

Abstract
Background
ACTA2 mutations are the major cause of familial thoracic aortic aneurysms and dissections. We sought to characterize these aortic diseases in a large case series of individuals with ACTA2 mutations.
Methods and Results
Aortic disease, management, and outcome associated with the first aortic event (aortic dissection or aneurysm repair) were abstracted from the medical records of 277 individuals with 41 various ACTA2 mutations. Aortic events occurred in 48% of these individuals, with the vast majority presenting with thoracic aortic dissections (88%) associated with 25% mortality. Type A dissections were more common than type B dissections (54% versus 21%), but the median age of onset of type B dissections was significantly younger than type A dissections (27 years, IQR 18–41 versus 36 years, IQR 26–45). Only 12% of aortic events were repair of ascending aortic aneurysms, which variably involved the aortic root, ascending aorta and aortic arch. Overall cumulative risk of an aortic event at age 85 years was 0.76 (95% CI 0.64, 0.86). After adjustment for intra-familial correlation, gender and race, mutations disrupting p.R179 and p.R258 were associated with significantly increased risk for aortic events, whereas p.R185Q and p.R118Q mutations showed significantly lower risk of aortic events compared to other mutations.
Conclusions
ACTA2 mutations are associated with high risk of presentation with an acute aortic dissection. The lifetime risk for an aortic event is only 76%, suggesting that additional environmental or genetic factors play a role in expression of aortic disease in individuals with ACTA2 mutations.
Keywords: thoracic aortic aneurysms, acute aortic dissections, ACTA2, smooth muscle α-actin
INTRODUCTION
Up to 20% of patients with thoracic aortic disease have a family history of the condition1,2. Familial thoracic aortic aneurysms and dissections (FTAAD) is an autosomal dominant disorder that is characterized by dilation or dissection of the thoracic aorta in families who do not have Marfan syndrome (MFS; OMIM #154700) or a related connective tissue disorder. ACTA2 (actin, alpha-2, smooth muscle, aorta; OMIM *102620) is the most frequently mutated gene causing FTAAD and is responsible for 12–21% of FTAAD cases3–6. ACTA2 encodes the smooth muscle-specific isoform of α-actin, which polymerizes to form the thin filament of the smooth muscle contractile filament. In addition to thoracic aortic aneurysms and dissections, preliminary studies have also shown significant correlations between specific ACTA2 mutations and increased risk for early onset stroke or coronary artery disease7. Additionally, ACTA2 missense mutations that disrupt arginine 179 lead to a distinctive multisystemic smooth muscle dysfunction syndrome characterized by aortic and cerebrovascular disease, fixed dilated pupils, hypotonic bladder, intestinal hypoperistalsis and pulmonary hypertension8. This particular mutation causes severe and early onset vascular disease and has only been identified as a de novo mutation in affected individuals. Mutations in genes encoding other proteins involved in smooth muscle cell contraction also cause an inherited predisposition to thoracic aortic disease, including MYH11 (myosin, heavy chain 11, smooth muscle; OMIM *160745), MYLK (myosin light chain kinase; OMIM *600922), and PRKG1 (protein kinase, cGMP-dependent, regulatory, type I; OMIM *176894)9–12.
MFS is caused by heterozygous mutations in a single gene, FBN1 (fibrillin 1; OMIM *134797), and predisposes to thoracic aortic disease with distinctive syndromic features. Approximately 64% of individuals with MFS are ascertained due to non-aortic abnormalities before they have an aortic event13. After FBN1 mutations were identified in MFS, it was recognized that mutations in other genes may confer overlapping MFS physical features, including aneurysms and dissections of the aorta, along with aneurysms of the aortic branch vessels and intracranial arteries. These disorders are all caused by mutations of genes in the canonical transforming growth factor-β (TGF-β) pathway, including TGFBR2 (transforming growth factor-beta receptor, type II; OMIM *190182)14–19.
Here we describe the aortic diseases in a large case series of individuals with ACTA2 mutations and provide probability estimates for aortic events, describe the clinical presentation of ACTA2-related aortic disease and identify the distinctive challenges in the diagnosis and management of these individuals. Appropriate integration of this data into genetic counseling, surveillance and medical management may lead to reduction of the extensive morbidity and mortality associated with ACTA2-related aortic disease.
MATERIALS AND METHODS
The University of Texas Health Science Center at Houston Institutional Review Board approved this study and informed consent was obtained from study participants. Index cases and their relatives who were identified with ACTA2 mutations by our research laboratory (53 families) and DNA diagnostic laboratories (28 families) were included in this study. 277 individuals from 81 families with 41 different ACTA2 mutations were studied, including 16 obligate carriers and 25 relatives, who had a 50% risk of inheriting the mutation and were affected with aortic or premature onset arterial disease.
Demographic data, diagnosis of aortic aneurysm or dissection, age at diagnosis or last aortic imaging, management and outcome of aortic disease were collected from medical records, autopsies and death records (E.S.R. and D.M.M.). When medical records were not available, aortic disease diagnoses were judged as highly probable based on medical history reported by the index case, i.e. history of surgical repair or doctor’s verbal report. Clinical data for individuals outside the USA were abstracted by collaborators (A.C., G.A., C.B., G.J., T.M., L.A., P.A., B.L.) using standard data collection forms. When available, computed tomographic (CT), magnetic resonance (MR) and echocardiographic images were requested and reviewed by physicians with expertise in aortic disease (S.P., A.E.) and aortic measurements at standard anatomical positions were obtained 20. Thoracic aortic dissections were described using the Stanford classification as type A (aortic dissections initiating in the ascending aorta that may extend to the descending aorta) or type B (initiating in the descending thoracic aorta just distal to the origin of the left subclavian artery or aortic arch). Aortic events were defined as aortic dissections or surgical repair of aortic aneurysms.
Sequencing and genotyping were performed according to previously described protocols3. There are no polymorphisms in ACTA2 that disrupt the translated protein in the NHLBI GO Exome Sequencing Project Exome Variant Server (http://evs.gs.washington.edu/EVS/), and thus all variants disrupting the protein sequence are classified as most likely deleterious. In addition, family studies to confirm co-segregation of variant with disease and bioinformatic evidence were used to evaluate pathogenicity of the variants. The ACTA2 variants included in this study alter evolutionary conserved residues and all are predicted to be damaging by the majority of protein prediction function tools. Individuals with mutations that alter the same amino acid residue were analyzed together, i.e. p.R39C, p.R39G and p.R39H were grouped together; p.R179C, p.R179H and p.R179L together; p.R198C and p.R198H together; and p.R258C and p.R258H together, and these groups were referred to based on the altered amino acid, i.e. p.R39, p.R179, p.R198 and p.R258 substitutions, respectively. Analyses of aortic events were performed for all ACTA2 mutations and recurrent mutations that occur in 2 or more families, i.e. p.R39, p.R179, and p.R258, p.R212Q, p.R149C, p.R185Q, p.R118Q, and p.G160D substitutions.
Categorical variables are presented in frequencies and percentages. Continuous variables are presented as median and interquartile range (IQR). Comparison of two-sample medians was performed using the Wilcoxon rank-sum test. Comparison of two-sample proportions was done using the Fisher’s exact test. Kaplan-Meier method was used to determine the cumulative risk of aortic event using first aortic event as endpoint and age at event or last follow up without aortic event as follow up time. Log-rank test was used to compare failure curves. Cox regression analyses were performed to adjust for intra-familial correlations and effects of potential moderators such as gender and race. Statistical analysis was performed using the STATA software program (Release 12; College Station, TX).
RESULTS
Description of study population
Table 1 summarizes the characteristics of the study population, including age at disease presentation or last follow-up, gender, race and ACTA2 mutation. Supplemental Table 1 lists all the mutations identified, predicted functional effects, the number of families and individuals assessed and frequency and age of aortic events for each mutation. In total, 41 ACTA2 mutations were studied, of which 40 were missense mutations predicted to produce a mutant α-actin monomer and one variant was predicted to lead to aberrant splicing of exon 8 and produce a protein truncated at glycine 270 and 35 aberrant amino acids added. The ACTA2 mutations were all heterozygous except for one homozygous missense mutation (p.W88R), which was identified in a 12-year-old female with patent ductus arteriosus and thoracoabdominal aneurysm due to a chronic descending dissection.
Table 1.
Demographic characteristics and ACTA2 mutations of the study population and groups with and without aortic events.
| Variable | All (n= 277) | With aortic events (n= 132) | Without aortic events (n= 145) | p value |
|---|---|---|---|---|
| Median age (IQR) | 34 (20–49) | 34.5 (24–45) | 34 (15–55) | 0.995 |
| Gender | 0.003 | |||
| Male | 146 (52.7%) | 82 (62.1%) | 64 (44.1%) | |
| Female | 131 (47.3%) | 50 (37.9%) | 81 (55.9%) | |
| Race | 0.03 | |||
| Caucasian | 220 (79.4%) | 112 (84.8%) | 108 (74.5%) | |
| Non-Caucasian | 57 (20.6%) | 20 (15.2%) | 37 (25.5%) | |
| ACTA2 substitutions | ||||
| p.R149C | 69 (24.9%) | 33 (25%) | 36 (24.8%) | 0.97 |
| p.R39 | 28 (10.1%) | 13 (9.8%) | 15 (10.3%) | 0.89 |
| p.R258 | 23 (8.3%) | 15 (5.4%) | 8 (5.5%) | 0.08 |
| p.R179 | 20 (7.2%) | 7 (5.3%) | 13 (9.0%) | 0.24 |
| p.R212Q | 16 (5.8%) | 6 (4.5%) | 10 (6.9%) | 0.4 |
| p.R118Q | 14 (5.0%) | 8 (6.1%) | 6 (4.1%) | 0.47 |
| p.G160D | 9 (3.2%) | 4 (3%) | 5 (3.4%) | 0.84 |
| p.R185Q | 9 (3.2%) | 2 (1.5%) | 7 (4.8%) | 0.12 |
| Other mutations | 89 (32.1%) | 44 (33.3%) | 45 (31.0%) | 0.68 |
Frequency and age of onset of aortic events
Aortic events, defined as aortic dissections or surgical repair of aortic aneurysms, occurred in 47.6% of individuals assessed, and the vast majority of these individuals presented with thoracic aortic dissections (87.9%) as shown in Table 2. Type A dissections occurred in 53.8% of individuals at a median age of 36 years (IQR 26–45). Type B dissections occurred less frequently (21.2%) at younger ages (median age 27 years; IQR 18–41; p= 0.01) compared to Type A dissections. The site of dissection was unspecified in 12.9% of individuals. Fewer individuals (12.1%) underwent elective surgical repair of ascending aortic aneurysms at a median age of 35.5 years (IQR 14.5–47.5).
Table 2.
Disease presentation and status of ACTA2 mutation carriers with and without aortic events (n= 277).
| Variable | Frequency (%) | Median age (IQR) | Min-max age |
|---|---|---|---|
| With aortic event | 132 (47.6) | 34.5 (24–45) | 11–76 |
| Thoracic aortic dissection | 116 (87.9) | 34.5 (25–45) | 12–76 |
| Type A | 71 (53.8) | 36 (26–45) | 15–69 |
| Type B | 28 (21.2) | 27 (18–41) | 12–49 |
| Unspecified site | 17 (12.9) | 37 (28–51) | 12–76 |
| Ascending aortic aneurysm repair | 16 (12.1) | 35.5 (14.5–47.5) | 11–67 |
| Without aortic event | 145 (52.4) | 34 (15–55) | 1–85 |
| Ascending aortic aneurysm | 25 (17.2) | 47 (9–57) | 1–77 |
| Descending aortic aneurysm | 1 (0.7) | 44 | |
| No aortic disease | 67 (46.2) | 33 (16–51) | 1–76 |
| Unknown aortic disease status | 52 (35.9) | 27 (14.5–56) | 1–85 |
More than half of the individuals with ACTA2 mutations (52.4%) did not report an aortic event. Of these individuals, 17.2% had ascending aortic aneurysm that did not require surgical repair, 46.2% were screened by thoracic imaging (CT, MRI or echocardiography) and were not found to have an aneurysm or dissection and 35.9% have unknown disease status.
Individuals with ACTA2 mutations who experienced aortic events were not significantly older than counterparts without an aortic event when analyzed in aggregate (p=0.995) or in subgroups with recurrent mutations. One notable exception was that individuals with mutations disrupting the R179 without aortic event were significantly younger than those with aortic events (5 vs. 14 years; p=0.001). Aortic events were more prevalent in men (62.1%) than in women (37.9%; p = 0.003), but the median age at the first aortic event did not differ significantly between men and women (33 versus 35 years; p= 0.4). Furthermore, the type of aortic dissection (A versus B dissection) was not influenced by gender (p=0.96). More aortic events were observed among individuals of European descent (p= 0.03).
Comparison of aortic disease presentation with FBN1 and TGFBR2 cohorts
Compared to cohorts of individuals with FBN1 and TGFBR2 mutations13, a greater percentage of probands with ACTA2 mutations (84%) were ascertained after presenting with an aortic event (Table 3). More individuals with FBN1 (80%) or TGFBR2 (91%) mutations were identified with aortic root dilation before developing an aortic event compared to only 17% of individuals with ACTA2 mutations. Among the individuals with aortic events, frequency of thoracic aortic dissections were lower for individuals with FBN1 (42%) and TGFBR2 (52%) mutations compared to patients with ACTA2 mutations (88%). Additionally, individuals with ACTA2 mutations presented with type B dissections more frequently and at younger ages compared to individuals with FBN1 and TGFBR2 mutations.
Table 3.
Comparison of aortic disease presentation among individuals with FBN1, TGFBR2, and ACTA2 mutations.
| Variable | FBN1 | TGFBR2 | ACTA2 |
|---|---|---|---|
| Number of individuals | 243 | 71 | 277 |
| Mean age | 30±16 | 29±17 | 38±20 |
| Number of probands | 122 | 26 | 81 |
| Percent of probands ascertained due to TAAD | 36% | 65% | 84% |
| With aortic event* | 74 (30%) | 25 (35%) | 132 (48%) |
| Thoracic aortic dissection | 31 (42%) | 13 (52%) | 116 (88%) |
| Type A | 23 (31%); 39±9 | 10 (40%); 38±12 | 71 (54%); 36±12 |
| Type B | 8 (11%); 44±10 | 3 (12%); 34±6 | 28 (21%); 29±12 |
| Aneurysm repair | 43 (58%); 39±13 | 12 (48%); 35±16 | 16 (12%); 33±18 |
| Without aortic event | 169 (70%) | 46 (65%) | 145 (52%) |
| Ascending aortic aneurysm | 135 (80%) | 42 (91%) | 25 (17%) |
Frequency of aortic event or diagnosis and mean age ± standard deviation are provided.
Cumulative risk of aortic event
The overall cumulative risk of an aortic event associated with ACTA2 mutations was 0.76 (95% CI 0.64, 0.86) at 85 years. Mutation-specific time to event analyses for 8 recurrent ACTA2 mutations showed that individuals who harbor mutations that disrupt p.R179 and R258 have significantly increased risks for aortic events at younger ages compared to all other mutations, with p-values <0.0001 and 0.02 respectively (Figure 1). The cumulative risk of aortic event among individuals with p.R179 substitutions was 1.0 by the age of 25 years and 0.90 (95% CI 0.68, 0.99) by age 63 for individuals with p.R258 substitutions. At the other end of the aortic disease risk spectrum, the cumulative risk of an aortic event for the p.R185Q substitution was only 0.57 by age 77 (95% CI 0.15, 0.99), which was significantly lower than the other ACTA2 mutations (p= 0.04). Cumulative risks of aortic events for p.R39, p.R212Q, p.R149C, p.R118Q, and p.G160D substitutions were not significantly different from all other ACTA2 mutations.
Figure 1.

Kaplan-Meier failure functions demonstrating (A) overall cumulative risk of aortic event for all ACTA2 mutations and significantly different cumulative probabilities of aortic events for (B) p.R179, (C) p.R258, and (D) p.R185Q substitutions compared to all other ACTA2 mutations.
Using Cox regression analysis with correction for intra-familial correlation, we found that individuals who carry mutations disrupting p.R179 (HR 26.39, 95% CI 10.40–66.95) and p.R258 (HR 2.05 (95% CI 1.17, 3.58) experienced significantly more aortic events than those with other ACTA2 mutations (Table 4). In contrast, the risk of aortic events associated with p.R185Q (HR 0.28, 95% CI 0.11–0.72) and p.R118Q (HR 0.79, 95% CI 0.64–0.98) substitutions were significantly lower than other ACTA2 mutations. These findings remained significant after adjustment for gender and race except for p.R258 substitutions. However, analysis without the individuals with p.R179 substitutions showed a significant hazard ratio for p.R258 (2.02, 95% CI 1.03, 3.95; p value 0.041) compared to other mutations.
Table 4.
Hazard ratios for aortic events associated with recurrent ACTA2 substitutions corrected for intra-familial correlations and adjusted for race and gender.
| Substitution | Unadjusted HR | 95% CI | p value | Adjusted HR | 95% CI | p value |
|---|---|---|---|---|---|---|
| p.R149C | 1.04 | 0.66, 1.65 | 0.853 | 0.95 | 0.59, 1.52 | 0.831 |
| p.R39 | 0.90 | 0.58, 1.37 | 0.608 | 1.11 | 0.70, 1.76 | 0.653 |
| p.R258 | 2.05 | 1.17, 3.58 | 0.012 | 1.85 | 0.98, 3.50 | 0.058* |
| p.R179 | 26.39 | 10.40, 66.95 | <0.001 | 27.86 | 11.87, 65.38 | <0.001 |
| p.R212Q | 1.00 | 0.60, 1.66 | 0.987 | 1.05 | 0.60, 1.82 | 0.868 |
| p.R118Q | 0.79 | 0.64, 0.98 | 0.033 | 0.71 | 0.55, 0.92 | 0.01 |
| p.G160D | 0.64 | 0.19, 2.16 | 0.469 | 0.52 | 0.15, 1.86 | 0.318 |
| p.R185Q | 0.28 | 0.11, 0.72 | 0.008 | 0.36 | 0.18, 0.69 | 0.002 |
Analysis of p.R258 substitutions without the individuals with p.R179 substitutions showed a significant adjusted hazard ratio of 2.02 (95% CI 1.03, 3.95, p value 0.041).
Outcome of aortic events
Of 71 individuals who had type A dissections, 75% (53) underwent successful surgical repair and 25% (18) died either prior to intervention or peri-operatively. Among 28 individuals with type B dissections, two died prior to intervention or post-operatively, 8 were treated medically, and 18 underwent surgery (14 open repairs, 4 endovascular repairs). In 9 cases with acute type B dissections, the indications for urgent surgery were visceral ischemia (3), limb ischemia (4) or aortic rupture (1). Of the 17 individuals with dissection at an unspecified site, 13 died at presentation. There were no deaths associated with elective repair of an aortic aneurysm. Therefore, the overall risk of mortality associated with the first aortic event in individuals with ACTA2 mutations was 25% (33/132) at a median age of 36 years (IQR 28–45). If 10 unexplained sudden deaths were included in the analysis as undiagnosed dissections, the risk of mortality would increase to 30%.
Eleven of the individuals with ACTA2 mutations who did not have an aortic event died at a median age of 45 years (IQR 28–63). Six of these deaths were due to other cardiovascular diseases: surgical complications related to moyamoya disease, intracranial aneurysm repair or coronary bypass, acute coronary syndrome, and acute thrombus on prosthetic aortic valve. Two individuals died from unrelated causes. The cause of death was not determined for three individuals, including a child with a p.R179H substitution who had an autopsy.
Clinical features associated with first aortic event
To characterize the first aortic event in individuals with ACTA2 mutations, we analyzed 70 available clinical records for presenting symptoms, medical histories prior to event, aortic disease, surgical approaches, and outcomes (summarized in Supplemental Table 2). This subgroup consisted of 10 elective ascending aortic aneurysm repairs, 41 acute type A dissections, 10 acute type B dissections and 9 chronic type B dissections.
Figure 2 shows representative images of ascending and aortic root disease associated with ACTA2 mutations. In 10 individuals who presented for elective repair of ascending aortic aneurysms, the maximum diameter was either in the aortic root (n=5) or in the ascending aorta (n=5) and ranged from 4.2–6.5 cm (median 5.2, IQR 5–5.5). Among 24 individuals with type A dissections whose aortic measurements were available, the maximum diameter of the dissected segment varied from 3.8- 9.5 cm (median 5.75, IQR 4.7–6.75) as shown in Figure 2C. One-third of these individuals (8/24; ages 27–60 years; 50% male) dissected when the maximum diameter was <5 cm. Three of the eight individuals who dissected at diameters <5 cm experienced the dissection during the peripartum period21. There were 9 individuals with aneurysms or dissections that involved other aortic segments.
Figure 2.
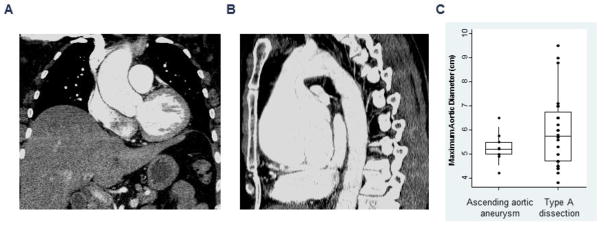
(A) Contrast-enhanced coronal CT image showing a 4.3 cm ascending aortic aneurysm and (B) sagittal view of the thoracic aorta showing marked aneurysmal dilatation of the aortic root and proximal ascending aorta measuring 8.8. cm. (C) Box-plots showing maximum aortic diameters in individuals undergoing elective repair of ascending aortic aneurysms and urgent surgical repair of type A aortic dissections. Circle symbols represent individual aortic measurements, and the middle, lower and upper lines of the box plots represent the median, 25th and 75th percentiles, respectively.
Among 10 individuals who presented for prophylactic repair of ascending aortic aneurysms, 4 had composite valve graft root replacement, 5 had valve-sparing aortic root replacement, 1 had ascending aortic replacement, and 3 also had partial or total aortic arch replacement. Of the 41 individuals with type A dissection, 42% (17) had replacement of the ascending aorta, 44% (18) had composite valve graft root replacement, and 5% (2) had a valve-sparing aortic root replacement; 30% (12) also had partial or total aortic arch replacement. Of the 10 individuals with acute type B dissection, 3 had open repair, 3 had endovascular repair, and 4 received medical treatment alone. Of the 9 individuals with chronic type B dissection, 8 had extent I or II thoracoabdominal aortic aneurysm (TAAA) repair.
The vast majority of these individuals reported symptoms prior to presentation. Among individuals with acute thoracic aortic dissections, the most common symptom was pain in 84% of individuals but up to 20% of individuals complained of various other symptoms besides pain (Supplemental Table 2). ACTA2 individuals with acute dissections were likely to report a family history of aortic disease (56%), but only 24% reported a personal history of hypertension.
Among individuals with acute aortic dissections, 46% had major in-hospital complications (14% in-hospital deaths), whereas only two of 10 individuals who presented for elective repair of aortic aneurysm had major complications. Median length of hospitalization was longest for individuals with chronic type B dissections (19, IQR 14–38 days), followed by individuals with acute type A or B dissections (11, IQR 8–18 days) and individuals who had elective aortic repairs (6, IQR 5–9 days).
DISCUSSION
Mutations in the ACTA2 gene are responsible for 12–21% of familial thoracic aortic disease without features of connective tissue disorders based on analyses of cohorts worldwide3–6. Individuals with MFS are also predisposed to thoracic aortic disease, but these individuals frequently come to medical attention due to their skeletal and ocular features, at which time approximately 80% of them are diagnosed with aortic dilatation13. In recent years, identification of MFS individuals and management of the thoracic aortic disease has led to decreased rates of acute dissections; about 40% of aortic events in MFS individuals were aortic dissections13. In contrast, most individuals with ACTA2 mutations do not have syndromic features and are often diagnosed upon presentation with aortic events, which are overwhelmingly acute aortic dissections. Nearly half of the individuals in this study population had experienced aortic events, and 88% of these events were thoracic aortic dissections associated with 25% mortality. These data imply that the absence of syndromic features leads to delayed recognition of patients with ACTA2 mutations, therefore making them more likely to present with acute aortic dissections than individuals with MFS.
Individuals with ACTA2 mutations present with both type A and type B aortic dissections at significantly younger ages than individuals enrolled in the International Registry of Acute Aortic Dissection study (IRAD)22 and are likely to report a family history of thoracic aortic disease. Type A dissections are more predominant among individuals with ACTA2 mutations, but type B aortic dissections occur at more than twice the rates reported for individuals with FBN1, TGFBR2, and SMAD3 mutations13, 23. Surprisingly, type B dissections due to ACTA2 mutations occur at significantly younger ages than type A dissections and are frequently complicated by aortic rupture and visceral and limb ischemia, requiring immediate surgical intervention. None of the individuals with type B dissections had prior diagnosis of aortic abnormalities. Therefore, anyone presenting with an acute aortic dissection without syndromic features and with a family history of the disease and young people with aortic dissections, especially type B aortic dissections, should raise a suspicion for an underlying ACTA2 mutation. Because these individuals present with complicated dissections often requiring surgical intervention, transfer to a tertiary care center should be considered.
The diameter of the aortic root and ascending aorta at the time of dissection varied widely, but one-third of the individuals experienced aortic dissections at diameters <5 cm. This is in contrast to individuals with MFS, who rarely experience dissection at diameters <5 cm24. Half of the prophylactic repairs of the ascending aorta were performed at aortic diameters between 5 and 5.5 cm, which follow the current recommendations established for MFS25. However, the significant number of individuals who had dissections at diameters <5.0 cm support surgical repair of the aorta when the diameter is 4.5 cm or greater. Future studies will focus on identifying the risk factors that predict dissections in individuals with minimal aortic enlargement so that factors can be modified to prevent these dissections or prophylactic aortic repair can be considered.
Ascending aortic aneurysms associated with ACTA2 mutations involved the aortic root and ascending aorta and often extended into the arch. Surgical repair often involved root replacement with a composite valve graft conduit or valve-sparing techniques, and nearly half of the individuals had concomitant partial or total aortic arch replacement, illustrating the extent of aneurysmal disease. In contrast, aortic aneurysms in individuals with MFS more often involve the aortic root at the sinuses of Valsalva, initially sparing the distal ascending aorta and aortic arch. Aortic surveillance in individuals with ACTA2 mutations should therefore include measurements of the entire ascending aorta at the level of the sinuses of Valsalva, the sinotubular junction, the mid-ascending aorta, and the aortic arch. Aortic imaging by CT, MR, or transesophageal echocardiography should be considered if the aorta is not adequately evaluated by routine echocardiography. The extent of involvement of the aortic root, ascending aorta and aortic arch should be taken into account when planning thoracic aortic repair, and consideration should be given to repairing all of these regions even if a specific location is not yet enlarged. Additionally, the extensive aortic disease in these individuals indicates that surveillance of the entire thoracic aorta should be performed routinely.
ACTA2 mutations demonstrate reduced penetrance for aortic disease and variability in disease presentation and age of onset3. The overall cumulative risk (i.e., penetrance) of an aortic event at age 85 years is 76%; conversely, the lifetime probability of not experiencing an aortic event even at advanced ages is 24%. However, certain mutations are associated with higher risk of aortic events. For example, the p.R179 and p.R258 substitutions have significantly higher risk of aortic events and have been associated with more severe vascular phenotype, including patent ductus arteriosus, early onset stroke, and cerebrovascular disease characterized by dilatation of the proximal internal carotid arteries and occlusion of the terminal portions, and dramatic tortuosity of the small arteries in the retina7, 26, 27.
ACTA2 mutations cause vascular disease that is different from MFS and is also distinct from the vascular disease associated with mutations disrupting TGF-β signaling pathways genes (TGFBR1, TGFBR2, SMAD3, and TGFB2). Mutations of these genes cause aortic root aneurysms leading to acute aortic dissections, but also aneurysms and dissections of other arteries, including the abdominal aorta, intracranial arteries, and branches arising from the aorta. A subset of mutations in TGFBR1 and TGFBR2 cause Loeys-Dietz syndrome associated with early onset and aggressive vascular disease28. In contrast, aneurysms and dissections of medium and small arteries are rare in individuals with ACTA2 mutations and only in the great arteries arising from the aortic arch in individuals with severe ACTA2 mutations such as mutations disrupting p.R179 and p.R25826. Additionally, intracranial artery aneurysms rarely occur in individuals with ACTA2 p.R258 and p.R39 substitutions7. Furthermore, mitral valve prolapse and regurgitation can occur in MFS individuals and in individuals with mutations in the TGF-β pathway genes13, 19, 23, 29 but are rare in individuals with ACTA2 mutations. Mitral valve prolapse was present in only 3% of individuals with ACTA2 mutations, which is significantly lower than rates reported in individuals with FBN1 (45%) and TGFBR2 (21%) mutations and similar to the population prevalence (2.4%)13, 30. The significant differences in clinical complications associated with these genes support that the clinical management of these individuals needs to be based on the underlying gene, and in the case of some ACTA2 mutations, the specific alteration.
In summary, this study demonstrates that ACTA2 mutations are associated with high risk of presentation with aortic dissections and death and variable disease presentation that is dictated in part by the mutation, but likely modified by other genetic or environmental factors. Testing for ACTA2 mutation should be strongly considered in individuals with the following: early onset thoracic aortic dissection, especially type B dissection; family history of thoracic aortic aneurysm or dissection; peripartum aortic dissection; and clinical features previously associated with ACTA2 mutations, such as early onset ischemic strokes and coronary artery disease, livedo reticularis, iris flocculi, and features of multisystemic smooth muscle cell dysfunction syndrome. Early diagnosis of mutation carriers and proper management, including aortic surveillance, blood pressure management, and timely surgical repair of ascending aortic aneurysms, may help reduce the risk of aortic complications and death associated with ACTA2 mutations.
The reduced penetrance and variable age of onset and severity suggest that some of these risks may be prevented by modifying risk factors and initiating therapies to decrease stresses on the aorta. Further studies are necessary to investigate environmental and genetic modifiers that increase the risk for early dissections as well as those that allow some individuals to live to advanced ages without aortic events. Future studies also need to address whether medical therapy such as β-adrenergic antagonists or angiotensin type I receptor blocking agents can mitigate the risk for aortic events.
Supplementary Material
Acknowledgments
We are grateful to the research participants and the physicians, genetic counselors and investigators who have referred individuals to this study and helped provide clinical data (Supplemental Material). We also thank Dr. Robert Lasky for his help with the data analysis and Dr. Regie L. Santos-Cortez for her help with the protein bioinformatics analyses.
FUNDING SOURCES
The following sources provided funding for these studies: RO1HL62594 (D.M.M.), P50HL083794-01 (D.M.M.), P01HL110869-01 (D.M.M.), UL1 RR024148 (University of Texas Health Science Center at Houston), Vivian L. Smith Foundation (D.M.M.), TexGen Foundation (D.M.M.), and the Richard T. Pisani Funds (D.M.M.).
Footnotes
DISCLOSURES
None.
References
- 1.Biddinger A, Rocklin M, Coselli J, Milewicz DM. Familial thoracic aortic dilatations and dissections: a case control study. J Vasc Surg. 1997;25:506–511. doi: 10.1016/s0741-5214(97)70261-1. [DOI] [PubMed] [Google Scholar]
- 2.Coady MA, Davies RR, Roberts M, Goldstein LJ, Rogalski MJ, Rizzo JA, et al. Familial patterns of thoracic aortic aneurysms. Arch Surg. 1999;134:361–367. doi: 10.1001/archsurg.134.4.361. [DOI] [PubMed] [Google Scholar]
- 3.Guo DC, Pannu H, Papke CL, Yu RK, Avidan N, Bourgeois S, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 4.Morisaki H, Akutsu K, Ogino H, Kondo N, Yamanaka I, Tsutsumi Y, et al. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD) Hum Mutat. 2009;30:1406–1411. doi: 10.1002/humu.21081. [DOI] [PubMed] [Google Scholar]
- 5.Disabella E, Grasso M, Gambarin FI, Narula N, Dore R, Favalli V, et al. Risk of dissection in thoracic aneurysms associated with mutations of smooth muscle alpha-actin 2 (ACTA2) Heart. 2011;97:321–326. doi: 10.1136/hrt.2010.204388. [DOI] [PubMed] [Google Scholar]
- 6.Renard M, Callewaert B, Baetens M, Campens L, MacDermot K, Fryns JP, et al. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFbeta signaling in FTAAD. Int J Cardiol. 2013;165:314–321. doi: 10.1016/j.ijcard.2011.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84:617–627. doi: 10.1016/j.ajhg.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milewicz DM, Ostergaard JR, la-Kokko LM, Khan N, Grange DK, Mendoza-Londono R, et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A. 2010;152A:2437–2443. doi: 10.1002/ajmg.a.33657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pannu H, Tran-Fadulu V, Papke CL, Scherer S, Liu Y, Presley C, et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum Mol Genet. 2007;16:3453–3462. doi: 10.1093/hmg/ddm201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu L, Vranckx R, Khau Van KP, Lalande A, Boisset N, Mathieu F, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, et al. Mutations in Myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010;87:701–707. doi: 10.1016/j.ajhg.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo DC, Regalado E, Casteel DE, Santos-Cortez RL, Gong L, Kim JJ, et al. Recurrent Gain-of-Function Mutation in PRKG1 Causes Thoracic Aortic Aneurysms and Acute Aortic Dissections. Am J Hum Genet. 2013;93:398–404. doi: 10.1016/j.ajhg.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Attias D, Stheneur C, Roy C, Collod-Beroud G, Detaint D, Faivre L, et al. Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in Marfan syndrome and related disorders. Circulation. 2009;120:2541–2549. doi: 10.1161/CIRCULATIONAHA.109.887042. [DOI] [PubMed] [Google Scholar]
- 14.Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, Harada N, Morisaki T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pannu H, Fadulu VT, Chang J, Lafont A, Hasham SN, Sparks E, et al. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–520. doi: 10.1161/CIRCULATIONAHA.105.537340. [DOI] [PubMed] [Google Scholar]
- 16.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 17.van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 18.Regalado ES, Guo DC, Villamizar C, Avidan N, Gilchrist D, McGillivray B, et al. Exome Sequencing Identifies SMAD3 Mutations as a Cause of Familial Thoracic Aortic Aneurysm and Dissection With Intracranial and Other Arterial Aneurysms. Circ Res. 2011;109:680–686. doi: 10.1161/CIRCRESAHA.111.248161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boileau C, Guo DC, Hanna N, Regalado ES, Detaint D, Gong L, et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44:916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE, Jr, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121:e266–e369. doi: 10.1161/CIR.0b013e3181d4739e. [DOI] [PubMed] [Google Scholar]
- 21.Regalado ES, Guo DC, Estrera AL, Buja LM, Milewicz DM. Acute aortic dissections with pregnancy in women with ACTA2 mutations. Am J Med Genet A. 2014;164:106–112. doi: 10.1002/ajmg.a.36208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagan PG, Nienaber CA, Isselbacher EM, Bruckman D, Karavite DJ, Russman PL, et al. The International Registry of Acute Aortic Dissection (IRAD): new insights into an old disease. JAMA. 2000;283:897–903. doi: 10.1001/jama.283.7.897. [DOI] [PubMed] [Google Scholar]
- 23.van dL I, van der LD, Oei EH, Bos PK, Bessems JH, Bierma-Zeinstra SM, et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J Med Genet. 2012;49:47–57. doi: 10.1136/jmedgenet-2011-100382. [DOI] [PubMed] [Google Scholar]
- 24.Jondeau G, Detaint D, Tubach F, Arnoult F, Milleron O, Raoux F, et al. Aortic event rate in the Marfan population: a cohort study. Circulation. 2012;125:226–232. doi: 10.1161/CIRCULATIONAHA.111.054676. [DOI] [PubMed] [Google Scholar]
- 25.Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE, Jr, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology,American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons,and Society for Vascular Medicine. J Am Coll Cardiol. 2010;55:e27–e129. doi: 10.1016/j.jacc.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 26.Munot P, Saunders DE, Milewicz DM, Regalado ES, Ostergaard JR, Braun KP, et al. A novel distinctive cerebrovascular phenotype is associated with heterozygous Arg179 ACTA2 mutations. Brain. 2012;135:2506–2514. doi: 10.1093/brain/aws172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moller HU, Fledelius HC, Milewicz DM, Regalado ES, Ostergaard JR. Eye features in three Danish patients with multisystemic smooth muscle dysfunction syndrome. Br J Ophthalmol. 2012;96:1227–1231. doi: 10.1136/bjophthalmol-2011-301462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 29.Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454–466. doi: 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Freed LA, Levy D, Levine RA, Larson MG, Evans JC, Fuller DL, et al. Prevalence and clinical outcome of mitral-valve prolapse. N Engl J Med. 1999;341:1–7. doi: 10.1056/NEJM199907013410101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.