

Abstract
Synucleinopathies, such as Parkinson’s disease and diffuse Lewy body disease, are progressive neurodegenerative disorders characterized by selective neuronal death, abnormal accumulation of misfolded α-synuclein, and sustained microglial activation. In addition to inducing neuronal toxicity, higher-ordered oligomeric α-synuclein causes proinflammatory responses in the brain parenchyma by triggering microglial activation, which may exacerbate pathogenic processes by establishing a chronic neuroinflammatory milieu. Here, we found that higher-ordered oligomeric α-synuclein induced a proinflammatory microglial phenotype by directly engaging the heterodimer TLR1/2 (Toll-like receptor 1 and 2) at the cell membrane, leading to the nuclear translocation of NF-κB (nuclear factor κB) and the increased production of the proinflammatory cytokines TNF-α and IL-1β in a MyD88-dependent manner. Blocking signaling by the TLR1/2 heterodimer with the small molecule inhibitor, CU-CPT22, reduced the expression and secretion of these inflammatory cytokines from cultured primary mouse microglia. Candesartan cilexetil, a drug approved for treating hypertension and that inhibits the expression of TLR2, reversed the activated proinflammatory phenotype of primary microglia exposed to oligomeric α-synuclein, supporting the possibility of repurposing this drug for synucleinopathies.
Introduction
Synucleinopathies, such as Parkinson’s disease and diffuse Lewy body disease, are progressive neurodegenerative disorders characterized by the loss of selective neurons and the accumulation of misfolded α-synuclein into hallmark pathological lesions, Lewy bodies and Lewy neurites. In addition to being the major constituent of Lewy bodies (1), α-synuclein is implicated in disease etiology since point mutations and overexpression of the gene encoding α-synuclein, SNCA, are associated with familial forms of Parkinson’s disease (2–6). Moreover, genome-wide association studies have linked SNCA polymorphisms with an increased risk of developing idiopathic Parkinson’s disease (7–9). Together, these data point to a central role for α-synuclein in disease pathophysiology.
Accumulating evidence from animal models, along with biochemical and biophysical studies, support the hypothesis that a key event in the pathogenesis of synucleinopathies is the process by which monomeric α-synuclein misfolds and self-assembles into oligomeric α-synuclein via a nucleated polymerization mechanism (10–16). Importantly, oligomeric α-synuclein has been shown to be cytotoxic, inciting neurodegeneration by disrupting proteosomal, lysosomal, and mitochondrial functions, while also increasing cell membrane conductance (17–21). Evidence also demonstrates that under pathological conditions, oligomeric α-synuclein can be released from neurons through non-classical exocytosis, enabling α-synuclein to propagate to neighboring neurons and glia, inducing inclusion-body formation, neuronal death, and neuroinflammation (22–33).
The present study focuses on this latter mechanism of inflammation, because the role of the innate immune response in the neurodegenerative processes underlying synucleinopathies and other diseases of the central nervous system has become increasingly evident (12, 34–38). Specifically, Parkinson’s disease patients demonstrate a marked increase in activated microglia (39–42) with increased expression and concentration of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-1β (IL-1β) in the substantia nigra pars compacta (SNpc), striatum, and cerebrospinal fluid as compared to control patients (43–47). In addition, α-synuclein leads to increased numbers of activated microglia in mouse models of protein overexpression prior to SNpc dopaminergic neuronal death and causes proinflammatory microglial activation in cell culture experiments (38, 48–55). Therefore, these observations suggest a close pathophysiological relationship between disease-associated α-synuclein and microglia-mediated neuroinflammation.
As the main contributors to inflammation within the brain parenchyma, microglia can be activated by engagement of membrane-bound pattern recognition receptors, such as toll-like receptors (TLRs), which respond to both pathogen-associated molecular patterns and danger or damage-associated molecular patterns (DAMPs) (56–62). The role of TLRs as modulators of neurological disorders has become more apparent; for example, TLR2 and TLR4 exacerbate tissue damage in animal models of stroke, and mediate the extracellular clearance of amyloid β (Aβ) peptide and Aβ-induced microglial activation (63–66). Linking TLRs with synucleinopathies, we previously showed that microglia exposed to misfolded α-synuclein upregulate the expression of genes encoding TLRs and the proinflammatory molecules TNF-α and IL-1β while undergoing morphological changes indicative of classical activation (48–50). Studies using cell culture and animal models have shown conflicting results regarding the requirement of TLRs in microglial activation in response to α-synuclein (55, 67). The discrepancy regarding the signaling mechanism represents the complexity of α-synuclein-mediated microglial activation, and elucidation of the intracellular molecular players involved in α-synuclein-mediated neuroinflammation enhances the probability of ameliorating disease progression.
In this study, we sought to identify the molecular mechanisms involved in α-synuclein-dependent microglial activation using mouse primary microglia, and we examined the possibility of using this knowledge to treat synucleinopathies.
Results
Misfolding of human α-synuclein produces different protein structures
To interrogate the molecular underpinnings of α-synuclein-mediated microglial activation, we separated misfolded human wild-type α-synuclein (SynTR) into higher-ordered oligomeric (SynO) and small oligomeric/monomeric (SynM) species using size-exclusion centrifugation. Resolution of these species using Western blot analysis under non-denaturing conditions confirmed the effective separation and enrichment of SynOand small SynM (Fig. 1A). Structural analysis of SynO and SynM using transmission electron microscopy revealed the presence of fibrils in SynO fractions, whereas fibrils were absent in SynM fractions (Fig. 1B).
Figure 1. Higher-ordered oligomeric α-synuclein induces a complex morphofunctional activation of microglia.

(A) Representative Western blot analysis of misfolded human α-synuclein (Syn) under non-denaturing conditions. Purified, recombinant Syn was misfolded (SynTR), and subsequently separated to isolate higher-ordered oligomeric conformers (SynO; bracket) from the monomeric and dimeric structures (SynM; arrowhead). (B) Representative transmission electron microscopy of SynO and SynM at 75,000× magnification. Fibrils are present in the SynO fraction (black arrows). Scale bar: 100 nm. (C) Immunohistochemistry for Iba-1 in microglia after a 24-hour exposure to SynM or SynO (80.0 ng/mL). Scale bar: 20 µm. (D) qRT-PCR for TNF-α and IL-1β expression in microglia after exposure to SynM or SynO (80.0 ng/mL). (E) ELISA for the concentration of TNF-α and IL-1β in the conditioned medium from microglia cultured for 2 and 24 hours with SynO or SynM (80.0 ng/mL). (F) qRT-PCR analysis for ARG-1 and IL-10 gene expression in microglia cultured with SynO or SynM (0.95 µg/mL) for 24 hours. Black dashed line: gene expression in vehicle control. (G) ELISA for the concentration of IL-10 in the supernatant of microglia cultured with either SynM or SynO (80 ng/mL) for 2 and 24 hours. All data are means ± SEM from 3 experiments. *P=0.01, ***P<0.0001. In (E), $$$P<0.0001 comparing 2- to 24-hour SynO data. Data were analyzed by unpaired Student’s t-test (D) or one-way ANOVA with Bonferroni post-hoc test (E to G). N.D., not detectable.
Oligomeric α-synuclein induces a complex activation phenotype in microglia
To explore the differential effects of the α-synuclein conformers on microglial activation, we exposed primary microglia to either SynM or SynO. We first characterized microglia morphology after exposure to these α-synuclein species by immunocytochemical staining for Iba-1, a microglia-specific calcium-binding protein. Microglia exposed to SynM retained a bipolar morphology, whereas microglia exposed to higher-ordered oligomers of Syn (SynO) undertook an amoeboid, phagocytic morphology indicative of classical activation (Fig. 1C). Furthermore, this SynO-induced morphological change was concurrent with a biochemical activation profile, such that these microglia had increased mRNA abundance and protein secretion of the proinflammatory cytokines TNF-α and IL-1β (Fig. 1, D and E). In contrast, the monomeric form of Syn had no effect on these proinflammatory molecules. We next probed for changes in the expression of genes encoding the immunoresolution or alternative activation molecules, arginase 1 and IL-10. The gene expression of these molecules was unchanged in the SynM-exposed microglia, but was increased upon exposure to higher-ordered oligomeric α-synuclein (SynO; Fig. 1F). Furthermore, using an ELISA specific for IL-10, SynO-exposed microglia demonstrated a significant increase in the secretion of this cytokine (Fig. 1G). However, unlike the robust upregulation of the proinflammatory cytokine, TNF-α, which was detectable after 2- and 24-hour exposures to SynO (Fig. 1E), IL-10 was detectable only after the longer exposure time. These data suggest a temporally distinct activation profile in response to SynO, consisting of an initial proinflammatory phase, followed by an upregulation of anti-inflammatory factors.
Because the expression of genes encoding TNF-α, IL-1β, arginase 1, and IL-10 are regulated in part by the transcription factor NF-κB (nuclear factor κB), we next interrogated whether SynO exposure causes nuclear translocation of the active p65 subunit of this sequence-specific DNA-binding protein. From our initial exploratory assays (fig. S1), we focused on the 2-hour time point to assess NF-κB translocation. Microglia exposed to SynO demonstrate a robust nuclear translocation of the NF-κB p65 subunit compared to cells exposed to SynM (Fig. 2A). Quantification of nuclear NF-κB p65 signal intensity by immunostaining demonstrated a significant translocation of p65 in the presence of SynO compared with SynM (Fig. 2B). We next used an ELISA-based activity assay to determine whether this nuclear p65 binds to its consensus DNA sequence, indicative of activity. Nuclear extracts from microglia exposed to SynO exhibited an increase in nuclear NF-κB activity compared with SynM-exposed cells (Fig. 2C). Together, these data suggest that the higher-ordered oligomeric form of α-synuclein incites a complex inflammatory activation profile that may be mediated through the NF-κB transcription pathway.
Figure 2. Higher-ordered oligomeric α-synuclein induces nuclear translocation of active NF-κB.

(A) Immunoctyochemistry for the p65 subunit of NF-κB in primary microglia after a 2-hour incubation with α-synuclein [SynO (arrows) or SynM (arrowheads); 80.0 ng/mL]. Scale bars: 20 µm. (B) Quantification of p65 nuclear signal intensity in (A). Data are means ± SEM from 3 independent replicates of 33 nuclei per condition. (C) Quantification of active nuclear p65 NF-κB. SynO exposure increases active nuclear p65 NF-κB in comparison to SynM. Data are means ± SEM from 3 experiments. ***P≤0.0001 comparing SynO to SynM, analyzed via unpaired Student’s t-test.
Oligomeric α-synuclein localizes to the microglial cell surface and signals through a MyD88-dependent pathway
Having established that higher-ordered oligomeric α-synuclein induces inflammatory activity in microglia and NF-κB nuclear translocation, we next probed the signaling mechanism mediating the observed proinflammatory response. Our data thus far supported a possible role for NF-κB-mediated transcriptional regulation, and it is well-established that this DNA binding protein is activated through microglial surface receptor signaling. Further linking surface receptors to α-synuclein-specific microglial activation, we previously report that microglia stimulated with misfolded α-synuclein containing higher-ordered oligomeric, small oligomeric, and monomeric conformers increase the expression of genes encoding the toll-like receptors (TLRs) and the TLR adaptor protein, MyD88 (50), encoding myeloid differentiation primary response gene 88. Together these data suggest that α-synuclein may initiate microglia activation at the cell surface by engaging TLRs.
First, to interrogate whether SynM and SynO localized to the microglial cell surface, microglia were exposed to the two α-synuclein species followed by biotinylation of cell surface proteins. Biotinylated proteins were subsequently isolated using an avidin-binding column and subjected to Western blot analyses under non-denaturing conditions. Successful isolation of cell membrane proteins was confirmed by probing for the highly expressed microglia surface receptors, TLR1 and TLR2 in all treatment groups (Fig 3, A and B). Probing for α-synuclein revealed that only the higher-ordered oligomeric form of α-synuclein interacted with the microglia membrane (Fig. 3C).
Figure 3. Higher-ordered oligomeric α-synuclein localizes to the surface of microglia and mediates MyD88-dependent signaling.

(A to C) Non-denaturing Western blot analyses of biotinylated surface TLR1 (A) and TLR2 (B) (arrowheads), and human α-synuclein (C) (bracket) from microglia exposed to SynM or SynO (0.8 µg/mL, 30 min). Blots are representative of 3 experiments. (D) Peptide inhibition of MyD88 homodimerization. SynO-induced TNF-α and IL-1β expression in microglia treated with either control peptide (CTLpep) or inhibitory MyD88 peptide (MyD88pep) for 30 min before being exposed to SynO (80.0 ng/mL, 2 hours). (E) ELISA for the concentration of TNF-α in the conditioned media from (D). Data are means ± SEM from 3 experiments. **P≤0.005 by unpaired Student’s t-test.
Our data support that higher-ordered oligomeric α-synuclein acts at the microglia cell surface and possibly in an NF-κB-dependent pathway suggestive of TLR engagement. Given that a subset of TLRs requires the homodimerization of MyD88 to promote nuclear translocation of NF-κB, we used a MyD88-peptide inhibitor to determine whether MyD88-dependent pathways are relevant to SynO-mediated microglial activation. We pretreated microglia with a control peptide (CTLpep) or an inhibitory peptide against MyD88 homodimerization (MyD88pep), and subsequently exposed the cells to SynO. Using quantitative real-time polymerase chain reaction (qRT-PCR), we found a significant reduction in TNF-α and IL-1β gene expression in cells treated with the MyD88 inhibitor compared to those treated with the control peptide (Fig. 3D). Moreover, in parallel with reduced proinflammatory gene expression, inhibition of MyD88 signaling decreased the amount of TNF-α released from microglia in response to stimulation with SynO (Fig. 3E). These data suggest that the higher-ordered oligomeric α-synuclein-induced proinflammatory response in microglia acts at the cell membrane through a MyD88-dependent mechanism.
Oligomeric α-synuclein interacts with TLR1/2
Thus far we have established that α-synuclein-induced microglial activation is dependent on both the higher-ordered oligomeric structure of α-synuclein and the homodimerization of MyD88, a critical adaptor protein for pattern recognition receptor signaling. Because our previous studies found that microglial-like murine cells (BV2 cells) and primary microglia have increased gene expression of TLR1, 2, and 3 upon exposure to misfolded α-synuclein (48, 50), we next inquired whether our isolated forms of α-synuclein induced differential expression of these pattern recognition receptors. We exposed microglia to oligomeric or monomeric α-synuclein for 24 hours and quantified the expression of these pattern recognition receptors. There was a significant increase in TLR1, TLR2, and TLR3 gene expression in microglia exposed to SynO compared to that in either SynM or vehicle conditions, with TLR 1 demonstrating the greatest increase in expression (Fig. 4A). There were no significant differences between SynM and vehicle-treated conditions for all gene expression measured.
Figure 4. SynO increases microglial TLR expression and interacts with hTLR2 and TLR1.

(A) qRT-PCR for TLR expression after a 24-hour exposure to SynO (80.0 ng/mL). Dashed line indicates vehicle gene expression level. Data are means ± SEM (n=2). (B) SEAP activity in HEK-hTLR cells. HEK-hTLR cells were exposed to vehicle, the TLR1/2 agonist Pam (1 µg/mL), the TLR3 agonist poly(I:C) [P(I:C); 1 µg/mL], SynM, or SynO (80.0 ng/mL/Syn treatment) for 20-hours followed by SEAP quantification. Data are means ± SEM (n=3). (C) Representative PLA images of microglia after exposure to vehicle, SynM, or SynO (0.8 µg/mL) for 10 minutes with probes against human Syn (LB509) and TLR1 antibodies (white dots indicate interaction). Nuclei were counterstained with DAPI. Insets represent enlarged image of one cell. Scale bar: 20 µm. (D) Quantification of SynO/TLR1 interaction. Dashed line represents vehicle signal intensity; normalized signal intensity determined from 4 random fields of view across two separate experiments. (A) **P=0.008, ***P<0.0001 comparing SynM and SynO; $$P=0.008, $$$P<0.0001 comparing vehicle and SynO; (B) ***P<0.0001; (D) ***P<0.0001 comparing SynM and SynO; $$$P<0.0001 comparing vehicle and SynO by one-way ANOVA with Bonferroni post-hoc test. For all data there was no significant difference between SynM and vehicle control.
Given both this robust increase in TLR1 expression and that TLR1 heterodimerizes with TLR2 to initiate MyD88-depedendent intracellular signaling, we used a secreted embryonic alkaline phosphatase (SEAP) reporter system in human embryonic kidney (HEK) 293 cells that overexpresses human TLR2 and expresses endogenous amounts of TLR1 (HEK-hTLR2) to determine whether α-synuclein directly interacted with these pattern-recognition receptors. We first established that HEK-hTLR2 reporter cells responded to a specific TLR1/2 agonist, Pam3CSK4, (Pam). HEK-hTLR2 cells exposed to Pam showed a robust increase in SEAP reporter activity (Fig. 4B), indicating that these cells are capable of forming and signaling through the TLR1/2 receptor heterodimer. We then used this system to test whether α-synuclein signaled through this same pathway. SynO-exposure induced TLR2-SEAP activity, whereas SynM had no detectable effect (Fig. 4B). Furthermore, because TLR3 mRNA abundance was also increased in microglia upon exposure to SynO, we used a similar SEAP reporter system, but with cells overexpressing human TLR3 (HEK-hTLR3) to investigate whether SynO engaged TLR3 homodimers. Only the TLR3-specific agonist, polyinosinic:polycytidylic acid (poly I:C), increased SEAP activity (Fig. 4B), indicating that neither form of α-synuclein directly interacted with TLR3, a MyD88-independent receptor.
To confirm that higher-ordered oligomeric α-synuclein interacts with TLR1 in primary microglia, we performed an in situ proximity ligation assay (PLA) using an antibody specific for human synuclein (LB509) in conjunction with a TLR1 antibody after microglial exposure to vehicle, SynM, or SynO. Confocal microscopy of microglia after in situ PLA revealed numerous punctate signals after SynO exposure, indicative of a robust interaction between SynO and TLR1 (Fig. 4C). In contrast, vehicle- and SynM-treated conditions elicited no appreciable interaction between α-synuclein and TLR1 (Fig. 4C). Quantification of the PLA further supported a significant interaction between the higher-ordered oligomeric form and TLR1 (Fig. 4D). Together these data show that α-synuclein-mediated microglial activation is specific to the higher-ordered oligomeric form of α-synuclein, which can directly signal through TLR1/2 heterodimers in a MyD88-dependent mechanism.
Pharmacological inhibition of TLR1/2 attenuates oligomeric α-synuclein-mediated effects
Because SynO directly interacted with the TLR1/2 receptor complex, we next asked whether blocking this interaction diminishes the higher-ordered oligomeric α-synuclein-mediated proinflammatory response. We previously developed a novel small molecule antagonist of the TLR1/2 complex, CU-CPT22 (68). To test the effectiveness of this compound in blocking the SynO-TLR1/2 interaction we used the HEK-hTLR2 reporter cells, which (as shown above) responds to the TLR1/2 activator, Pam, and SynO. Incubation of these reporter cells with CU-CPT22 and either Pam or SynO caused a significant decrease in reporter activity (Fig. 5A), further supporting the hypothesis that SynO can signal through the TLR1/2 complex.
Figure 5. CU-CPT22 attenuates SynO-mediated signaling and activation.

(A) SEAP activity of HEK-hTLR2 cells (n=3) following simultaneous exposure to Pam (1 ng/mL) or SynO (8.0 ng/mL) and CU-CPT22 or vehicle. Data are means ± SEM (n=3); ***P<0.0001 comparing CU-CPT22 condition to vehicle condition for Pam and SynO analyzed by unpaired Student’s t-test. (B) Nuclear NF-κB translocation following treatment of microglia with SynO (8.0 ng/mL) and vehicle (white arrows) or CU-CPT22 (white arrow heads). Scale bar: 20 µm. (C) Quantification of the change in nuclear NF-κB p65 shown in panel (B). (D) ELISA for the concentration of TNF-α in conditioned medium from microglia simultaneously treated with SynO (80.0 ng/mL; 72 hours) and vehicle or CU-CPT22 (10 µM). Data are means ± SEM (n=3); *P<0.01 by one-way ANOVA with Bonferroni post-hoc test; N.D., not detectable.
We next sought to translate these results to our primary cell culture model by exposing microglia to SynO while simultaneously inhibiting TLR1/2. To isolate the effect of TLR1/2 signaling on nuclear NF-κB translocation after SynO exposure, we treated primary microglia with CU-CPT22, the TLR1/2 antagonist, and simultaneously exposed the cells to SynO. Immunocytochemical analysis of NF-κB confirmed that SynO enhanced the nuclear translocation of p65 (Fig. 5B) but that blocking TLR1/2 with CU-CPT22 significantly decreased this effect (Fig. 5, B and C). Treatment with CU-CPT22 also significantly decreased the SynO-mediated release of TNF-α (Fig. 5D). Together, these data support that TLR1/2 engagement is important for oligomeric synuclein-induced microglial activation.
We tested a second pharmacologic agent, candesartan cilexetil (candesartan), for its ability to attenuate synuclein-mediated activation of microglia. Candesartan is an angiotensin II receptor blocker (ARB) that also inhibits TLR2 receptor expression and activity in both in vitro and in vivo models of inflammation (69). Given its reported effect on the expression of the gene encoding TLR2, and due to the ability of higher-ordered oligomeric α-synuclein to activate the TLR1/2 complex, we reasoned that candesartan might prove effective in dampening SynO-mediated microglial activation. To test this hypothesis, we pretreated microglia with increasing concentrations of candesartan and subsequently exposed these cells to higher-ordered oligomeric α-synuclein. We observed a marked morphological and functional change in candesartan-treated microglia (Fig. 6). Specifically, increasing concentrations of candesartan more potently reversed the prototypical ambeoid morphology indicative of classically activated microglia (Fig. 6A) and significantly decreased SynO-induced TNF-α secretion (Fig. 6B). These data show that this FDA-approved drug attenuates the SynO-induced microglial proinflammatory phenotype, and that TLR1/2 is a potential target to suppress synuclein-mediated glial activation.
Figure 6. Candesartan diminishes SynO-mediated morphofunctional responses in microglia.
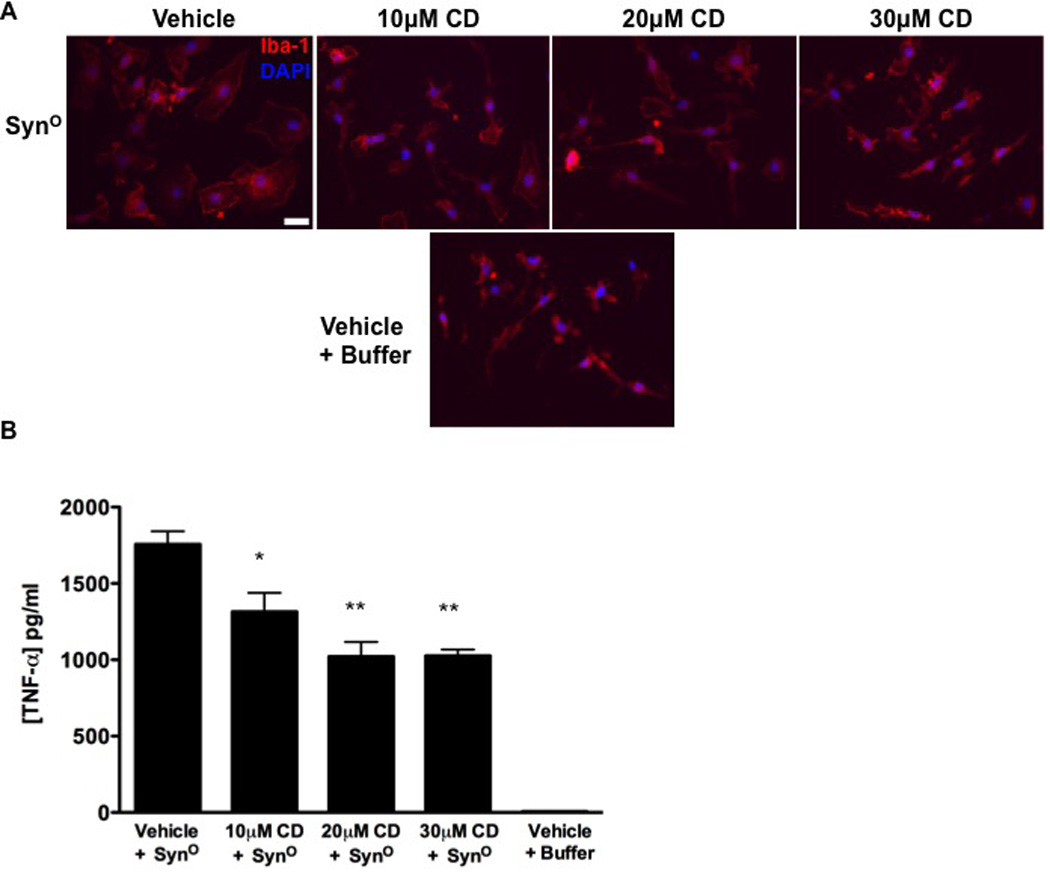
(A) Iba-1 immunocytochemistry (red) of microglia pretreated with 10, 20, or 30 µM of candesartan cilexetil (CD) or vehicle (equivalent to 30 µM CD) for 2 hours and subsequently exposed to SynO (8.0 ng/mL) or buffer for 12 hours. Cell nuclei were stained with DAPI (blue). Scale bar: 20 µm. (B) ELISA for the concentration of TNF-α in conditioned medium of microglia treated as in (A). Data are means ± SEM (n=3); *P=0.01, **P=0.001; comparing 10, 20, or 30 µM conditions to Vehicle + SynO by via one-way ANOVA with Bonferonni post-hoc test.
Discussion
We previously showed that higher-ordered oligomeric α-synuclein causes a complex morphofunctional response in microglia (48–50). Interestingly, this response is conformation-specific, because monomeric or smaller oligomeric species of α-synuclein do not incite a similar activation profile. Here we identified the mechanism by which higher-ordered oligomeric α-synuclein induces microglial activation through a MyD88-dependent TLR1/2 pathway. Together, our findings suggest that this pattern recognition receptor complex is a druggable target to treat neuroinflammation in synucleinopathies.
The idea that higher-ordered α-synuclein acts as a DAMP promoting neuroinflammation has implications for disease pathogenesis. In support of this, studies show oligomeric α-synuclein is released from neurons and subsequently propagates to neighboring neurons and glia (26, 28–33, 70). Human pathology studies also support the concept that oligomeric α-synuclein spreads throughout the nervous system with evidence for α-synuclein pathology in the gut, olfactory system, brain stem, midbrain and cortical regions; however, there has been debate as to the exact mechanism of this “prion-like” pathology (71–78). Injection of pre-formed recombinant human α-synuclein fibrils into mice results in a spreading of Lewy body-like pathology, lending further support to the propagation theory for synucleinopathies (79–84). Likewise, the injection of Lewy body extracts from the brains of Parkinson’s disease patients into those of either mice or rhesus monkeys demonstrates the ability of this misfolded protein to incite disease pathology (85). Collectively these studies support a role for oligomeric α-synuclein in Parkinson’s disease pathogenesis.
The present work supports a role for higher-ordered oligomeric α-synuclein in the initiation of neuroinflammation, a common feature of synucleinopathies (86–89). Although we are unable to pinpoint the exact molecular structure of the inflammation-mediating α-synuclein, we have demonstrated that only the higher-ordered oligomeric fibrillar conformers (>720 kD) cause a robust inflammatory response; the smaller oligomeric and monomeric forms of this protein do not incite a measurable response in microglia. It would be useful in future studies to further classify the oligomeric α-synuclein subspecies, because this information could be used for the development of additional drug therapies. Furthermore, work by others suggests that not all oligomeric species are toxic to cells, because only higher-ordered oligomers cause a microglial response [reviewed in (16)], and here we support this hypothesis.
As the innate immune cells of the central nervous system, microglia work to maintain homeostasis, responding to changes in the microenvironment with complex and variable immunofunctional profiles. However, activation states are not simply categorical, as microglial cells can adopt a mixed phenotype within the spectrum of “resting” to “fully activated.” In fact, here we demonstrated that in a nearly pure population of cultured microglia there is evidence for both “classically” activated (such as TNF-α and IL-1β abundance and release) and “immune-resolution” or “alternatively” activated phenotypes [such as IL-10 and arginase 1 abundance; reviewed in (90)]. Previous studies using recombinant human α-synuclein report detectable mRNA expression of proinflammatory molecules in microglia 45 min after exposure (52). Herein we captured early and late mRNA expression changes and found that proinflammatory TNF-α was detectable as early as 2 hours after exposure to higher-ordered oligomeric α-synuclein, whereas proinflammatory IL-1β as well as anti-inflammatory IL-10 were not evident until 24 hours after. Previous studies show that microglial IL-10 abundance is upregulated during proinflammatory events and subsequently inhibits the expression of genes encoding cytokines (such as TNF-α) and cytokine receptors (91). Thus, our results suggest that IL-10 expression is increased in response to microglial activation in an attempt to maintain homeostasis.
The signaling pathway leading to these varied activation states are controlled by the engagement of surface receptors by specific ligands (59, 60, 92–96). Our findings suggest that higher-ordered oligomeric α-synuclein is a DAMP that interacts with a specific TLR complex. Although our work substantiates a role for TLR1/2 in Parkinson’s disease neuroinflammation, we recognize that other pattern recognition and surface receptors also play a role in synucleinopathies. Previous work suggests that CD36 is important for α-synuclein-mediated microglial activation (52). However, that study did not isolate different α-synuclein structures, and primary microglia from CD36 knockout mice did not completely abrogate the synuclein-mediated effect, suggesting that multiple receptors could be important for microglial activation or that the response is α-synuclein structure-specific. Others have implicated TLR4 in Parkinson’s disease: Fellner et al. (55) demonstrate that microglial phagocytosis, NF-κB nuclear translocation, release of proinflammatory molecules, and nitric oxide production all decrease after α-synuclein exposure in microglial cells isolated from TLR4 knockout mice. In a mouse model of α-synuclein overexpression (Thy1-aSyn mice), the expression of TLR1, TLR4 and TLR8 in the substantia nigra are upregulated at approximately 6 months of age whereas only TLR2 is increased at 14 months of age (105), suggesting a temporally-specific TLR effect. In cell culture, oligomeric α-synuclein released from SH-SY5Y cells activates microglia, and this effect is reduced in microglia derived from TLR2 knockout mice (67). Other studies also support an involvement of TLR2 in synuclein-mediated inflammation (106, 107). Overall there is strong support for TLR signaling in α-synuclein-mediated activation of microglia converging on TLR2 and TLR4. Interestingly other DAMPs, including fibrillar β-amyloid, signal through both TLR2 and TLR4, suggesting that that these receptors are promiscuous [reviewed in (99) and (64, 108, 109)].
One outstanding question is whether TLRs and subsequently activated microglia are the cause or consequence of neuronal death in synulceinopathies. Work by Doorn et al. shows that TLR2 co-localizes with microglia and is increased in post-mortem brains of incidental Lewy body disease cases as well as those from Parkinson’s disease patients (110). Furthermore, these findings support that α-synuclein is the trigger for microglial activation in synucleinopathies, because activated microglia were only present in brain regions displaying α-synuclein deposits. Importantly, Doorn et al. (115) also demonstrated that neuronal death was not required for microglial activation, as activated glia were evident in areas that had no measurable neuronal death. This pathology-based study suggests that activated microglia are dynamic contributors to pathogenesis rather than simply harbingers of neuronal death.
Our identification of a TLR1/2-dependent mechanism in a Lewy body disease-relevant inflammatory pathway provides us the opportunity to target this pathway with new or repurposed therapies (110–113), such as with CU-CPT22, a benzotropolone analogue with specificity and potency for TLR1/2, or candesartan cilexetil, an FDA-approved drug that is already used to treat cerebrovascular disorders, hypertension, and chronic heart failure [reviewed in (114)]. The group of drugs to which candesartan belongs (called sartans) are also potent anti-atherosclerotic and neuroprotective agents that decrease vascular inflammation, autoimmune neuroinflammation, dopaminergic neuronal death, and α-synuclein aggregation (115–126). Evidence suggests that the anti-atherosclerotic activity of sartans is mediated through activation of the peroxisome proliferator-activated receptor-γ (PPARγ) (127–129). However, candesartan is not a potent agonist of PPARγ (130), suggesting the effect of this drug on α-synuclein-induced microglial activation is not mediated through PPARγ. Interestingly, candesartan can decrease the abundance of TLR2 and the signaling activity of the TLR1/2 receptor in monocytes (69). Future studies are needed to decipher the exact mechanism of candesartan’s effect on α-synuclein-induced microglial activation (and presumably neuronal inflammation), but we hypothesize that it does so by suppressing overall TLR1/2 signaling in the microglia. Thus, our work suggests that inhibitors of TLR1/2 may suppress neuroinflammation and improve disease pathology in patients with synucleinopathies.
Materials and Methods
Preparation and misfolding of α-synuclein
Human α-synuclein (herein, Syn) was prepared as previously described (131, 132) except that after purification the Syn was lyophilized and stored at −80°C until use. To misfold Syn, the lyophilized protein was resuspended in TEN buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 20 mM NaCl) to a final concentration of 1 mg/mL and sonicated at 20 Hz (2 × 10 s bursts with a 10-s rest between bursts). Syn was subsequently incubated for 5 days at 37°C (T) with rotation at 1000 RPM (R), resulting in the SynTR fraction (Eppendorf Thermomixer; Eppendorf North America).
SynTR was then separated via size-exclusion centrifugation to isolate high molecular weight oligomers from the monomeric conformer (48). Specifically, 0.3 mg of SynTR was placed onto a 150 kD molecular weight cut-off concentrator (Thermo Scientific; Waltham, MA) and centrifuged at 2,000 × g for 30 min. The concentrate (SynO) was resuspended in 100 µL of TEN buffer and used for subsequent analyses. The flow through was processed using a 20 kDa concentrator (Thermo Scientific) and centrifuged at 3,000 × g for 30 min; the concentrate (SynM) was resuspended in 100 µL of TEN buffer and used for subsequent analyses.
This method of isolation results in recombinant synuclein fractions with an endotoxin content that is below the detection limit of the E-TOXATE test kit (0.03 Endotoxin Units (EU)/mL; Sigma-Aldrich; Cat #: ET0200; 1 EU/mL ~ 0.1 to 0.2 ng endotoxin/mL), consistent with our previous reports (48, 52).
Western blot
SynTR, SynO, and SynM fractions (0.1µg) were subjected to native polyacrylamide gradient (3–12% Bis-Tris) gel electrophoresis utilizing the NativePAGE Novex Bis-Tris gel system (Life technologies). Proteins were transferred to PVDF membrane and probed for Syn using the Syn211 clone antibody (1:1000, Thermo Scientific). Immune complexes were visualized on film (Amersham Hyperfilm ECL; General Electric Healthcare) following incubation with horseradish peroxidase (HRP)-conjugated goat anti-mouse 2° antibody (1:20,000; Chemicon) and Super Signal West Femto Chemiluminescent Substrate (Thermo Scientific).
To probe for synuclein in cell surface isolation experiment, microglia were treated, and cell surface proteins were isolated as described below. Isolated biotinylated proteins were subjected to native polyacrylamide gradient (3–12% Bis-Tris) gel electrophoresis utilizing the NativePAGE Novex Bis-Tris gel system. Proteins were transferred to a PVDF membrane, and membranes were blocked for 1 hour at room temperature in TBST/NFDM [20mM Tris-HCl pH 7.5, 150mM NaCl, 0.1% (v/v) Tween, 5% (w/v) non-fat dry milk (NFDM)], followed by incubation with primary antibody (Syn211; 1:1000). Immune complexes were visualized on film after incubation with HRP-conjugated goat anti-mouse 2° antibody (1:5000; Chemicon) and Amersham Chemiluminescent Substrate (General Electric Healthcare). Membranes were reprobed using rabbit anti-TLR2 (1:1000; LifeSpan BioSciences; Cat #: LS-C98268) and rabbit anti-TLR1 (1:250; Imgenex; Cat #: IMG-5012), which served as controls for successful isolation of surface proteins.
Electron microscopy
SynO or SynM (1.0 µg) was placed on a carbon formavor coated 300 mesh copper grid (Electron Microscopy Sciences). Samples were allowed to adhere to each grid for 20 min, followed by staining with 2% uranyl acetate for 2 min. Grids were imaged using Sciences Hitachi H-7600 W filament transmission electron microscope with a Hammamatsu Orca-HR CCD camera in collaboration with Dr. Anastas Popratiloff at The George Washington University.
Preparation and treatment of primary microglia
Primary microglia cultures derived from C57/Bl6 mouse cortices were prepared as previously described (133). Microglia were plated at a density of 1 × 105 cells per well (24-well plates) in 0.5 mL of Microglia Growth Media (MGM; Minimum Essential Medium Earle’s (MEM), supplemented with 1mM sodium pyruvate, 0.6% (v/v) D-(+)-glucose, 1mM L-glutamine, 100 µg/mL penicillin/streptomycin, and 5% v/v Fetal Bovine Serum). For immunocytochemistry experiments, microglia were plated on sterile glass coverslips using the same procedure. For isolation of surface proteins experiments, microglia were plated at 6 × 105 cells per well (6-well plate) format in 2.0 mL MGM. For in situ Proximity Ligation Assay experiments, microglia were plated at 2 × 104 cells per well in 16-well chamber slides (Nunc Lab-Tek; Thermo Scientific; Cat #: 178599). For quantification of nuclear NF-κB activity, microglia were plated at 8.5 × 105 cells per well (6-well plate). Microglia were subsequently treated with SynO, SynM, or 20mM TEN buffer (Vehicle) in MGM according to the figure legends.
Isolation of surface proteins
Microglia were plated as described above and treated with 0.8 µg/mL SynO or SynM for 30 min at 4°C. Media was subsequently removed and microglia were washed with ice-cold PBS (1×) and surface proteins were biotinylated and isolated according to manufacturer’s instructions (Thermo Scientific; Cat #: 89881). This procedure was performed in two separate experiments with three biological replicates.
Immunocytochemistry
Microglia were plated on glass coverslips (12 mm; Deckglaser), treated as described, and subsequently processed for immunocytochemistry. More specifically, following treatment, cells were washed with PBS for 5 min, fixed with PBS containing 4% (w/v) paraformaldehyde and 4% (w/v) sucrose pH 7.4 at room temperature for 15 min, permeabilized in PBS containing 0.1% (v/v) triton X-100 for 5 min, and blocked for 1 hour with PBS containing 10% (v/v) goat serum. Cells were subsequently incubated overnight at 4°C with either rabbit Iba-1 antibody (anti-Iba-1; 1:750; Wako) or rabbit anti-NF-κB (1:1000; α-p65; Abcam) in blocking buffer. Antibody:antigen complexes were visualized following incubation with Alexa Fluor 594 conjugated goat anti-rabbit IgG secondary antibody (1:1000) in PBS containing 0.1% (v/v) triton X-100 and 1% goat serum. Unbound 2° antibody was removed by washing with PBS containing 0.1% (v/v) triton X-100. Cells were counterstained with 4',6-diamidino-2-phenylindole (DAPI; 13.0 ng/µL) in PBS for 5 min followed by two washes with PBS. Coverslips were mounted with Citifluor (Ted Pella) and sealed with nail polish. Cells were imaged using a Zeiss Axioskop fluorescent microscope (Carl Zeiss) and images were captured using an AxioCam HRm camera (Carl Zeiss).
RNA extraction
Following treatment, RNA was harvested from microglia using an RNeasy mini Kit with on-column DNase I digestion, according to manufacturer’s instructions (Qiagen). RNA concentrations were measured using a NanoDrop 1000 spectrophotometer (Thermo Scientific).
Quantitative real-time PCR
RNA (1 µg) was reverse transcribed in a 20 µL reaction volume using a High-Capacity cDNA Archive Kit (Life Technologies). β-actin RT-PCR was subsequently performed to verify the quality of the cDNA. Gene expression was quantified by qRT-PCR in a 96-well format. Specifically, cDNA (2.5 µL) was combined with 17.5 µL of master mix containing the appropriate primer/probe pairs and TaqMan® Universal PCR master mix. Quantitative RT-PCR reactions were performed using the ABI Prism 7900HT Sequence Detection System (Life Technologies). Data were analyzed utilizing the relative quantification ΔΔCt method, normalizing target gene expression to a GAPDH endogenous control, followed by normalization to either SynM or TEN buffer-treated controls. Primers/Probes used: ARG-1 Mm00833903_m1, IL-10 Mm0434228_m1, TNF-α Mm00443258_m1, IL-1β Mm0434228_m1, TLR1 Mm0120884_m1, TLR2 Mm00442346_m1, TLR3 Mm00446577_g1, GAPDH 4352339E. Gene expression changes are represented as fold change (2− ΔΔCt). Statistical analyses were performed on ΔCt values as specified in the figure legends and the significance threshold was set at P < 0.05. All measurements were performed in three separate experiments with three biological replicates and duplicate technical replicates, unless otherwise noted in figure legends.
ELISA
TNF-α, IL-1β, and IL-10 protein concentrations in cell culture supernatants were quantified using an enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (R&D Systems). All measurements were performed in three separate experiments with three biological replicates and duplicate technical replicates.
MyD88 homodimerization inhibition
Primary microglia were plated as described above. Cells were then pre-treated with 200 µM of control peptide or MyD88 inhibitory peptide (Imgenex) for 30 min before a 2 hour exposure to 80.0 ng/mL SynO. RNA was then harvested from the treated cells and the gene expression of TNF-α and IL-1β was quantified using qRT-PCR as described above. All measurements were performed in three separate experiments with three biological replicates and duplicate technical replicates.
In situ proximity ligation assay (PLA)
Microglia were plated as described above in 16-well chamber slides. Microglia were exposed to vehicle, SynM, or SynO (0.8 µg/mL) for 10 minutes at 37°C. After the exposure time, media was removed; cells were washed twice with ice-cold PBS (1×) and fixed with PBS containing 4% (w/v) paraformaldehyde and 4% (w/v) sucrose pH 7.4 at room temperature for 15 minutes. PLA was performed according to manufacturer’s instructions (Duolink; Sigma Aldrich). In brief, microglia were permeabilized in PBS containing 0.1% (v/v) triton X-100 for 5 min; and blocked for 1 hour with PBS containing 10% (v/v) normal goat serum. Cells were subsequently incubated in a drop-wise reaction for 1 hour in a humidified chamber at 37°C with mouse synuclein antibody (anti-synuclein LB509; 1:1000; Sigma Aldrich) and rabbit anti-TLR1 (IMG-5012; 1:250; Imgenex) in blocking buffer. Removal of unbound primary antibody was performed via washes in PBS containing 0.1% (v/v) triton x-100 and 0.1% (v/v) normal goat serum. Donkey anti-rabbit (+) PLA probe (Sigma; DUO92002) and Donkey anti-mouse (−) PLA probe (DUO92004) were diluted 1:5 in blocking buffer and applied to cells in a drop-wise reaction. Cells were incubated with PLA probes for 1 hour in a humidified chamber at 37°C. Cells were subsequently washed in wash buffer; PLA probes were ligated in 1× ligation buffer with ligase (DUO92013) for 30 min, followed by rolling circular amplification in 1× amplification buffer with polymerase (DUO92013); amplified signal was visualized with far-red fluorescent detection molecule (DUO92013). Cells were mounted with in situ mounting media containing DAPI (DUO82040). In situ PLA signals were imaged using an LSM510 confocal microscope. Images are representative Z-stack projections (7 µm stack; 1 µm/section). This procedure was repeated in three separate experiments with three biological replicates.
HEK-Blue TLR reporter assay
HEK293 reporter cells stably expressing human TLR2 (HEK-hTLR2) and human TLR3 (HEK-hTLR3) were used according to manufacturer’s instructions (Invivogen). HEK-hTLR2 cells were exposed to either Pam3CSK4 (1.0 µg/mL), or the different forms of Syn (80.0 ng/mL) and allowed to incubate for 20 hours. Similarly, HEK-hTLR3 cells were exposed to either Poly (I:C) (1.0 µg/mL), or the different forms of Syn (80.0 ng/mL) and allowed to incubate for 20 hours. All measurements were performed in two separate experiments in three biological replicates with duplicate technical replicates.
CU-CPT22 treatment
A previously described TLR1/2 inhibitor, CU-CPT22 (68), was used to block signaling in primary microglia and HEK-hTLR2 cells. HEK-hTLR2 cells were simultaneously treated with 100 µM CU-CPT22 and either SynM (8.0 ng/mL), SynO (8.0 ng/mL), or the TLR1/2 agonist, Pam3CSK4 (1 µg/mL) for 20-hours, followed by SEAP reporter activity quantification according to manufacturer’s instructions (Invivogen). Primary microglia were simultaneously treated with 400 µM CU-CPT22 and 8.0 ng/mL SynO for 10 or 20 mins then NF-κB nuclear translocation was quantified as described below (Quantification of nuclear NF-κB activity). For experiments quantifying TNF-α release, primary microglia were simultaneously treated with 10 µM CU-CPT22 and 80.0 ng/mL SynO for 72 hours. TNF-α was quantified as described above (ELISA). All measurements were performed in two separate experiments in duplicate biological replicates with duplicate technical replicates.
Microglial nuclear extraction
Microglial cells were plated as described above and exposed to either SynM (80.0 ng/mL) or SynO (80.0 ng/mL) for 2 hours. Preparation of nuclear extracts was subsequently performed according to manufacturer’s instructions (Nuclear Extraction Kit; Cat #: 40010; Active Motif; Carlsbad, CA). In brief, cell culture supernatant was removed and cells were washed with 2 mL of PBS/Phosphatase Inhibitor solution (125 mM NaF, 250 mM β-glycerophosphate, 250 mM p-nitrophenyl phosphate, 25 mM NaVO3). Cells were subsequently harvested in 1 mL of PBS/Phosphatase Inhibitor solution via cell scraping and cell suspension was centrifuged for 5 min at 200 × g in a microcentrifuge pre-cooled at 4°C. The supernatant was discarded and the cell pellet was resuspended in 125 µL of 1× Hypotonic Buffer (20 mM HEPES pH 7.5, 5 mM NaF, 10 µM Na2MoO4, 0.1 mM EDTA) and incubated for 15 min on ice to allow cells to swell. In order to release cell nuclei, cells were lysed by adding detergent (5 µL 10% NP-40) and vortexed for 10 s. This suspension was centrifuged for 30 s at 14,000 × g in a microcentrifuge pre-cooled at 4°C to pellet cell nuclei. Supernatant (cytoplasmic fraction) was collected and stored at −80°C. Nuclear pellets were resuspended in 50 µL Complete Lysis Buffer (Lysis Buffer AM1 supplemented with 1mM DTT and 1.0% [v/v] Protease Inhibitor Cocktail) and vortexed for 10 s. Suspensions were incubated for 30 min on ice with rotary shaking, vortexed for 30 s, and subsequently centrifuged for 10 min at 14,000 × g in a microcentrifuge pre-cooled at 4°C. The nuclear fractions were stored at −80°C until utilized for quantification of nuclear NF-κB Activity.
Quantification of nuclear NF-κB activity
Activation of nuclear NF-κB was measured in nuclear protein extracts utilizing the TransAM p65 NF-κB Kit (Active Motif; Cat #: 40596), an ELISA-based method designed to quantify NF-κB p65 subunit activation, according to the manufacturer’s instructions. Absorbances were measured at 450 nm using a microplate absorbance reader model 680 (Biorad). All measurements were performed in two separate experiments in triplicate biological replicates with duplicate technical replicates.
Nuclear NF-κB translocation quantification
Cells were processed for NF-κB immunocytochemistry as described above. Images of NF-κB and DAPI were captured as described above from 10 distinct, randomly selected regions of each treatment condition by a blinded observer. Quantification of nuclear NF-κB intensity was completed as previously described (134, 135) utilizing Image J software (National Institute of Health). A total of 52 cells were analyzed for each condition. The percent change in nuclear NF-κB was calculated by normalizing the raw data obtained from microglia treated with inhibitor to their respective vehicle control. All measurements were performed in two separate experiments in duplicate biological replicates. Percent decrease from both experiments was plotted as mean ± S.E.M.
Candesartan treatment
Primary microglia were plated as described above on glass coverslips followed by pre-treatment with 10, 20, or 30 µM of candesartan cilexetil (CD; Sigma; SML0245) or vehicle (DMSO) for 2-hours. Pre-treated cells were subsequently exposed to 8.0 ng/mL SynO for 12-hours and processed for Iba-1 immunocytochemistry as described above. All measurements were performed in triplicate in three separate biological replicates.
Statistical analyses
All statistical analyses were performed using Graphpad Prism 5 (Graphpad Software Inc.). For qRT-PCR, all statistical analyses were performed on ΔCt values. One-way analysis of variance (ANOVA) was performed followed by an appropriate post hoc test, as specified in the figure legends. All data are reported as mean ± S.E.M and significance was set at P ≤ 0.05; all P-values for each statistical analysis are reported in the appropriate figure legend.
Supplementary Material
Acknowledgements
We thank Ms. Amanda Edwards for technical assistance and Dr. Anastas Popratiloff for assistance with electron microscopy.
Funding: This work was supported in part by a Parkinson’s Disease Foundation Summer Student Fellowship PDF-SFW-1350 (CD), NIH/NINDS T32NS041218 (DB), NIH R01GM101279 (HY), NIH R01GM103843 (HY), NIH/NIEHS R01ES014470 (KMZ), and a Parkinson’s Movement Disorder Foundation grant (KMZ).
Footnotes
Supplementary Materials
Fig. S1. Preliminary time course of p65 staining in microglia.
Author contributions: S.G.D. and K.A.M.-Z. designed the research, analyzed the data and wrote the paper; S.G.D. performed most of the experiments; D.B. performed the electron microscopy and qRT-PCR for TLR1–3; C.D. performed the HEKhTLR experiments; S.G.D., D.B., C.D. and K.A.M.-Z. prepared and analyzed the synuclein; K.C. and H.Y. prepared the CU-CPT22 and advised on the use of this compound; all authors edited the paper.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: Patents exist related to this work: “Antagonists of the Toll-Like Receptor 1/2 Complex”, Yin, H.; Cheng, K.; PCT Int. Appl. 2013, US2013/52517; USA Appl. 2014, 14/417,676; EU Appl. 2015, 13826030.2.
References and Notes
- 1.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Athanassiadou A, Voutsinas G, Psiouri L, Leroy E, Polymeropoulos MH, Ilias A, Maniatis GM, Papapetropoulos T. Genetic analysis of families with Parkinson disease that carry the Ala53Thr mutation in the gene encoding alpha-synuclein. Am J Hum Genet. 1999;65:555–558. doi: 10.1086/302486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 4.Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iorio G, Sanges G, Stenroos ES, Pho LT, Schaffer AA, Lazzarini AM, Nussbaum RL, Duvoisin RC. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science. 1996;274:1197–1199. doi: 10.1126/science.274.5290.1197. [DOI] [PubMed] [Google Scholar]
- 5.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 6.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 7.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 8.Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, Melchers A, Paudel R, Gibbs JR, Simon-Sanchez J, Paisan-Ruiz C, Bras J, Ding J, Chen H, Traynor BJ, Arepalli S, Zonozi RR, Revesz T, Holton J, Wood N, Lees A, Oertel W, Wullner U, Goldwurm S, Pellecchia MT, Illig T, Riess O, Fernandez HH, Rodriguez RL, Okun MS, Poewe W, Wenning GK, Hardy JA, Singleton AB, Del Sorbo F, Schneider S, Bhatia KP, Gasser T. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol. 2009;65:610–614. doi: 10.1002/ana.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 11.Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, Bertoncini CW, Wood NW, Knowles TP, Dobson CM, Klenerman D. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell. 2012;149:1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oueslati A, Fournier M, Lashuel HA. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: implications for Parkinson's disease pathogenesis and therapies. Prog Brain Res. 2010;183:115–145. doi: 10.1016/S0079-6123(10)83007-9. [DOI] [PubMed] [Google Scholar]
- 14.Taschenberger G, Garrido M, Tereshchenko Y, Bahr M, Zweckstetter M, Kugler S. Aggregation of alphaSynuclein promotes progressive in vivo neurotoxicity in adult rat dopaminergic neurons. Acta Neuropathol. 2012;123:671–683. doi: 10.1007/s00401-011-0926-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsigelny IF, Sharikov Y, Miller MA, Masliah E. Mechanism of alpha-synuclein oligomerization and membrane interaction: theoretical approach to unstructured proteins studies. Nanomedicine. 2008;4:350–357. doi: 10.1016/j.nano.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uversky VN. Mysterious oligomerization of the amyloidogenic proteins. Febs J. 2010;277:2940–2953. doi: 10.1111/j.1742-4658.2010.07721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biasini E, Fioriti L, Ceglia I, Invernizzi R, Bertoli A, Chiesa R, Forloni G. Proteasome inhibition and aggregation in Parkinson's disease: a comparative study in untransfected and transfected cells. Journal of Neurochemistry. 2004;88:545–553. doi: 10.1046/j.1471-4159.2003.02152.x. [DOI] [PubMed] [Google Scholar]
- 18.Emmanouilidou E, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol Aging. 2010;31:953–968. doi: 10.1016/j.neurobiolaging.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Feng LR, Federoff HJ, Vicini S, Maguire-Zeiss KA. Alpha-synuclein mediates alterations in membrane conductance: a potential role for alpha-synuclein oligomers in cell vulnerability. Eur J Neurosci. 2010;32:10–17. doi: 10.1111/j.1460-9568.2010.07266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 21.Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One. 2009;4:e5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem. 2010;113:1263–1274. doi: 10.1111/j.1471-4159.2010.06695.x. [DOI] [PubMed] [Google Scholar]
- 24.Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol. 2008;40:1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 26.Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010;285:9262–9272. doi: 10.1074/jbc.M109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danzer KM, Ruf WP, Putcha P, Joyner D, Hashimoto T, Glabe C, Hyman BT, McLean PJ. Heat-shock protein 70 modulates toxic extracellular alpha-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 2011;25:326–336. doi: 10.1096/fj.10-164624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42. doi: 10.1186/1750-1326-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Angot E, Steiner JA, Lema Tome CM, Ekstrom P, Mattsson B, Bjorklund A, Brundin P. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS ONE. 2012;7:e39465. doi: 10.1371/journal.pone.0039465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, Desplats P, Masliah E, Lee SJ. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32:13454–13469. doi: 10.1523/JNEUROSCI.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nature reviews. Molecular cell biology. 2010;11:301–307. doi: 10.1038/nrm2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee SJ, Desplats P, Lee HJ, Spencer B, Masliah E. Cell-to-cell transmission of alpha-synuclein aggregates. Methods Mol Biol. 2012;849:347–359. doi: 10.1007/978-1-61779-551-0_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Appel SH. Inflammation in Parkinson's disease: cause or consequence? Mov Disord. 2012;27:1075–1077. doi: 10.1002/mds.25111. [DOI] [PubMed] [Google Scholar]
- 35.Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 36.Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson's disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S210–S212. doi: 10.1016/S1353-8020(11)70065-7. [DOI] [PubMed] [Google Scholar]
- 37.Marques O, Outeiro TF. Alpha-synuclein: from secretion to dysfunction and death. Cell death & disease. 2012;3:e350. doi: 10.1038/cddis.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reynolds AD, Glanzer JG, Kadiu I, Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, Cerny R, Gelman B, Thomas MP, Mosley RL, Gendelman HE. Nitrated alpha-synuclein-activated microglial profiling for Parkinson's disease. J Neurochem. 2008;104:1504–1525. doi: 10.1111/j.1471-4159.2007.05087.x. [DOI] [PubMed] [Google Scholar]
- 39.Bartels AL, Leenders KL. Neuroinflammation in the pathophysiology of Parkinson's disease: evidence from animal models to human in vivo studies with [11C]-PK11195 PET. Mov Disord. 2007;22:1852–1856. doi: 10.1002/mds.21552. [DOI] [PubMed] [Google Scholar]
- 40.Gerhard A, Neumaier B, Elitok E, Glatting G, Ries V, Tomczak R, Ludolph AC, Reske SN. In vivo imaging of activated microglia using [11C]PK11195 and positron emission tomography in patients after ischemic stroke. Neuroreport. 2000;11:2957–2960. doi: 10.1097/00001756-200009110-00025. [DOI] [PubMed] [Google Scholar]
- 41.Ouchi Y, Yagi S, Yokokura M, Sakamoto M. Neuroinflammation in the living brain of Parkinson's disease. Parkinsonism Relat Disord. 2009;15(Suppl 3):S200–S204. doi: 10.1016/S1353-8020(09)70814-4. [DOI] [PubMed] [Google Scholar]
- 42.Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T. Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol. 2005;57:168–175. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- 43.Blum-Degen D, Muller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer's and de novo Parkinson's disease patients. Neurosci Lett. 1995;202:17–20. doi: 10.1016/0304-3940(95)12192-7. [DOI] [PubMed] [Google Scholar]
- 44.Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch EC. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson's disease. Neurosci Lett. 1994;172:151–154. doi: 10.1016/0304-3940(94)90684-x. [DOI] [PubMed] [Google Scholar]
- 45.Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, Agid Y, Dugas B, Hirsch EC. FcepsilonRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci. 1999;19:3440–3447. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180:147–150. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
- 47.Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994;165:208–210. doi: 10.1016/0304-3940(94)90746-3. [DOI] [PubMed] [Google Scholar]
- 48.Beraud D, Hathaway HA, Trecki J, Chasovskikh S, Johnson DA, Johnson JA, Federoff HJ, Shimoji M, Mhyre TR, Maguire-Zeiss KA. Microglial activation and antioxidant responses induced by the Parkinson's disease protein alpha-synuclein. J Neuroimmune Pharmacol. 2013;8:94–117. doi: 10.1007/s11481-012-9401-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beraud D, Maguire-Zeiss KA. Misfolded alpha-synuclein and Toll-like receptors: therapeutic targets for Parkinson's disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S17–S20. doi: 10.1016/S1353-8020(11)70008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beraud D, Twomey M, Bloom B, Mittereder A, Ton V, Neitzke K, Chasovskikh S, Mhyre TR, Maguire-Zeiss KA. alpha-Synuclein Alters Toll-Like Receptor Expression. Front Neurosci. 2011;5:80. doi: 10.3389/fnins.2011.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Su X, Federoff HJ, Maguire-Zeiss KA. Mutant alpha-Synuclein Overexpression Mediates Early Proinflammatory Activity. Neurotox Res. 2009;16:238–254. doi: 10.1007/s12640-009-9053-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson's disease. Neurobiol Aging. 2008;29:1690–1701. doi: 10.1016/j.neurobiolaging.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Theodore S, Cao S, McLean PJ, Standaert DG. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol. 2008;67:1149–1158. doi: 10.1097/NEN.0b013e31818e5e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 55.Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, Wenning GK, Stefanova N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. 2013;61:349–360. doi: 10.1002/glia.22437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Current opinion in immunology. 2005;17:338–344. doi: 10.1016/j.coi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 60.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 61.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 62.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia. 2010;58:253–263. doi: 10.1002/glia.20928. [DOI] [PubMed] [Google Scholar]
- 64.Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Richard KL, Filali M, Prefontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1–42 and delay the cognitive decline in a mouse model of Alzheimer's disease. J Neurosci. 2008;28:5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, Fassbender K. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2007;20:947–956. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- 67.Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nature communications. 2013;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng K, Wang X, Zhang S, Yin H. Discovery of small-molecule inhibitors of the TLR1/TLR2 complex. Angew Chem Int Ed Engl. 2012;51:12246–12249. doi: 10.1002/anie.201204910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dasu MR, Riosvelasco AC, Jialal I. Candesartan inhibits Toll-like receptor expression and activity both in vitro and in vivo. Atherosclerosis. 2009;202:76–83. doi: 10.1016/j.atherosclerosis.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Angot E, Steiner JA, Hansen C, Li JY, Brundin P. Are synucleinopathies prion-like disorders? Lancet Neurol. 2010;9:1128–1138. doi: 10.1016/S1474-4422(10)70213-1. [DOI] [PubMed] [Google Scholar]
- 71.Braak H, Bohl JR, Muller CM, Rub U, de Vos RA, Del Tredici K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov Disord. 2006;21:2042–2051. doi: 10.1002/mds.21065. [DOI] [PubMed] [Google Scholar]
- 72.Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease-related brain pathology. Neurosci Lett. 2006;396:67–72. doi: 10.1016/j.neulet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 73.Braak H, Del Tredici K. Invited Article: Nervous system pathology in sporadic Parkinson disease. Neurology. 2008;70:1916–1925. doi: 10.1212/01.wnl.0000312279.49272.9f. [DOI] [PubMed] [Google Scholar]
- 74.Braak H, Rub U, Gai WP, Del Tredici K. Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110:517–536. doi: 10.1007/s00702-002-0808-2. [DOI] [PubMed] [Google Scholar]
- 75.Burke RE, Dauer WT, Vonsattel JP. A critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol. 2008;64:485–491. doi: 10.1002/ana.21541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dickson DW, Uchikado H, Fujishiro H, Tsuboi Y. Evidence in favor of Braak staging of Parkinson's disease. Mov Disord. 2010;25(Suppl 1):S78–S82. doi: 10.1002/mds.22637. [DOI] [PubMed] [Google Scholar]
- 77.Jellinger KA. Critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol. 2010;67:550. doi: 10.1002/ana.21638. [DOI] [PubMed] [Google Scholar]
- 78.Sacino AN, Brooks M, Thomas MA, McKinney AB, McGarvey NH, Rutherford NJ, Ceballos-Diaz C, Robertson J, Golde TE, Giasson BI. Amyloidogenic alpha-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol. 2014;127:645–665. doi: 10.1007/s00401-014-1268-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC, Lee VM. Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha-synuclein and neurodegeneration. Cell reports. 2014;7:2054–2065. doi: 10.1016/j.celrep.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sacino AN, Brooks M, McKinney AB, Thomas MA, Shaw G, Golde TE, Giasson BI. Brain Injection of alpha-Synuclein Induces Multiple Proteinopathies, Gliosis, and a Neuronal Injury Marker. J Neurosci. 2014;34:12368–12378. doi: 10.1523/JNEUROSCI.2102-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S, Regenhardt RW, McGarvey NH, Ayers JI, Notterpek L, Borchelt DR, Golde TE, Giasson BI. Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci U S A. 2014;111:10732–10737. doi: 10.1073/pnas.1321785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sacino AN, Brooks M, McGarvey NH, McKinney AB, Thomas MA, Levites Y, Ran Y, Golde TE, Giasson BI. Induction of CNS alpha-synuclein pathology by fibrillar and non-amyloidogenic recombinant alpha-synuclein. Acta neuropathologica communications. 2013;1:38. doi: 10.1186/2051-5960-1-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Recasens A, Dehay B, Bove J, Carballo-Carbajal I, Dovero S, Perez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, Farinas I, Obeso JA, Bezard E, Vila M. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75:351–362. doi: 10.1002/ana.24066. [DOI] [PubMed] [Google Scholar]
- 86.McGeer EG, Klegeris A, McGeer PL. Inflammation, the complement system and the diseases of aging. Neurobiol Aging. 2005;26(Suppl 1):94–97. doi: 10.1016/j.neurobiolaging.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 87.McGeer PL, McGeer EG. Inflammation and the degenerative diseases of aging. Ann N Y Acad Sci. 2004;1035:104–116. doi: 10.1196/annals.1332.007. [DOI] [PubMed] [Google Scholar]
- 88.McGeer PL, McGeer EG. Glial reactions in Parkinson's disease. Mov Disord. 2008;23:474–483. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- 89.Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7:354–365. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Colton CA, Wilcock DM. Assessing activation states in microglia. CNS Neurol Disord Drug Targets. 2010;9:174–191. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- 91.Sawada M, Suzumura A, Hosoya H, Marunouchi T, Nagatsu T. Interleukin-10 inhibits both production of cytokines and expression of cytokine receptors in microglia. J Neurochem. 1999;72:1466–1471. doi: 10.1046/j.1471-4159.1999.721466.x. [DOI] [PubMed] [Google Scholar]
- 92.Kawai T, Akira S. Toll-like receptor downstream signaling. Arthritis research & therapy. 2005;7:12–19. doi: 10.1186/ar1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kumagai Y, Takeuchi O, Akira S. Pathogen recognition by innate receptors. Journal of infection and chemotherapy : official journal of the Japan Society of Chemotherapy. 2008;14:86–92. doi: 10.1007/s10156-008-0596-1. [DOI] [PubMed] [Google Scholar]
- 94.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. International reviews of immunology. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 95.Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annual review of biochemistry. 2007;76:447–480. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- 96.Lee SJ, Lee S. Toll-like receptors and inflammation in the CNS. Curr Drug Targets Inflamm Allergy. 2002;1:181–191. doi: 10.2174/1568010023344698. [DOI] [PubMed] [Google Scholar]
- 97.Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85–95. doi: 10.1016/s0165-2478(02)00228-6. [DOI] [PubMed] [Google Scholar]
- 98.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 99.Trotta T, Porro C, Calvello R, Panaro MA. Biological role of Toll-like receptor-4 in the brain. J Neuroimmunol. 2014;268:1–12. doi: 10.1016/j.jneuroim.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 100.Brown V, Brown RA, Ozinsky A, Hesselberth JR, Fields S. Binding specificity of Toll-like receptor cytoplasmic domains. Eur J Immunol. 2006;36:742–753. doi: 10.1002/eji.200535158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen H, Jiang Z. The essential adaptors of innate immune signaling. Protein & cell. 2013;4:27–39. doi: 10.1007/s13238-012-2063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bryant CE, Symmons M, Gay NJ. Toll-like receptor signalling through macromolecular protein complexes. Molecular immunology. 2014 doi: 10.1016/j.molimm.2014.06.033. [DOI] [PubMed] [Google Scholar]
- 103.Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol. 2014;14:546–558. doi: 10.1038/nri3713. [DOI] [PubMed] [Google Scholar]
- 104.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Watson MB, Richter F, Lee SK, Gabby L, Wu J, Masliah E, Effros RB, Chesselet MF. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp Neurol. 2012;237:318–334. doi: 10.1016/j.expneurol.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Lachaud CC, Guilliams T, Fernandez-Montesinos R, Benitez-Rondan A, Robledo G, Hmadcha A, Delgado M, Dobson CM, Pozo D. Preconditioning of microglia by alpha-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS ONE. 2013;8:e79160. doi: 10.1371/journal.pone.0079160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, de Bernard M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE. 2013;8:e55375. doi: 10.1371/journal.pone.0055375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Oblak A, Jerala R. Toll-like receptor 4 activation in cancer progression and therapy. Clinical & developmental immunology. 2011;2011:609579. doi: 10.1155/2011/609579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi K. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J Neuroinflammation. 2008;5:23. doi: 10.1186/1742-2094-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Doorn KJ, Moors T, Drukarch B, van de Berg W, Lucassen PJ, van Dam AM. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson inverted question marks disease patients. Acta neuropathologica communications. 2014;2:90. doi: 10.1186/s40478-014-0090-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dunne A, Marshall NA, Mills KH. TLR based therapeutics. Curr Opin Pharmacol. 2011 doi: 10.1016/j.coph.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 112.Gambuzza M, Licata N, Palella E, Celi D, Foti Cuzzola V, Italiano D, Marino S, Bramanti P. Targeting Toll-like receptors: emerging therapeutics for multiple sclerosis management. J Neuroimmunol. 2011;239:1–12. doi: 10.1016/j.jneuroim.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 113.Hennessy EJ, Parker AE, O'Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov. 2010;9:293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- 114.Michel MC, Foster C, Brunner HR, Liu L. A systematic comparison of the properties of clinically used angiotensin II type 1 receptor antagonists. Pharmacological reviews. 2013;65:809–848. doi: 10.1124/pr.112.007278. [DOI] [PubMed] [Google Scholar]
- 115.Benicky J, Sanchez-Lemus E, Honda M, Pang T, Orecna M, Wang J, Leng Y, Chuang DM, Saavedra JM. Angiotensin II AT1 receptor blockade ameliorates brain inflammation. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2011;36:857–870. doi: 10.1038/npp.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Grammatopoulos TN, Outeiro TF, Hyman BT, Standaert DG. Angiotensin II protects against alpha-synuclein toxicity and reduces protein aggregation in vitro. Biochem Biophys Res Commun. 2007;363:846–851. doi: 10.1016/j.bbrc.2007.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Joglar B, Rodriguez-Pallares J, Rodriguez-Perez AI, Rey P, Guerra MJ, Labandeira-Garcia JL. The inflammatory response in the MPTP model of Parkinson's disease is mediated by brain angiotensin: relevance to progression of the disease. J Neurochem. 2009;109:656–669. doi: 10.1111/j.1471-4159.2009.05999.x. [DOI] [PubMed] [Google Scholar]
- 118.Rodriguez-Pallares J, Parga JA, Joglar B, Guerra MJ, Labandeira-Garcia JL. Mitochondrial ATP-sensitive potassium channels enhance angiotensin-induced oxidative damage and dopaminergic neuron degeneration. Relevance for aging-associated susceptibility to Parkinson's disease. Age (Dordr) 2012;34:863–880. doi: 10.1007/s11357-011-9284-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Saavedra JM. Angiotensin II AT(1) receptor blockers as treatments for inflammatory brain disorders. Clin Sci (Lond) 2012;123:567–590. doi: 10.1042/CS20120078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Saavedra JM. Angiotensin II AT(1) receptor blockers ameliorate inflammatory stress: a beneficial effect for the treatment of brain disorders. Cell Mol Neurobiol. 2012;32:667–681. doi: 10.1007/s10571-011-9754-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Saavedra JM, Sanchez-Lemus E, Benicky J. Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation and ischemia: Therapeutic implications. Psychoneuroendocrinology. 2011;36:1–18. doi: 10.1016/j.psyneuen.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Villar-Cheda B, Dominguez-Meijide A, Joglar B, Rodriguez-Perez AI, Guerra MJ, Labandeira-Garcia JL. Involvement of microglial RhoA/Rho-kinase pathway activation in the dopaminergic neuron death. Role of angiotensin via angiotensin type 1 receptors. Neurobiol Dis. 2012;47:268–279. doi: 10.1016/j.nbd.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 123.Grammatopoulos TN, Ahmadi F, Jones SM, Fariss MW, Weyhenmeyer JA, Zawada WM. Angiotensin II protects cultured midbrain dopaminergic neurons against rotenone-induced cell death. Brain Res. 2005;1045:64–71. doi: 10.1016/j.brainres.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 124.Grammatopoulos TN, Jones SM, Ahmadi FA, Hoover BR, Snell LD, Skoch J, Jhaveri VV, Poczobutt AM, Weyhenmeyer JA, Zawada WM. Angiotensin type 1 receptor antagonist losartan, reduces MPTP-induced degeneration of dopaminergic neurons in substantia nigra. Mol Neurodegener. 2007;2:1. doi: 10.1186/1750-1326-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Platten M, Youssef S, Hur EM, Ho PP, Han MH, Lanz TV, Phillips LK, Goldstein MJ, Bhat R, Raine CS, Sobel RA, Steinman L. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc Natl Acad Sci U S A. 2009;106:14948–14953. doi: 10.1073/pnas.0903958106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lanz TV, Ding Z, Ho PP, Luo J, Agrawal AN, Srinagesh H, Axtell R, Zhang H, Platten M, Wyss-Coray T, Steinman L. Angiotensin II sustains brain inflammation in mice via TGF-beta. J Clin Invest. 2010;120:2782–2794. doi: 10.1172/JCI41709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Erbe DV, Gartrell K, Zhang YL, Suri V, Kirincich SJ, Will S, Perreault M, Wang S, Tobin JF. Molecular activation of PPARgamma by angiotensin II type 1-receptor antagonists. Vascular pharmacology. 2006;45:154–162. doi: 10.1016/j.vph.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 128.Garrido-Gil P, Joglar B, Rodriguez-Perez AI, Guerra MJ, Labandeira-Garcia JL. Involvement of PPAR-gamma in the neuroprotective and anti-inflammatory effects of angiotensin type 1 receptor inhibition: effects of the receptor antagonist telmisartan and receptor deletion in a mouse MPTP model of Parkinson's disease. J Neuroinflammation. 2012;9:38. doi: 10.1186/1742-2094-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Villapol S, Yaszemski AK, Logan TT, Sanchez-Lemus E, Saavedra JM, Symes AJ. Candesartan, an angiotensin II AT(1)-receptor blocker and PPAR-gamma agonist, reduces lesion volume and improves motor and memory function after traumatic brain injury in mice. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2012;37:2817–2829. doi: 10.1038/npp.2012.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Benson SC, Iguchi R, Ho CI, Yamamoto K, Kurtz TW. Inhibition of cardiovascular cell proliferation by angiotensin receptor blockers: are all molecules the same? Journal of hypertension. 2008;26:973–980. doi: 10.1097/HJH.0b013e3282f56ba5. [DOI] [PubMed] [Google Scholar]
- 131.Giasson BI, Uryu K, Trojanowski JQ, Lee VM. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- 132.Maguire-Zeiss KA, Wang CI, Yehling E, Sullivan MA, Short DW, Su X, Gouzer G, Henricksen LA, Wuertzer CA, Federoff HJ. Identification of human alpha-synuclein specific single chain antibodies. Biochem Biophys Res Commun. 2006;349:1198–1205. doi: 10.1016/j.bbrc.2006.08.127. [DOI] [PubMed] [Google Scholar]
- 133.Daniele SG, Edwards AA, Maguire-Zeiss KA. Isolation of cortical microglia with preserved immunophenotype and functionality from murine neonates. Journal of visualized experiments : JoVE. 2014:e51005. doi: 10.3791/51005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Noursadeghi M, Tsang J, Haustein T, Miller RF, Chain BM, Katz DR. Quantitative imaging assay for NF-kappaB nuclear translocation in primary human macrophages. J Immunol Methods. 2008;329:194–200. doi: 10.1016/j.jim.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fuseler JW, Merrill DM, Rogers JA, Grisham MB, Wolf RE. Analysis and quantitation of NF-kappaB nuclear translocation in tumor necrosis factor alpha (TNF-alpha) activated vascular endothelial cells. Microscopy and microanalysis : the official journal of Microscopy Society of America, Microbeam Analysis Society, Microscopical Society of Canada. 2006;12:269–276. doi: 10.1017/S1431927606060260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.