AIMLESS for merging (Winter et al., 2013 ▸). Data-collection and processing statistics for the native SepL crystal grown in NaBr, which was subsequently refined and used in this study, are shown in Table 3 ▸. Data-collection and processing statistics for the selenomethionine-derivative crystal (which was used to obtain initial phases and was not subsequently refined) are given in Table S2.
Table 3. Data collection and processing.
Values in parentheses are for the outer shell.
| Diffraction source | Synchrotron |
| Wavelength () | 0.9202 |
| Temperature (K) | 100 |
| Space group | P43212 |
| a, b, c () | 84.32, 84.32, 238.96 |
| , , () | 90, 90, 90 |
| Resolution range () | 84.323.21 (3.433.21) |
| Total No. of reflections | 275631 (50201) |
| No. of unique reflections | 15064 (2621) |
| Completeness (%) | 100.0 (100.0) |
| Multiplicity | 18.5 (19.2) |
| I/(I) | 23.1 (4.6) |
| R merge † | 0.121 (0.823) |
| R p.i.m. ‡ | 0.028 (0.270) |
| Overall B factor from Wilson plot (2) | 81.5 |
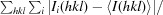
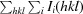
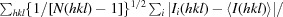
2.4. Structure solution and refinement
AutoSHARP (Vonrhein et al., 2007 ▸) was used to obtain initial phases for the selenomethionine-derivative protein, using peak and inflection data sets as the remote data set had significant radiation damage. Six selenomethiones were located in the phasing procedure. Automatic model building was performed with ARP/wARP within autoSHARP. ARP/wARP built 235 residues and assigned the majority of the sequence. The selenomethionine-derivative structure was only used to obtain initial phases and was not refined. The resulting model was used as a search model for the NaBr-derivative data by MR-SAD using Phaser in PHENIX (McCoy et al., 2007 ▸) (Table 4 ▸). Phaser located four bromides using this initial molecular-replacement model as a partial solution. Buccaneer (from the CCP4 suite) was used for model building (Winn et al., 2011 ▸), although three loop regions were deleted and manually rebuilt. Real-space refinement and rebuilding of loop regions was performed using Coot (Emsley et al., 2010 ▸). REFMAC5 was used for rigid-body refinement and a normal refinement was then performed using autoBUSTER (Bricogne et al., 2011 ▸; Emsley et al., 2010 ▸; Winn et al., 2011 ▸; Murshudov et al., 2011 ▸). BUSTER was used to give the final statistics in Table 4 ▸. Structure validation was performed using Coot and MolProbity (Emsley et al., 2010 ▸; Chen et al., 2010 ▸). Figures were generated using Chimera (Pettersen et al., 2004 ▸).
Table 4. Structure solution and refinement.
Values in parentheses are for the outer shell.
| Final R cryst † | 0.211 (0.246) |
| Final R free | 0.256 (0.347) |
| No. of non-H atoms | |
| Protein | 4271 |
| Ligand | 4 |
| Solvent | 9 |
| Total | 4283 |
| R.m.s. deviations | |
| Bonds () | 0.010 |
| Angles () | 1.250 |
| Average B factors (2) | |
| Protein | 96.3 |
| Ligand | 44.8 |
| Ramachandran plot | |
| Most favoured (%) | 95.5 |
| Allowed (%) | 4.0 |
R
cryst = 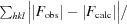
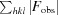 .
.
2.5. Structural solution of SepL
Initially, SepL (70–351) in complex with SepD (6–151) was set up for crystallization trials. An early hit condition that produced small needle-like crystals was subsequently used as an additive for further screening, eventually resulting in the growth of diffraction-quality crystals in numerous crystallization conditions. Data sets were collected from selenomethionine-derivative crystals, as well as native protein crystals grown in the presence of NaBr. Starting phases were solved using multi-wavelength anomalous dispersion data from the selenomethionine-derivative crystal (space group P422) collected at peak and inflection wavelengths (Supplementary Table S2). This model, which contained density for one copy of SepL in the asymmetric unit, was then used for molecular-replacement single-wavelength anomalous dispersion (MR-SAD) phasing of data collected at the bromide peak wavelength from a native protein crystal grown in NaBr. This crystal form (space group P43212) had two copies of SepL (chain A and chain B) in the asymmetric unit and diffracted to a resolution of 3.2 Å. This structure was refined and used for all further analyses. Although crystallization trials were performed with the SepL (70–351)–SepD (6–151) complex, the resulting model did not contain density for SepD, indicating that only SepL is present in the crystal lattice. Data-collection and refinement statistics are listed in Tables 3 ▸ and 4 ▸. Of note, attempts to crystallize SepL (70–351) in the absence of SepD were unsuccessful, and crystals of SepL (70–351) could only be grown by setting up crystallization trials using the complex.
3. Results and discussion
3.1. Overall structure of SepL
The model of SepL encompasses residues 80–348 and forms three X-bundle domains (Fig. 1 ▸). Chain A has missing density for residues 196–200 and chain B has missing density for residues 194–199, a region which forms a loop between α-helices 6 and 7. Chains A and B adopt a similar conformation, with a root-mean-square deviation (r.m.s.d.) of 0.461 Å over 259 aligned Cα atoms. The first domain (residues 80–172) consists of five α-helices and an extended loop region encompassing residues 103–125 (Fig. 1 ▸). The second domain (residues 173–266) and third domain (residues 267–350) also consist of five α-helices. Each domain is connected by a hinge-like loop (Fig. 2 ▸). The SepL domains do not align along the same axis, creating curvature in the overall shape of SepL (Fig. 1 ▸).
Figure 1.

Structural overview of SepL. SepL X-bundle domains are coloured blue (domain 1), orange (domain 2) and green (domain 3). α-Helices are labelled 1–15, and N- and C-termini are demarcated. The SepL surface is represented in grey. The bottom structure is a 90° rotation around the horizontal axis of the SepL structure shown above. Images were made using SepL chain A.
Figure 2.

Comparison of the overall SepL structure with homologue structures. Each of the gatekeeper structures consists of three X-bundle domains (schematic). The helices of domains 2 and 3 on the nearest face of each gatekeeper are coloured orange. The central helix (shown in red) of SepL and YopN/TyeA is broken into two helices by a hinge. SepL domains 1 and 2 are also connected by a hinge, creating more curvature in the arrangement of domains. The YopN chaperone-binding region is shown in blue. Regions of the gatekeepers proposed to interact with T3SS components are marked and labelled. PDB codes: YopN/TyeA, 1xl3, 1xkp (Schubot et al., 2005 ▸); MxiC, 2vj5 (Deane et al., 2008 ▸); CopN, 4p40 (Nawrotek et al., 2014 ▸).
3.2. Comparison of SepL to gatekeeper homologues
Homologues of SepL from Chlamydia, Shigella and Yersinia have been structurally characterized (Schubot et al., 2005 ▸; Deane et al., 2008 ▸; Nawrotek et al., 2014 ▸; Archuleta & Spiller, 2014 ▸), and a DALI search with SepL revealed these homologues to be the top-scoring structural homologues (Supplementary Table S1). Structurally characterized gatekeeper proteins all have three X-bundle domains with domains 2 and 3 connected by a long central helix (Fig. 2 ▸). The central helix of SepL is broken into two helical segments, connected by a hinge, as also seen in the central helix region of YopN/TyeA (Fig. 2 ▸). Conversely, the central helix of MxiC and CopN is rigid. Domains 1 and 2 of SepL are connected by a hinge-like loop, whereas in YopN/TyeA, MxiC and CopN domains 1 and 2 are connected by a kinked helix (Fig. 2 ▸). The hinge-like regions that connect the SepL domains create a curved appearance by offsetting domains, while MxiC and CopN appear to have a more rigid structure and a linear overall shape (Fig. 2 ▸). This difference in curvature is reflected in the relatively high r.m.s.d. values (3.4–4.8 Å) between SepL and its homologues (Supplementary Table S1). When individual domains are aligned, such as domain 3 of SepL and TyeA, the r.m.s.d. values are lower (0.9–1.6 Å). The variation in curvature of the gatekeeper structure could represent species-specific differences, or the gatekeepers may be generally flexible between domains and the differences observed between species represent ‘snapshots’ of conformations captured during crystallization.
The gatekeeper is likely to act as a scaffold, providing multiple protein–protein interaction sites on each of its three X-bundle domains. Residues or regions of the gatekeeper implicated in binding a component of the T3SS are noted in Fig. 2 ▸. SepL has been shown to interact with Tir through its C-terminal domain. The translocon chaperone Scc3 was shown to interact with two regions of CopN bridging domains 2 and 3. MxiC residue Phe206 (shown in green) located on the surface of domain 2 is required for interaction with the inner rod, and a number of residues (Glu201, Glu276 and Glu293 shown in red) form an electronegative surface on domains 2 and 3 and are required for the regulation of translocon/effector secretion hierarchy in Shigella (Cherradi et al., 2013 ▸; Roehrich et al., 2013 ▸). So far, the gatekeeper domain 1 has not been implicated in interaction with any other T3SS component. SepL, like all of the gatekeepers, has one surface that is more negatively charged than the opposite face when rotated 180° about the horizontal axis (Fig. 3 ▸). It was previously noted that this charged surface might be involved in protein–protein interactions and the mutation of MxiC residues in this region causes premature secretion (Deane et al., 2008 ▸; Roehrich et al., 2013 ▸).
Figure 3.

Surface charge of gatekeeper homologues. Electrostatic surfaces of the gatekeeper homologues were calculated using Chimera and are displayed on a red–white–blue scale (red is negatively charged, blue is positively charged). PDB codes: YopN/TyeA, 1xl3 (Schubot et al., 2005 ▸); MxiC, 2vj5 (Deane et al., 2008 ▸); CopN, 4p40 (Nawrotek et al., 2014 ▸).
While the gatekeeper proteins have a conserved X-bundle domain structure, they have low overall sequence identity (Supplementary Table S1), with only three SepL residues (Tyr188, Phe327 and Arg333) out of 280 residues absolutely conserved with the corresponding residues from CopN, YopN/TyeA and MxiC. Comparison of SepL domains 2 and 3 with the corresponding domains of CopN, YopN/TyeA and MxiC reveal three notable points of sequence conservation (Fig. 4 ▸). One region corresponds to the binding site of the translocon chaperone (as identified in the Chlamydia CopN/Scc3 structure) located at the loop region connecting SepL α14 and α15 (Fig. 4 ▸, top panel). An absolutely conserved arginine (Arg333 in SepL), required for binding of CopN to Scc3, and the neighbouring Phe327 and Asp329 are within this region. While the interaction between the Chlamydia gatekeeper CopN and the translocon chaperone Scc3 has been structurally characterized, we have yet to observe an interaction between SepL and the corresponding translocon chaperone in EPEC. An interaction between SepL and translocon-specific chaperones may not have been observed owing to a weaker affinity, or perhaps additional proteins (such as the translocon proteins) are required for the interaction to occur. Testing the effect of the mutation of the conserved SepL Arg333 could provide evidence as to whether this chaperone-binding region of CopN is functionally conserved in SepL. However, mutation of Arg333 in SepL, as well as the neighbouring Asp329, to alanine had no effect on the secretion of Tir or EspB in vivo, suggesting that this particular double mutant was not sufficient to disrupt SepL function (data not shown) and that additional mutations may be required to disrupt any putative binding interface.
Figure 4.

Structural alignment of SepL domains 2 and 3 with gatekeeper homologues. Alignment of SepL (cyan) domain 3 with the corresponding region of MxiC (purple), CopN (orange) and TyeA (yellow) is shown in the top and middle panels. Top inset: an absolutely conserved arginine required for interaction of CopN with the translocon chaperone and conserved surrounding residues are shown. Labelled residues use SepL numbering. Middle inset: conserved residues in domain 3 are shown as sticks. The bottom panel shows the alignment of SepL with domain 2 of gatekeeper homologues. Bottom inset: an absolutely conserved tyrosine and surrounding conserved residues are shown as sticks. PDB codes: YopN/TyeA, 1xl3 (Schubot et al., 2005 ▸); MxiC, 2vj5 (Deane et al., 2008 ▸); CopN, 4p40 (Nawrotek et al., 2014 ▸).
Another interesting region of sequence conservation in the third gatekeeper domain surrounds a conserved bulky hydrophobic residue (Trp288), which is located in a surface-exposed loop between α12 and α13, as well as the surface of α15 (Fig. 4 ▸, middle panel). Neighbouring conserved residues include a methionine (Met339), an asparagine (Asn335) and two negatively charged residues (Glu331 and Thr290). To our knowledge, mutation of this residue has not been tested in any SepL homologues. Owing to its proximity to the translocon chaperone-binding site in domain 3, this region may contact the translocon when it is bound to its specific chaperone.
Domain 2 has one region of sequence conservation surrounding an absolutely conserved tyrosine (Tyr188 in SepL) in α7 (Fig. 4 ▸, bottom panel). Interestingly, in MxiC, CopN and YopN/TyeA this region is located within a cleft formed between domains 1 and 2, whereas in SepL, owing to the position of domain 1 relative to domain 2, this conserved tyrosine is more accessible. Tyr188 is situated near a conserved aspartate and phenylalanine (Asp229 and Phe228), both on the same side of α9. A loop connecting α7 and α8 has two conserved negatively charged residues (Ser201 and Asp248) and α7 also has a conserved surface-exposed glutamine (Gln182).
Domain 1 has the least sequence conservation among all of the domains, and we failed to find any notable regions of conservation localizing to the protein surface. The sequence, and also the structure of domain 1, is much less well conserved than the other domains (Pallen et al., 2005 ▸), which may indicate a divergence in binding partners between species. SepD, which has been shown to interact with SepL and is critical for the regulation of hierarchy translocator and effector secretion (Deng et al., 2005 ▸), is likely to interact with domain 1 and/or 2 of SepL (Wang et al., 2008 ▸). Interestingly, if SepD does function as a chaperone, as suggested by Younis et al. (2010 ▸), it may interact with a different region of SepL than the region that corresponds to the chaperone-binding region of YopN. Characterization of the interaction of SepL and SepD by methods such as isothermal titration calorimetry has proven challenging owing to the instability of SepD in the absence of SepL.
4. Conclusions
In order to understand how the gatekeeper regulates the secretion hierarchy of translocon proteins and effectors, a more detailed knowledge of how various T3SS components bind to the scaffolding domains of the gatekeeper will be required. The challenge in characterizing these interactions is the seemingly weak affinity of the gatekeeper for its binding partners. In Chlamydia (but not in other species), interaction between the gatekeeper and translocon chaperone can be observed between the purified proteins, perhaps owing to an extended region of the translocon chaperone Scc3 that provides an additional surface area for interaction that is not present in homologues (Archuleta & Spiller, 2014 ▸). In Salmonella, the gatekeeper only binds a chaperone–translocon complex and not empty chaperone (Kubori & Galán, 2002 ▸). It is likely that the interactions between the gatekeeper and its binding partners are weak and transient, as they must assemble and disassemble prior to secretion. Alternatively, interaction may require multiple binding partners, such as is the case in Salmonella (Kubori & Galán, 2002 ▸). It is likely that characterization of SepL binding interactions will require further structure-based mutational analysis to test the effect on function, as identification of interactions between purified recombinant proteins have proven difficult, perhaps owing to weak interactions or the requirement of additional unknown partners.
In this study, we have structurally characterized SepL, the T3SS gatekeeper protein from EPEC. Structure-based alignment of SepL with its homologues CopN, MxiC and YopN/TyeA reveals three regions of sequence conservation across domains 2 and 3. Deletion of the gatekeeper has varying phenotypes between species. In EPEC the phenotype for ΔsepL or ΔsepD mutants is very striking, with abrogated secretion of translocator proteins (EspA, EspB and EspD) and increased secretion of effector proteins (Deng et al., 2005 ▸). Because of the clear phenotype of the EPEC ΔSepL mutant, structure-based mutational analysis of SepL could potentially help to determine which domains are required to prevent premature secretion of effectors, to facilitate translocon secretion and to interact with SepD and CesL.
Supplementary Material
PDB reference: SepL, 5c9e
Supporting Information.. DOI: 10.1107/S2053230X15016064/ub5079sup1.pdf
Acknowledgments
We thank the beamline staff at the Advanced Light Source beamline 5.0.2 at Lawrence Berkeley National Laboratory for their assistance. We thank Wanyin Deng for insight into SepL and preliminary secretion assays. BB was funded by a University of British Columbia Centre for Blood Research graduate student fellowship. SS was funded by the Science without Borders program, a joint effort of the Brazilian Ministry of Education and the Ministry of Science and Technology. NCJS is a Canada Research Chair in Antibiotic Discovery. This work was funded by a CIHR operating grant and operating funding from HHMI International Scholar program awarded to NCJS.
References
- Archuleta, T. L., Du, Y., English, C. A., Lory, S., Lesser, C., Ohi, M. D., Ohi, R. & Spiller, B. W. (2011). J. Biol. Chem. 286, 33992–33998. [DOI] [PMC free article] [PubMed]
- Archuleta, T. L. & Spiller, B. W. (2014). PLoS Pathog. 10, e1004498. [DOI] [PMC free article] [PubMed]
- Arndt, U. W., Crowther, R. A. & Mallett, J. F. (1968). J. Phys. E Sci. Instrum. 1, 510–516. [DOI] [PubMed]
- Botteaux, A., Sory, M. P., Biskri, L., Parsot, C. & Allaoui, A. (2009). Mol. Microbiol. 71, 449–460. [DOI] [PubMed]
- Bricogne, G., Blanc, E., Brandl, M., Flensburg, C., Keller, P., Paciorek, W., Roversi, P., Sharff, A., Smart, O. S., Vonrhein, C. & Womack, T. O. (2011). BUSTER v.2.10.0. Cambridge: Global Phasing Ltd.
- Chatterjee, S., Chaudhury, S., McShan, A. C., Kaur, K. & De Guzman, R. N. (2013). Biochemistry, 52, 2508–2517. [DOI] [PMC free article] [PubMed]
- Chen, G.-J., Qiu, N., Karrer, C., Caspers, P. & Page, M. G. (2000). Biotechniques, 28, 498–505. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cherradi, Y., Schiavolin, L., Moussa, S., Meghraoui, A., Meksem, A., Biskri, L., Azarkan, M., Allaoui, A. & Botteaux, A. (2013). Mol. Microbiol. 87, 1183–1199. [DOI] [PubMed]
- Day, J. B., Ferracci, F. & Plano, G. V. (2003). Mol. Microbiol. 47, 807–823. [DOI] [PubMed]
- Deane, J. E., Roversi, P., King, C., Johnson, S. & Lea, S. M. (2008). J. Mol. Biol. 377, 985–992. [DOI] [PMC free article] [PubMed]
- Deng, W., Li, Y., Hardwidge, P. R., Frey, E. A., Pfuetzner, R. A., Lee, S., Gruenheid, S., Strynakda, N. C. J., Puente, J. L. & Finlay, B. B. (2005). Infect. Immun. 73, 2135–2146. [DOI] [PMC free article] [PubMed]
- Deng, W., Puente, J. L., Gruenheid, S., Li, Y., Vallance, B. A., Vázquez, A., Barba, J., Ibarra, J. A., O’Donnell, P., Metalnikov, P., Ashman, K., Lee, S., Goode, D., Pawson, T. & Finlay, B. B. (2004). Proc. Natl Acad. Sci. USA, 101, 3597–3602. [DOI] [PMC free article] [PubMed]
- Diepold, A. & Wagner, S. (2014). FEMS Microbiol. Rev. 38, 802–822. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Ferracci, F., Schubot, F. D., Waugh, D. S. & Plano, G. V. (2005). Mol. Microbiol. 57, 970–987. [DOI] [PubMed]
- Fields, K. A. & Hackstadt, T. (2000). Mol. Microbiol. 38, 1048–1060. [DOI] [PubMed]
- Journet, L., Agrain, C., Broz, P. & Cornelis, G. R. (2003). Science, 302, 1757–1760. [DOI] [PubMed]
- Kubori, T. & Galán, J. E. (2002). J. Bacteriol. 184, 4699–4708. [DOI] [PMC free article] [PubMed]
- Marlovits, T. C., Kubori, T., Sukhan, A., Thomas, D. R., Galan, J. E. & Unger, V. M. (2004). Science, 306, 1040–1042. [DOI] [PMC free article] [PubMed]
- Martinez-Argudo, I. & Blocker, A. J. (2010). Mol. Microbiol. 78, 1365–1378. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Mota, L. J., Journet, L., Sorg, I., Agrain, C. & Cornelis, G. R. (2005). Science, 307, 1278. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nawrotek, A., Guimarães, B. G., Velours, C., Subtil, A., Knossow, M. & Gigant, B. (2014). J. Biol. Chem. 289, 25199–25210. [DOI] [PMC free article] [PubMed]
- O’Connell, C. B., Creasey, E. A., Knutton, S., Elliott, S., Crowther, L. J., Luo, W., Albert, M. J., Kaper, J. B., Frankel, G. & Donnenberg, M. S. (2004). Mol. Microbiol. 52, 1613–1625. [DOI] [PubMed]
- Pallen, M. J., Beatson, S. A. & Bailey, C. M. (2005). BMC Microbiol. 5, 9. [DOI] [PMC free article] [PubMed]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed]
- Raymond, B., Young, J. C., Pallett, M., Endres, R. G., Clements, A. & Frankel, G. (2013). Trends Microbiol. 21, 430–441. [DOI] [PubMed]
- Roehrich, A. D., Guillossou, E., Blocker, A. J. & Martinez-Argudo, I. (2013). Mol. Microbiol. 87, 690–706. [DOI] [PMC free article] [PubMed]
- Sal-Man, N., Setiaputra, D., Scholz, R., Deng, W., Yu, A. C. Y., Strynadka, N. C. J. & Finlay, B. B. (2013). J Bacteriol. 195, 2481–2489. [DOI] [PMC free article] [PubMed]
- Schubot, F. D., Jackson, M. W., Penrose, K. J., Cherry, S., Tropea, J. E., Plano, G. V. & Waugh, D. S. (2005). J. Mol. Biol. 346, 1147–1161. [DOI] [PubMed]
- Slabinski, L., Jaroszewski, L., Rychlewski, L., Wilson, I. A., Lesley, S. A. & Godzik, A. (2007). Bioinformatics, 23, 3403–3405. [DOI] [PubMed]
- Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. (2007). Methods Mol. Biol. 364, 215–230. [DOI] [PubMed]
- Wang, D., Roe, A. J., McAteer, S., Shipston, M. J. & Gally, D. L. (2008). Mol. Microbiol. 69, 1499–1512. [DOI] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst. 34, 130–135.
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Winter, G., Lobley, C. M. C. & Prince, S. M. (2013). Acta Cryst. D69, 1260–1273. [DOI] [PMC free article] [PubMed]
- Worrall, L. J., Lameignere, E. & Strynadka, N. C. J. (2011). Curr. Opin. Microbiol. 14, 3–8. [DOI] [PubMed]
- Yang, W., Zhang, L., Lu, Z., Tao, W. & Zhai, Z. (2001). Protein Expr. Purif. 22, 472–478. [DOI] [PubMed]
- Younis, R., Bingle, L. E., Rollauer, S., Munera, D., Busby, S. J., Johnson, S., Deane, J. E., Lea, S. M., Frankel, G. & Pallen, M. J. (2010). J. Bacteriol. 192, 6093–6098. [DOI] [PMC free article] [PubMed]
- Zarivach, R., Deng, W., Vuckovic, M., Felise, H. B., Nguyen, H. V., Miller, S. I., Finlay, B. B. & Strynadka, N. C. J. (2008). Nature (London), 453, 124–127. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: SepL, 5c9e
Supporting Information.. DOI: 10.1107/S2053230X15016064/ub5079sup1.pdf