

Abstract
Oligomeric forms of the Parkinson’s disease-causing protein α-synuclein are suspected to mediate neurodegeneration, but the mechanisms are not understood. The present study of Shrivastava et al (2015) provides a fresh insight into this old mystery. α-Synuclein oligomers are shown by a combination of top state-of-the-art biochemical and super-resolution microscopy methods to sequester the neuronal sodium–potassium pump. Such perturbation of ion currents would ultimately lead to Ca2+ excitotoxicity.
See also: AN Shrivastava et al (October 2015)
α-Synuclein is a conformationally dynamic protein abundant in synapses of the central nervous system. Its physiological role is related to synaptic vesicle cycling, but the exact mode of action is not well understood. α-Synuclein became infamous when it was recognized as a major genetic risk factor of Parkinson’s disease (PD) as well as the building block of the characteristic neuropathological lesions. Despite enormous research efforts over the past 15 years, it is still unknown how α-synuclein causes neurological deficits in PD and related disorders. PD is one of the common age-dependent neurodegenerative proteinopathies. Therefore, it is likely that the aggregation process of α-synuclein into amyloid-like material plays a role in the pathomechanisms. Standard histological methodology allows practically only detection of the aggregation end products, namely intraneuronal inclusions called Lewy bodies. Mature α-synuclein aggregates are water-insoluble and proteinase-resistant, thus biologically quite inert. Therefore, it is believed that aggregation intermediates are the culprits causing neuronal dysfunction and degeneration in α-synucleinopathies.
The amyloidogenic pathway of α-synuclein is populated by a variety of conformers and oligomeric assemblies until reaching the mature amyloid fibrils. We are only beginning to understand the biological activities of α-synuclein oligomers (ASOs). Seminal work indicated that protofibrillar α-synuclein has pore-forming activity in vitro, potentially causing membrane Ca2+ permeabilization (Volles et al, 2001). Early cell culture studies confirmed that overexpression of α-synuclein or application of certain ASO species enhanced Ca2+ permeability, apparently via pore-forming or leak channel mechanisms (Furukawa et al, 2006; Danzer et al, 2007; Feng et al, 2010). On the other hand, it was noted that α-synuclein could also enhance voltage-operated Ca2+ channel activity (Hettiarachchi et al, 2009; Melachroinou et al, 2013). Thus, it is debated whether α-synuclein alters neuronal Ca2+ homeostasis via pores or by influencing Ca2+ channels. The present study of Shrivastava et al (2015) adds a novel aspect to this issue. They report that ASOs inhibit the neuronal sodium/potassium pump leading to inefficient membrane repolarization and hence more Ca2+ influx.
The authors added S-tagged monomeric, oligomeric, and fibrillar α-synuclein preparations to pure rat cortical neuron cultures for pull-down experiments. Interacting proteins were analyzed by mass spectrometry. One interactor was particularly intriguing: the neuron-specific subunit α3 of the Na+/K+-ATPase (NKA). It was found that ASOs formed clusters at the surface of neurons, which appeared to sequester α3-NKA. This was correlated with reduced Na+ efflux and enhanced glutamate-induced Ca2+ influx. ASOs acted like the classical NKA inhibitor ouabain, leading to the conclusion that ASOs inhibit neuronal membrane repolarization and hence could sensitize to Ca2+ excitotoxicity, as had been shown elsewhere (Hüls et al, 2011).
Several aspects deserve closer attention in the context of this interesting study. It is remarkable that the relatively large ASOs bind to a defined binding site at α3-NKA (see Fig1). Of course, the exact ASO structure in this cellular paradigm is not known and probably alters in the observed clusters. Nevertheless, the interaction sites roughly mapped in crosslinking experiments would fit with the dimensions of synthetic ASOs, as estimated from small-angle X-ray scattering measurements (Giehm et al, 2011; Lorenzen et al, 2014). ASOs could cover the edge of the ouabain-binding site, extend contacts along the transmembrane domain (assuming that ASOs can integrate into the plasma membrane bilayer) and even reach the β-NKA subunit in the case of larger α-synuclein fibrils. However, more contacts were crosslinked, so the structural details of ASO binding to NKA, and perhaps even other members of the structurally related family of P-type ATPases need to be worked out more systematically. However, conventional biochemistry techniques become limiting when dealing with membrane channels that comprise large hydrophobic regions and the metastable ASOs. Entering such a complex protein level shows the benefit of super-resolution microscopy, whereby transcending the diffraction limit imposed on classical light microscopy allows precise localization of minute oligomeric and fibrillar clusters. The authors employ stochastic optical reconstruction microscopy (STORM) by which hundreds to thousands of images of the sample, each containing only a few arbitrarily switched-on fluorescent molecules, are amalgamated into one image of superb clarity. With an effective resolution increase of up to 10-fold, precise localization of structures as small as ASOs relative to the plasma membrane is made possible. Taking advantage of the single molecule localization inherent in the method, the authors made use of the technique to detect α3-NKA spatial redistribution, showing increased density of α3-NKA clusters in the presence of ASOs. From a technical point of view, this work constitutes a fine example where the application of super-resolution microscopy is used to obtain spatial information conventionally veiled by light diffraction, ultimately leading to an understanding of complex cellular mechanisms or their malfunction.
Figure 1.
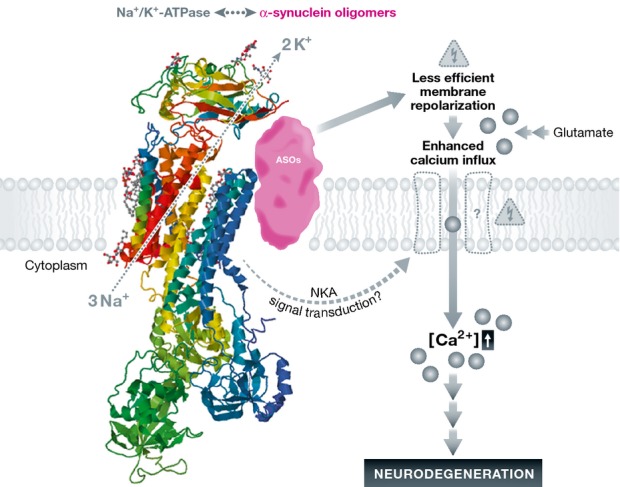
Inhibition of the neuronal α3-NKA isoform by ASOs would reduce membrane repolarization and hence sensitize to glutamate-induced Ca2+ neurotoxicity
The present report shows that ASOs bind to α3-NKA and cause their clustering. As a consequence, NKA activity is reduced. This leads to less efficient membrane repolarization and therefore enhanced Ca2+ influx after glutamate stimulation. Whether this occurs through specific Ca2+ channels remains to be established. One might speculate that such a mechanism could even have a physiological relevance to facilitate neurotransmission, but in excess ASOs may contribute to excitotoxic dying back neurodegeneration. The detail image shows the crystal structure (PDB 4HYT) of NKA complexed with the inhibitor ouabain (ball and stick structure between the yellow and blue transmembrane helices). Synthetic ASO dimensions are roughly drawn to scale (purple shape), revealing appropriate dimensions to roughly cover a loop next to the ouabain binding site (peptide 884–901) and transmembrane helix α8 (peptides 903–928/930), shown in orange. Larger ASOs or fibrils may extend to the β-NKA subunit on top. Additional crosslinks (peptides 603–615, 620–637, 689–697) were observed in the phosphorylation domain (light green) and (peptide 436–448) in the nucleotide-binding domain (green) as well as (peptides 203–217, 218–230, 44–57) in the activation domain (cyan-blue). Chemical crosslinking of lysates is a rather crude method to map such complex interactions, but it cannot be ruled out that α-synuclein interactions occur also at the cytosolic domains of NKA, leading to direct inhibition. Further structural and functional work both in cell-autonomous α-synuclein overexpression models as well as after exogenous ASO administration shall elucidate the effects of α-synuclein assemblies on neuronal ion channels.
ASOs prepared here elicit α3-NKA effects at the given concentrations. Given the conflicting previous results whereby α-synuclein alters Ca2+ fluxes via membrane pores or Ca2+ channels (see above), it is paramount that the authentic nature of patient ASOs is defined, as well as their concentrations in the interstitial fluid and in synapses. Another, related, aspect is the source of ASOs. Exogenous additions of ASOs simulate in first approximation juxtacrine effects. If ASOs come from adjacent neurons, then they must be secreted in unconventional pathways (Marques & Outeiro, 2012). What are the exact conformations of cell-derived ASOs, and are they pure protein assemblies or do they contain additional (exosomal) lipid material and/or transport proteins in vivo? It will be interesting to follow up the present study in co-culture experiments and ex vivo experiments. Alternatively, the effects of ASOs might be autocrine. Indeed, α-synuclein is enriched in pre-synapses. In affected neurons, ASOs could directly bind to α3-NKA immediately after secretion, or even during membrane passage. That way they would enhance Ca2+ influx via channels that remain to be characterized in detail. Autocrine ASO-enhanced Ca2+ influx could account for a direct dying back mechanism. Less efficient spread to the post-synaptic membrane would leave striatal neurons unaffected, accounting for the selective vulnerability observed in PD brain.
In addition to the question where ASOs come from is where they go to. They are taken up into cells quite efficiently. The present study addressed effects on the plasma membrane with short observation times of ≤ 60 min. What happens in more protracted time courses? Note that PD lasts over decades. The neurotoxic mechanism proposed here does not require α-synuclein internalization. Would that indicate that α-synuclein endocytosis acts to clear ASOs from the cell surface, followed by endolysosomal degradation or isolation in solid aggregates? Does the clustering prevent ASO internalization, or on the contrary promote NKA internalization and removal from the neuronal membrane? Either way would be consistent with the authors’ model of NKA downregulation by membrane-clustered ASOs. Finally, is NKA inhibition the prevailing mechanism, or is it restricted to the acute effects studied here? Could a pore-forming activity of ASOs develop more slowly, but then provide the final blow to the damaged neuron? As typical for a great study, the work of Shrivastava et al (2015) pumps a whole host of fresh thoughts and ideas into the field of neurodegenerative proteinopathies.
References
- Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of α-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng LR, Federoff HJ, Vicini S, Maguire-Zeiss KA. α-synuclein mediates alterations in membrane conductance: a potential role for α-synuclein oligomers in cell vulnerability. Eur J Neurosci. 2010;32:10–17. doi: 10.1111/j.1460-9568.2010.07266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Matsuzaki-Kobayashi M, Hasegawa T, Kikuchi A, Sugeno N, Itoyama Y, Wang Y, Yao PJ, Bushlin I, Takeda A. Plasma membrane ion permeability induced by mutant α-synuclein contributes to the degeneration of neural cells. J Neurochem. 2006;97:1071–1077. doi: 10.1111/j.1471-4159.2006.03803.x. [DOI] [PubMed] [Google Scholar]
- Giehm L, Svergun DI, Otzen DE, Vestergaard B. Low-resolution structure of a vesicle disrupting α-synuclein oligomer that accumulates during fibrillation. Proc Natl Acad Sci USA. 2011;108:3246–3251. doi: 10.1073/pnas.1013225108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettiarachchi NT, Parker A, Dallas ML, Pennington K, Hung C-C, Pearson HA, Boyle JP, Robinson P, Peers C. α-Synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells. J Neurochem. 2009;111:1192–1201. doi: 10.1111/j.1471-4159.2009.06411.x. [DOI] [PubMed] [Google Scholar]
- Hüls S, Högen T, Vassallo N, Danzer KM, Hengerer B, Giese A, Herms J. AMPA-receptor-mediated excitatory synaptic transmission is enhanced by iron-induced α-synuclein oligomers. J Neurochem. 2011;117:868–878. doi: 10.1111/j.1471-4159.2011.07254.x. [DOI] [PubMed] [Google Scholar]
- Lorenzen N, Nielsen SB, Buell AK, Kaspersen JD, Arosio P, Vad BS, Paslawski W, Christiansen G, Valnickova-Hansen Z, Andreasen M, Enghild JJ, Pedersen JS, Dobson CM, Knowles TP, Otzen DE. The role of stable α-synuclein oligomers in the molecular events underlying amyloid formation. J Am Chem Soc. 2014;136:3859–3868. doi: 10.1021/ja411577t. [DOI] [PubMed] [Google Scholar]
- Marques O, Outeiro TF. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. 2012;3:e350. doi: 10.1038/cddis.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melachroinou K, Xilouri M, Emmanouilidou E, Masgrau R, Papazafiri P, Stefanis L, Vekrellis K. Deregulation of calcium homeostasis mediates secreted α-synuclein-induced neurotoxicity. Neurobiol Aging. 2013;34:2853–2865. doi: 10.1016/j.neurobiolaging.2013.06.006. [DOI] [PubMed] [Google Scholar]
- Shrivastava AN, Redeker V, Fritz N, Pieri L, Almeida LG, Spolidoro M, Liebmann T, Bousset L, Renner M, Lena C, Aperia A, Melki R, Triller A. α-synuclein assemblies sequester neuronal α3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 2015;34:2408–2423. doi: 10.15252/embj.201591397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, Lee S-J, Rochet J-C, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT., Jr Vesicle permeabilization by protofibrillar α-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]