

Abstract
Primary cilia start forming within the G1 phase of the cell cycle and continue to grow as cells exit the cell cycle (G0). They start resorbing when cells re-enter the cell cycle (S phase) and are practically invisible in mitosis. The mechanisms by which cilium biogenesis and disassembly are coupled to the cell cycle are complex and not well understood. We previously identified the centrosomal phosphoprotein NDE1 as a negative regulator of ciliary length and showed that its levels inversely correlate with ciliogenesis. Here, we identify the tumor suppressor FBW7 (also known as FBXW7, CDC4, AGO, or SEL-10) as the E3 ligase that mediates the destruction of NDE1 upon entry into G1. CDK5, a kinase active in G1/G0, primes NDE1 for FBW7-mediated recognition. Cells depleted of FBW7 or CDK5 show enhanced levels of NDE1 and a reduction in ciliary length, which is corrected in cells depleted of both FBW7 or CDK5 and NDE1. These data show that cell cycle-dependent mechanisms can control ciliary length through a CDK5-FBW7-NDE1 pathway.
Keywords: CDK5, cilia, FBW7, NDE1, p35
See also: AS Nikonova & EA Golemis (October 2015)
Introduction
The primary cilium is an antenna-like organelle present in virtually every cell type of the human body. It is indispensable for human health, as structural defects in primary cilia result in pathological conditions as diverse as kidney cysts and cancer to retinal degeneration and brain malformations (Hildebrandt et al, 2011). It functions as a signaling center for receptor tyrosine kinases and G protein-coupled receptors, as well as the Hedgehog, Notch, and Wnt pathways (Goetz & Anderson, 2010; Christensen et al, 2012). A unique feature of the primary cilium is that its biogenesis and resorption are intimately associated with the cell cycle, as primary cilia form during quiescence and resorb before mitosis, providing an exquisite means to integrate multiple signaling pathways with the cell cycle (Quarmby & Parker, 2005; Pan & Snell, 2007; Plotnikova et al, 2008; Seeley & Nachury, 2010; Kim & Tsiokas, 2011; Sung & Li, 2011). The cellular program that triggers the initiation of cilia formation is not completely understood, but depends on the combinatorial actions of numerous positive and negative regulators of ciliogenesis (Kobayashi & Dynlacht, 2011; Kim & Dynlacht, 2013; Sung & Leroux, 2013). Activity and/or levels of positive regulators of ciliogenesis reach and maintain high levels, whereas inhibitors of ciliogenesis are removed from pre-ciliary structures or destructed at the onset of ciliogenesis (Ishikawa & Marshall, 2011; Kobayashi & Dynlacht, 2011). Ciliogenesis proceeds in an orchestrated fashion initiated by the attachment of Golgi-derived ciliary vesicles to the distal ends of the mother centriole inside the cell, followed by centriole docking to the plasma membrane. The docked mother centriole elongates and the axonemal extension follows. Finally, cilium assembly and maintenance are achieved by intraflagellar transport (IFT) mediated by the concerted action of anterograde kinesins (IFT-B complex) and retrograde dynein (IFT-A complex) (Ishikawa & Marshall, 2011; Kobayashi & Dynlacht, 2011; Nigg & Stearns, 2011; Sung & Leroux, 2013). Three negative regulators of ciliogenesis have been shown to be destructed or removed from ciliary structures at the onset of ciliogenesis. First, CP110, a mother centriole capping protein (Spektor et al, 2007; Tsang & Dynlacht, 2013), which blocks centriole elongation, is removed following the action of the Tau tubulin kinase 2 (TTBK2) (Goetz et al, 2012). Second, trichoplein, a protein preventing axonemal extension, is recognized by the Cullin 3-RING E3 ligase, KCTD17, and is degraded through the ubiquitin–proteasome system (UPS) (Kasahara et al, 2014). Cells depleted of CP110 or trichoplein show increased ciliary length. Third, the Oral-facial-digital syndrome 1 (OFD1) gene product is removed from centriolar satellites through selective autophagy induced by nutrient deprivation (Tang et al, 2013). These mechanisms seem to impact different stages of ciliogenesis in a complementary fashion, allowing for proper cilium biogenesis and function. However, exactly how the removal, destruction, and/or mislocalization of negative regulators of ciliogenesis are integrated within the cell cycle remains poorly understood.
We have identified the centrosomal phosphoprotein, NDE1, as a suppressor of the later stages of cilium formation affecting ciliary length by modulating the IFT process (Kim et al, 2011). We also showed that its level of expression is markedly reduced when cells enter G1 and peaks when cells are in mitosis. This oscillatory pattern of NDE1 during the cell cycle, which has been reported not only in cells in culture, but also in the human brain (Bakircioglu et al, 2011), suggests that high levels of NDE1 may not be compatible with cellular functions in quiescent cells, which could be consistent with its negative effect on ciliary length. Here, we show that CDK5, a kinase active in G1/G0 (Hellmich et al, 1992; Tsai et al, 1994; Nikolic et al, 1996; Su & Tsai, 2011; Shah & Lahiri, 2014), phosphorylates NDE1. Subsequently, CDK5-primed NDE1 is recognized by the FBW7 E3 ubiquitin ligase (Davis et al, 2014) and degraded through the UPS. Thus, a low level of NDE1 is maintained during G1 and G0 through the actions of CDK5 and FBW7, allowing cilia to form and function properly. Downregulation CDK5 or FBW7 leads to markedly decreased ciliary length that is dependent on increased levels of NDE1.
Results
FBW7 physically interacts with NDE1
The mechanisms responsible for the oscillatory expression of NDE1 during the cell cycle are entirely unknown. Treatment of serum-starved RPE1-hTERT cells with MG132 or lactacystin, known inhibitors of the UPS, minimized the overall decrease in NDE1 induced by serum starvation (Fig1A). A similar effect was seen in the centro-somal pool of NDE1 (Fig1B). These results indicated that the downregulation of NDE1 upon serum starvation is mediated through the UPS. Examination of the primary structure of NDE1 for potential degradation motifs revealed the presence of KEN and D-box motifs, both recognized by the anaphase promoting complex/cyclosome (APC/C), multiple RxL motifs recognized by cyclin F, a β-TrCP1-specific phosphodegron, and an FBW7-specific phosphodegron (Fig1C). A high-affinity FBW7-specific recognition motif consists of the core Threonine(T)Proline(P)XXSerine(S) motif. Phosphorylation of T at position 1 is required for binding to FBW7, whereas the phosphorylation of S at position 4 or the presence of a negatively charged residue such as glutamic acid (E) enhances binding, but is not absolutely required (Hao et al, 2007). T is often phosphorylated by GSK3, after priming at the secondary phosphorylation site. In some instances, P-directed S/T kinases such as CDKs may mediate T phosphorylation at position 1 (Hao et al, 2007; Welcker & Clurman, 2008). Deviations from this di-phosphorylated motif have been reported (Mao et al, 2008).
Figure 1.
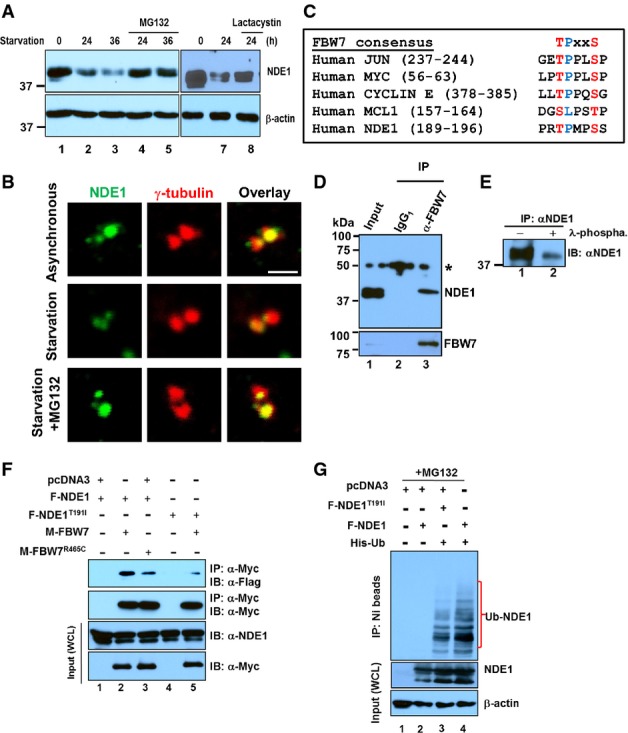
- A Expression levels of NDE1 (upper panels) or β-actin (lower panels) in exponentially growing RPE1-hTERT cells (lanes 1 and 6) or serum-starved cells for the indicated time points (lanes 2–5 and 7 and 8). Cells were treated with proteasomal inhibitors, MG132 (10 μM, lanes 4 and 5) or lactacystin (5 μM, lane 8), for 4 h before the indicated collection time points.
- B Expression levels of NDE1 at the centrosome in exponentially growing (asynchronous), 24-h serum starvation or 24-h serum-starved RPE1-hTERT cells treated for 4 h prior to fixation with MG132 (10 μM). Cells were double stained with rabbit α-NDE1 (green) and mouse γ-tubulin (red). Scale bar: 1 μm.
- C FBW7 phosphodegron sequences in human JUN, MYC, CYCLIN E, MCL1, and NDE1.
- D Physical interaction of endogenous FBW7 and NDE1 in RPE1-hTERT cells. Control mouse IgG1 (lane 2) or mouse α-FBW7 (lane 3) was added to cell lysates, and captured immunocomplexes (lanes 2 and 3) were immunoblotted with rabbit α-NDE1 (upper panel). Expression of NDE1 (upper panel) or FBW7 (lower panel) in lysates is shown in lane 1. Immunoprecipitated FBW7 is shown in the lower panel (lane 3). Asterisk indicates non-specific band.
- E NDE1 is endogenously phosphorylated in hRPE1-hTERT cells. Immunoprecipitated NDE1 was left untreated (lane 1) or treated with λ-phosphatase.
- F F-Box pathogenic mutation R456C in FBW7 or T191I within the FBW7 recognition site in NDE1 suppresses interaction between FBW7 and NDE1. HEK293T cells were transfected with indicated plasmids and serum starved for 24 h before lysis. Cells were treated with MG132 (10 μM) for 4 h prior to cell lysis. Wild-type and mutant form of Myc-tagged FBW7 was immunoprecipitated with α-Myc, and complexes were blotted with α-Flag to detect wild-type or mutant Flag-tagged NDE1. Expression levels of Myc- or Flag-tagged proteins in whole cell lysates (WCL) are shown in the lower panels.
- G NDE1 is ubiquitylated in vivo. HEK293T cells were co-transfected with Flag-tagged wild-type F-NDE1 or NDE1T191I (F-NDE1T91I) and His-tagged ubiquitin (His-Ub). Cells were lysed in a denaturing buffer, and His-Ub was purified over a nickel column (NI beads). Protein complexes covalently bound to His-Ub were immunoblotted with α-NDE1 (upper panel). Expression levels of wild-type and mutant NDE1 or β-actin are shown in middle or lower panel, respectively. Cells were treated with 10 μM MG132 for 4 h before lysis.
The role of APC/C in ciliary disassembly and suppression of centriole amplification through the regulation of Nek1 and STIL, respectively, have been shown (Arquint & Nigg, 2014; Wang et al, 2014). Thus, we tested whether NDE1 could also be targeted for degradation through the APC/C. Incubation of in vitro transcribed/translated NDE1 with Xenopus egg extracts in the presence of Cdh1 or non-degradable cyclin B failed to promote NDE1 degradation (Appendix Fig S1A). Similarly, NDE1 failed to physically interact with cyclin F or β-TrCP1 in transfected cells, suggesting that these E3 ligases are unlikely to mediate the ubiquitin-dependent degradation of NDE1 (Appendix Fig S1B). In contrast, endogenous NDE1 co-immunoprecipitated with endogenous FBW7 in RPE1-hTERT cell lysates. Interestingly, only the slower migrating species of NDE1 interacted with FBW7 (Fig1D), which would be consistent with the possibility that only phosphorylated NDE1 interacted with FBW7. Indeed, phosphatase treatment of immunoprecipitated NDE1 resulted in the elimination of the slower migrating species (Fig1E), suggesting that phospho-NDE1 interacted with FBW7 under physiological conditions. This should be expected for an FBW7-specific target (Davis et al, 2014). To test whether NDE1 could be a direct target of FBW7, we mutated the primary phosphorylation site within the predicted FBW7 phosphodegron in NDE1 as shown in Fig1C from T191 to isoleucine (I) and tested for its interaction with wild-type FBW7α. Conversely, we used the FBW7R465C mutant, which was identified in colorectal cancers abrogating the interaction and destruction of cyclin E (Rajagopalan et al, 2004), and tested for its interaction with wild-type NDE1. These mutations in NDE1 or FBW7 reduced the strength of interactions with wild-type FBW7 or NDE1, respectively (Fig1F), suggesting that T191 in NDE1 and R465 in FBW7 are critical for the interaction of NDE1 and FBW7. We confirmed that overexpressed NDE1T191I accumulated at the centrosome/basal body of transfected RPE1-hTERT cells resulting in abnormally short, if not invisible cilia (Appendix Fig S2). To test whether NDE1 can be directly conjugated with ubiquitin (Ub) in cells, we co-transfected 6× His-tagged Ub (His-Ub) with wild-type or NDE1T191I in HEK293T cells. Cells were lysed in a denaturing solution, and the covalent conjugation of Ub to NDE1 was tested by immunoprecipitation using nickel beads followed by immunoblotting. NDE1 was heavily ubiquitylated under these conditions (Fig1G). The NDE1T191I mutant that showed reduced association with FBW7 (Fig1F, lane 5) also reduced ubiquitylation compared to wild-type NDE1 (Fig1G, lane 3), suggesting that impaired recognition of NDE1T191I by endogenous FBW7 in HEK293T cells may account for its reduced level of ubiquitylation.
Next, we examined whether depletion of FBW7 would lead to increased protein stability of NDE1 in G1/G0. Transfection of RPE1-hTERT cells with FBW7-siRNA#1 or #2 resulted in efficient silencing of FBW7 and increased the expression of steady-state NDE1 levels in serum-starved cells (Fig2A). Downregulation of FBW7 did not cause significant changes in levels of Aurora A kinase, a known partner and target of FBW7 (Mao et al, 2004; Otto et al, 2009) with an indispensable role in ciliary disassembly (Pugacheva et al, 2007). This is not entirely unexpected since the degree of FBW7-mediated targeting is cell- and context-specific (Mao et al, 2004; Hoeck et al, 2010; Sancho et al, 2014). Nevertheless, Aurora A kinase was downregulated in serum-starved cells consistent with its degradation by the APC/C in G1 (Littlepage & Ruderman, 2002). Depletion of FBW7 did not affect CDK5 levels. Thus, these data showed that both FBW7-specific siRNAs downregulated FBW7, resulting in the upregulation of NDE1. To test whether the upregulation of NDE1 occurred at the centrosome/basal body, we quantified NDE1 signal intensity at this structure in the presence or absence of serum in wild-type and FBW7-depleted cells. Upon serum starvation, there was a significant accumulation of NDE1 at the centrosome/basal body in cells depleted of FBW7 compared to wild-type cells (Fig2B). The negative regulation of NDE1 by FBW7 was also confirmed in human colon adenocarcinoma DLD-1-FBXW7−/− cells, where both alleles of FBXW7 (formal gene name) were inactivated by homologous recombination (Rajagopalan et al, 2004). NDE1 steady-state levels were increased upon serum starvation in cells lacking FBW7 in contrast to a marked downregulation in wild-type cells (Fig2C). Expression of C-MYC, which is a known target of FBW7, was used as a positive control (Welcker et al, 2004; Yada et al, 2004). Taken together, these results demonstrate that FBW7 can serve as an effective E3 ligase for NDE1.
Figure 2.
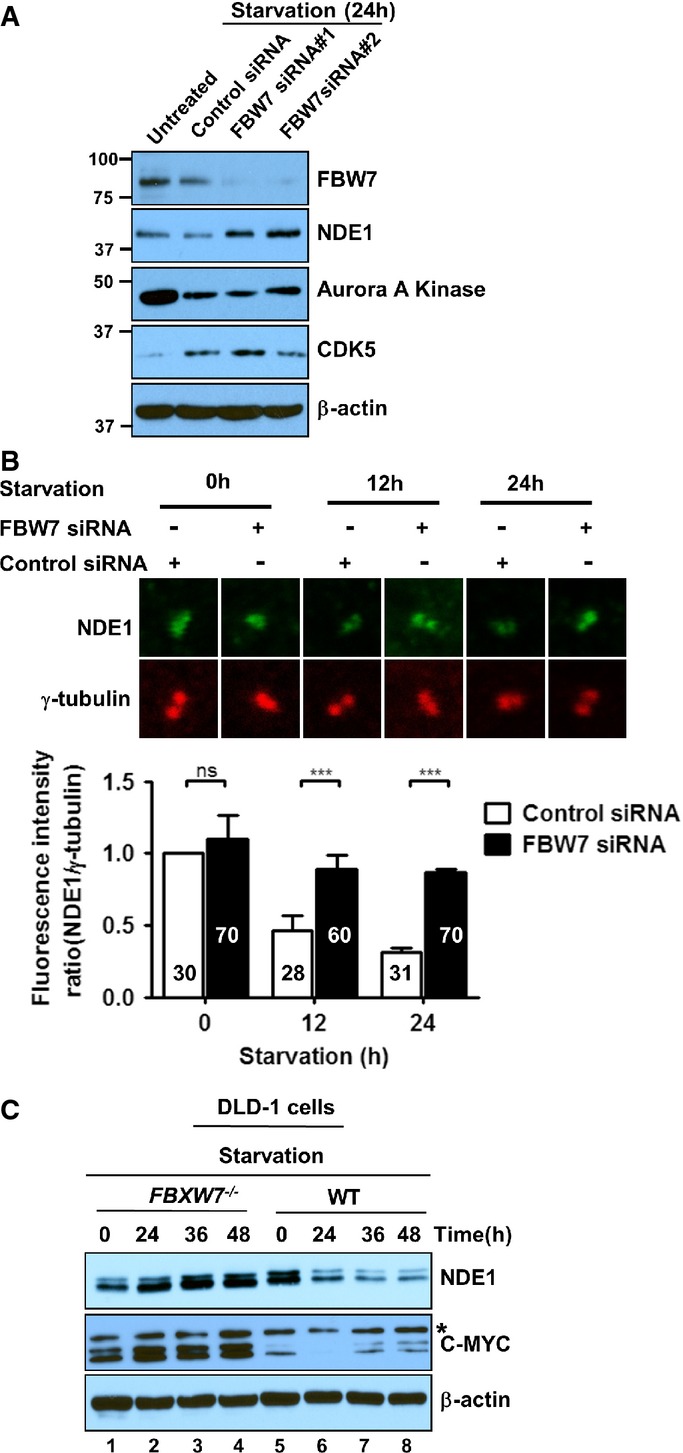
- A Expression levels of FBW7, NDE1, Aurora A kinase, and CDK5 in wild-type and FBW7-depleted RPE1-hTERT cells in exponentially growing un-trans-fected cells (untreated, lane 1) or 24-h serum-starved cells transfected with a scrambled siRNA (control siRNA, lane 2) or separately with two FBW7-specific siRNAs (siRNA#1 or siRNA#2) (lanes 3 and 4).
- B Quantification of fluorescence intensity ratio of NDE1/γ-tubulin (green/red) signals at the centrosome in 0-, 12-, and 24-h serum-starved transfected cells with a scrambled (control) or FBW7-specific siRNA (FBW7 siRNA#1). Fluorescence intensity of coinciding green or red pixels within the box area (inset) was measured. The range of fluorescence intensity per pixel in a box was 0–255 (total number of cells from three independent transfections is indicated on graph). Data represent mean ± SEM. One-way ANOVA followed by Newman–Keuls post-test was used to determine significant difference among groups. ***P < 0.005, ns indicates no significance.
- C Upregulation of steady-state levels of NDE1 in DLD-1 FBW7−/− cells. Lysates prepared from exponentially growing (lanes 1 and 5) and serum-starved FBW7−/− (lanes 2–4) or wild-type DLD-1 cells (lanes 6–8) were immunoblotted with rabbit α-NDE1 (upper panel), mouse α-C-MYC (middle panel), or α-β-actin (lower panel).
FBW7 co-localizes with NDE1 at the centrosome
While NDE1 shows strong expression in the centrosome (Feng et al, 2000; Bakircioglu et al, 2011; Kim et al, 2011), the presence of FBW7 in this structure in intact ciliated cells has not been examined. Double staining of FBW7 and γ-tubulin or NDE1 showed co-localization of FBW7 and γ-tubulin or NDE1 in the centrosome of exponentially growing hRPE1-hTERT cells (Fig3Aa–g). In addition to the centrosome, FBW7 showed punctate cytoplasmic and nuclear staining, consistent with previous reports (Welcker et al, 2004; Bhaskaran et al, 2013). In ciliated, serum-starved RPE1-hTERT cells (arrested in G1), FBW7 was not present in the cilium (Fig3Ba–c), but co-localized with γ-tubulin in the two centrioles (Fig3Bd–f), ninein, a marker of the distal and proximal regions of the mother centriole and proximal region of daughter centriole (Fig3Bg–i), or centrin-2, a marker of the two centrioles, but not pericentriolar material (Fig3Bj–l). FBW7 also showed co-localization with CEP164, a marker of mature mother centriole present at the distal appendages (Appendix Fig S3). These data indicated that FBW7 was present in both centrioles in exponentially growing RPE1-hTERT cells or in ciliated cells. The presence of FBW7 in the pericentriolar material cannot be excluded. To confirm the specificity of the mouse antibody used to detect FBW7 by immunofluorescence (Fig3Aa–g and Ba–f) and immunoprecipitation (Fig1D), wild-type DLD-1 and DLD-1-FBXW7−/− cells were immunostained with the same antibody. No FBW7 signal was detected in the red channel in cells lacking FBW7 (Fig3C). NDE1 is expressed in cortical and hippocampal neurons (Pei et al, 2014). Thus, we tested whether it would co-localize with FBW7 at the centrosome in primary post-mitotic hippocampal neurons identified as such by Tau-positive staining (Fig3Da). FBW7 co-localized with γ-tubulin (Fig3Db–d) and NDE1 (Fig3De–g) in these cells. These data showed that FBW7 co-localizes with NDE1 at the centrosome and basal body of diverse cell types.
Figure 3.
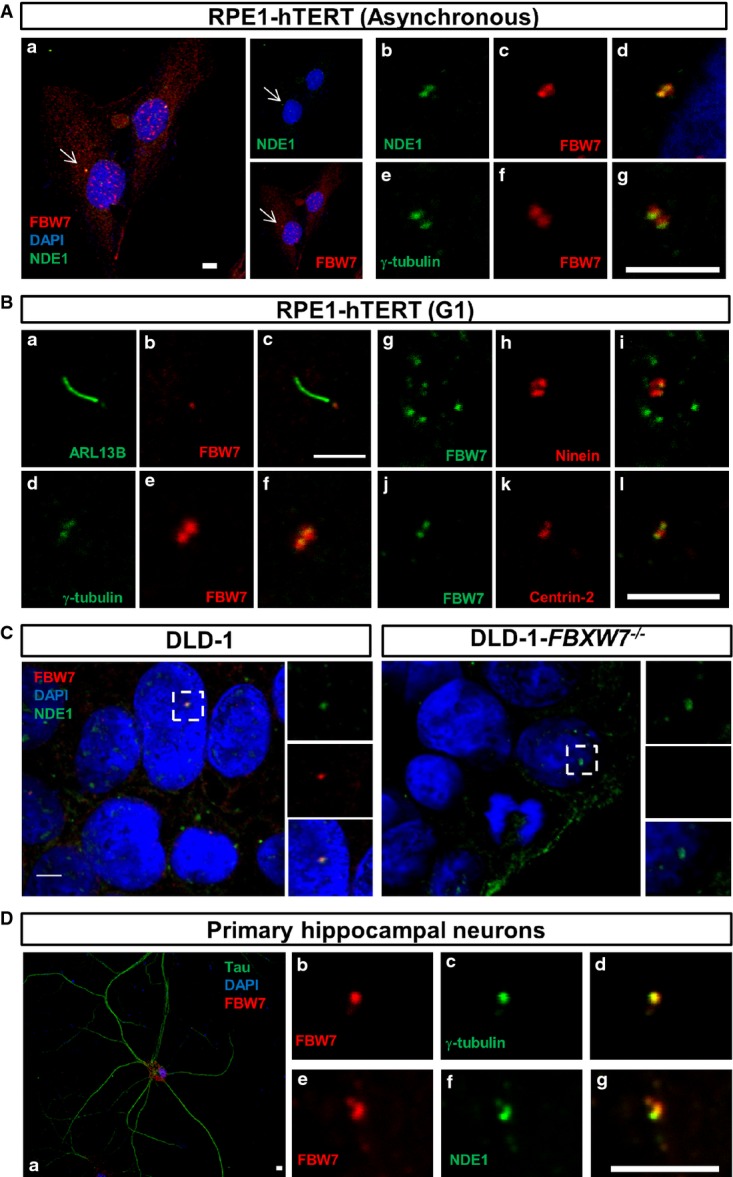
- A, B Co-localization of NDE1 and FBW7 at the centrosome of exponentially growing (asynchronous, A) or serum-starved (24 h, G1-arrested, B) RPE1-hTERT cells. (A) Cells were double stained with mouse α-FBW7 (red) and rabbit α-NDE1 (green) (a-d) or mouse α-FBW7 (green) and rabbit γ-tubulin (red) (e–g). Nuclei were stained with DAPI (blue). Scale bars: 10 μm (a) and 5 μm (b–g). (B) Presence of FBW7 in the basal body of serum-starved RPE1-hTERT cells visualized by double staining with cilia (ARL13B, a–c)- or centrosome [(γ-tubulin, d–f), (ninein, g–i), (centrin-2, j–l)]-specific markers. Rabbit or mouse primary antibodies are shown in green or red, respectively. Scale bar: 5 μm.
- C Co-localization of NDE1 and FBW7 in the centrosome of DLD-1 cells. Wild-type or FBXW7−/− DLD-1 cells were double stained with rabbit α-NDE1 (green) and mouse α-FBW7 (red). Scale bar: 5 μm.
- D Presence of FBW7 in the centrosome of mouse primary hippocampal neurons. Hippocampal neurons were identified by Tau-positive staining (a), and the expression of FBW7 in the centrosome and its co-localization with NDE1 were determined by double labeling with mouse α-FBW7 (red) and γ-tubulin (green) (b–d) or NDE1 (green) (e–g). Scale bar: 5 μm.
FBW7 regulates ciliary length
Given that FBW7 controls the stability of NDE1 and NDE1 negatively regulates ciliary length (Kim et al, 2011), we hypothesized that FBW7 and NDE1 may function antagonistically in the regulation of ciliary length. To test this hypothesis, FBW7 or NDE1 was knocked down separately or together in RPE1-hTERT cells and the effects of these depletions on ciliary length were determined at 24 h following serum starvation (Fig4A). Efficiency of siRNAs in silencing respective targets was confirmed (Fig4B). As we have shown earlier, RPE1-hTERT cells depleted of NDE1 show an increased ciliary length, but not a further increase in the number of ciliated cells at 24 h following serum starvation, as at this time point, ciliation has been completed (Kim et al, 2011). Knockdown of FBW7 had a strong effect on cilium formation, as only ˜20% of cells had visible cilia (Fig4C and D). To test whether depletion of FBW7 caused a block in ciliogenesis or a marked reduction in ciliary length that would make cilia invisible for counting, we performed a time course experiment in which cells depleted of FBW7 were scored for the presence of cilia at 12, 24, or 48 h following serum starvation. If depletion of FBW7 had blocked ciliogenesis, the number of ciliated cells should not increase over time. Figure4C shows that instead, the number of ciliated cells depleted of FBW7 increased over time (Fig4D), suggesting that FBW7 did not block cilium formation, but it rather had a strong effect on ciliary length. To test whether this effect was partially or entirely contributed to the upregulation of NDE1, cells were co-transfected with FBW7- and NDE1-specific siRNAs. Mock-, FBW7-, or NDE1-specific siRNAs were co-transfected with GFP, and the percentage of ciliated cells was determined only from GFP-positive cells. Knockdown of FBW7 caused a reduction in the number of ciliated cells by ˜3-fold at 24 h following serum starvation, and this effect was partially rescued in cells depleted of both FBW7 and NDE1 (Fig4C), suggesting that NDE1 can function downstream of FBW7 in the regulation of ciliary length.
Figure 4.
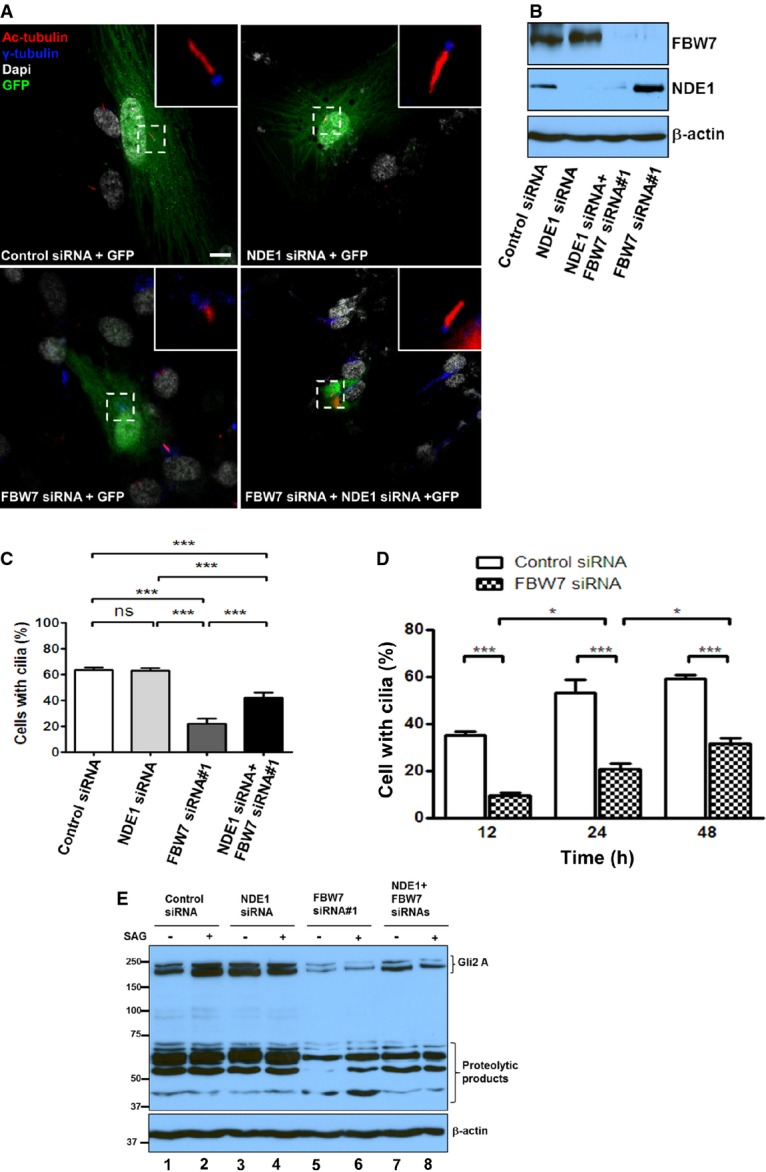
- A RPE1-hTERT cells were transfected with a scrambled siRNA + GFP (mock), an NDE1-specific siRNA + GFP, FBXW7-specific siRNA#1 + GFP, or NDE1-specific and FBW7-specific siRNAs + GFP, serum starved for 24 h and immunostained with acetylated α-tubulin (mouse monoclonal, 6-11B, red) and γ-tubulin (rabbit polyclonal, blue). Scale bar: 5 μm.
- B Expression levels of FBW7 and NDE1 in RPE1-hTERT cells transfected with scrambled siRNA, NDE1-specific siRNA, FBW7-specific siRNA#1, or NDE1- and FBW7-specific siRNAs.
- C Quantification of ciliated wild-type (transfected with control scrambled siRNA) and FBW7-depleted (transfected with FBW7-specific siRNA#1) GFP-positive cells 12, 24, and 48 h following serum starvation from 3 independent experiments. One hundred GFP-positive cells per group were counted per experiment. Data represent mean ± SEM. One-way ANOVA followed by Newman–Keuls post-test was used to determine significant difference among groups. ***P < 0.005, ns indicates no significance.
- D Quantification of ciliated wild-type (transfected with control scrambled siRNA), NDE1-depleted (transfected with an NDE1-specific siRNA), FBW7-depleted (transfected with FBW7-specific siRNA#1), or FBW7- and NDE1-depleted (co-transfected with FBW7-specific siRNA#1 and NDE1-specific siRNA) GFP-positive cells 24 h following serum starvation from three independent experiments. One hundred GFP-positive cells per group were counted per experiment. Data represent mean ± SEM. One-way ANOVA followed by Newman–Keuls post-test was used to determine significant differences among groups. *P < 0.05, ***P < 0.005, ns indicates no significance.
- E Effect of FBW7 and NDE1 on expression levels of GLI2. Wild-type (transfected with control scrambled siRNA, lanes 1 and 2), NDE1-depleted (transfected with an NDE1-specific siRNA, lanes 3 and 4), FBW7-depleted (transfected with FBW7-specific siRNA#1, lanes 5 and 6), or FBW7- and NDE1-depleted (co-transfected with FBW7-specific siRNA#1 and NDE1-specific siRNA, lanes 7 and 8) cells were serum starved for 24 h and stimulated for an additional 24 h by SAG (0.4 μM). Cells were lysed, and expression levels of GLI2 were determined by immunoblotting.
To provide independent evidence that NDE1 is negatively regulated by FBW7, we examined the effect of single and double depletions on the activation of the Hedgehog pathway, which is dependent on cilia for proper expression and processing of Glioma-Associated Oncogene Homolog 1 (GLI) transcription factors (Goetz & Anderson, 2010). Activation of the pathway at the level of Smoothened (SMO) using Smoothened Agonist (SAG) caused a significant accumulation of GLI2 in mock-transfected cells, as expected (Fig4E, lanes 1 and 2). However, cells depleted of NDE1 showed enhanced steady-state levels of GLI2 that could not be increased further by SAG (Fig4E, lanes 3 and 4). Conversely, cells depleted of FBW7 showed the opposite effect (Fig4E, lanes 5 and 6). The reduced expression of GLI2 was partially rescued in cells depleted of both proteins (Fig4E, lanes 7 and 8). These data supported the hypothesis that FBW7 negatively controls NDE1 levels and this downregulation might be important for the regulation of the Hedgehog pathway in ciliated cells.
NDE1 is phosphorylated by CDK5
Substrate phosphorylation is required for FBW7-mediated recognition (Koepp et al, 2001; Yada et al, 2004; Davis et al, 2014). Thus, we attempted to identify the kinase that would prime NDE1 for recognition by FBW7. We tested the following kinases: CDK1/cyclin A, CDK2/cyclin E, CDK5/p25, and GSK3β. CDK1/cyclin A has been shown to phosphorylate NDE1 (Alkuraya et al, 2011), whereas CDK5 has been shown to phosphorylate NDEL1 (Niethammer et al, 2000; Sasaki et al, 2005), a homolog of NDE1. In addition, CDK5 is most active in post-mitotic cells (Su & Tsai, 2011) and its expression is inversely correlated with that of NDE1 during cell cycle exit or entry to G1 (Appendix Fig S4). CDK5 requires interaction with p35 (or CDK5R1) or its proteolytic fragment, p25, for activation (Lew et al, 1994; Tsai et al, 1994; Patrick et al, 1999). GSK3β is a priming kinase for known FBW7 substrates (Welcker & Clurman, 2008; Busino et al, 2012), and its activity can be induced by an interaction with p25 (Chow et al, 2014). It should be noted that GSK3 and CDK5 show similar overall folding (Chow et al, 2014). The effect of CDK2/cyclin E on NDE1 phosphorylation was unknown. Cell lysates were separated in SDS–PAGE supplemented with 50 μM Phos-tag™ reagent that allows step-wise separation of phosphorylated species. The majority of transfected NDE1 migrated as a non-phosphorylated protein (Appendix Fig S5). Co-transfection with CDK5/p25 increased the abundance of the di-phosphorylated species by about 5-fold (indicated by two asterisks in Fig5A, lane 2). It also induced phosphorylation at a single residue (indicated by one asterisk, Fig5A, lane 2). Co-transfection with none of the other kinases induced di-phosphorylation to levels seen by CDK5/p25 (Fig5B; Appendix Fig S5). Co-transfection of NDE1T191I with CDK5/p25 led to an almost similar level of di-phosphorylation compared to wild-type NDE1, but there was no detectable mono-phosphorylation (Fig5A, lane 4). These data suggested that phosphorylation at T191 required CDK5/p25. To test whether mono-phosphorylation of NDE1 occurs in native conditions such as in endogenous NDE1 in RPE1-hTERT cells, lysates from these cells were analyzed side by side with lysates from HEK293T cells transfected with NDE1 alone or NDE1 plus CDK5/p25. Indeed, endogenous NDE in RPE1-hTERT cells migrated as a mono-phosphorylated species (Fig5C), indicating that this species is likely to interact with FBW7 in RPE1-hTERT cells (Fig1D, lanes 1 and 3).
Figure 5.
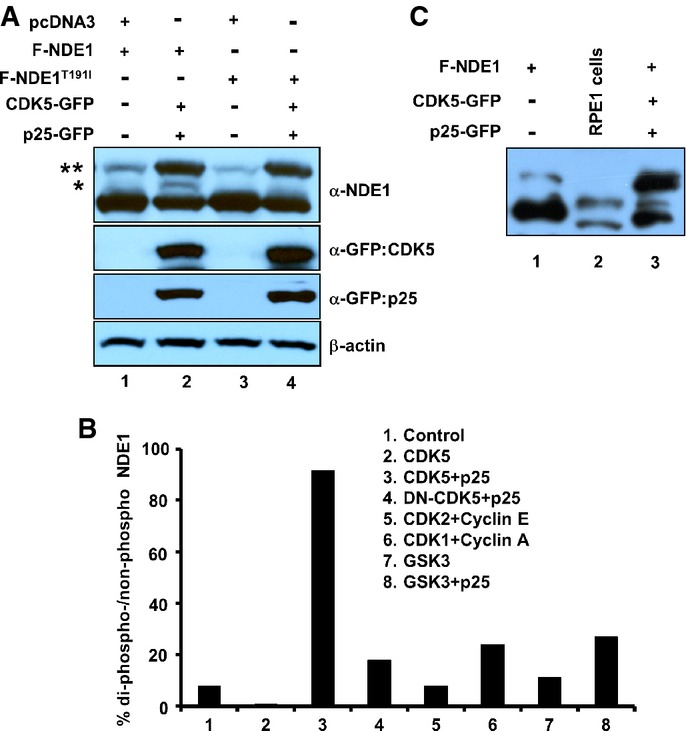
- A HEK293T cells were transfected with indicated plasmids, and total cell lysates were separated through SDS–PAGE containing 50 μM Phos-tag™ reagent. Two asterisks indicate phosphorylated species in two sites, whereas one asterisk indicates phosphorylated species in one site.
- B Quantification of NDE1 phosphorylation in two sites (shown by two asterisks in A) co-transfected with indicated kinases and activators.
- C Mono-phosphorylation of endogenous NDE1 in RPE1-hTERT cells. Cell lysates from HEK293T cells transfected with Flag-tagged NDE1 (F-NDE1, lane 1) or F-NDE1 + CDK5 + p25-GFP (lane 3) or untransfected RPE1-hTERT cells (lane 2) were separated by SDS–PAGE containing 50 μM Phos-tag™ reagent and immunoblotted with rabbit α-NDE1.
CDK5 induces downregulation of NDE1
We reasoned that if CDK5 phosphorylates NDE1 at T191 within the FBW7 phosphodegron, co-transfection of activated CDK5 should reduce the steady-state levels of wild-type NDE1, but not NDE1T191I. Co-transfection of wild-type NDE1 with CDK5/p25 or p35 (both activators of CDK5) led to downregulation of wild-type NDE1 (Fig6A, lanes 4 and 7). However, this was not the case with NDE1T191I, whose levels remained unchanged in cells co-transfected with mutant NDE1 and CDK5/p25 (Fig6B, lane 4). Next, we tested whether NDE1 could physically interact with p35 or p25. Despite a significant downregulation of NDE1 in whole cell lysates transfected with CDK5/p25 or CDK5/p35, NDE1 co-immunoprecipitated with p35 or p25 (Fig6C, lanes 4 and 7). In addition to p35/p25, CDK5 can be activated by p39 (or CDK5R2) (Su & Tsai, 2011; Shah & Lahiri, 2014). Thus, we tested whether CDK5/p39 could destabilize NDE1 and whether p39 could interact with NDE1. Interestingly, CDK5/p39 did not have an effect on NDE1 levels and p39 did not physically interact with NDE1 in transfected cells (Fig6D). Consistent with co-immunoprecipitation data, endogenous p35/p25 co-localized with ninein in the centrosome in cycling RPE1-hTERT cells (Fig6E). Depletion of p35 resulted in the loss of p35-specific signal in the green channel (Fig6E) and an upregulation of NDE1 levels in serum-starved cells (Fig6F). These data suggest that CDK5 may be recruited to NDE1 through its activators, p25 or p35, but not p39, to prime NDE1 for FBW7-mediated recognition.
Figure 6.

- A HEK293T cells were transfected with indicated plasmids, and total cell lysates prepared 24 h following transfections were immunoblotted with α-Flag, α-GFP, α-Myc, and α-β-actin.
- B Activated CDK5 does not affect steady-state levels of NDE1T191I. HEK293T cells were transfected with indicated plasmids, and total cell lysates prepared 24 h following transfections were immunoblotted with α-Myc, α-GFP, and α-β-actin.
- C NDE1 physically interacts with p25/p35. HEK293T cells were transfected with indicated plasmids, and F-NDE1 was immunoprecipitated with α-Flag. Immunocomplexes were immunoblotted with α-p25/p35 or α-NDE1. Expression levels of p25-GFP, p35-Myc, F-NDE1, and CDK5-GFP of β-actin in whole cell lysates (WCL) were determined by immunoblotting.
- D NDE1 does not interact with p39. HEK293T cells were transfected with indicated plasmids, and F-NDE1 was immunoprecipitated with α-Flag. Immunocomplexes were immunoblotted with mouse α-turboGFP to detect turboGFP-tagged p39 (upper panel) or rabbit α-Flag. Expression levels of p39-tGFP or F-NDE1 in whole cell lysates (WCL) were determined by immunoblotting.
- E p35 is present in the centrosome of RPE1-hTERT cells. Wild-type cells or cells transfected with a p35-specific siRNA were double-labeled with rabbit α-p35 (green) or mouse α-ninein (red) and visualized by indirect immunofluorescence. Scale bar: 5 μm.
- F Upregulation of NDE1 in RPE1-hTERT cells depleted of p35. Cells were transfected with a scrambled siRNA (Control siRNA) or a p35-specific siRNA (p35 siRNA), and total cell lysates from 24-h serum-starved (starvation 24 h) or exponentially growing cells (untreated) were immunoblotted using α-p35 (upper panel), α-NDE1 (middle panel), or α-β-actin (lower panel).
To determine whether the downregulation of NDE1 in cells co-transfected with CDK5/p25 was due to decreased stability of NDE1, we measured the half-life of wild-type NDE1 or NDE1T191I in the presence or absence of CDK5/p25. NDE1 levels remained unchanged for 6 h after the addition of emetine, a protein synthesis inhibitor, in cells transfected with a dominant negative form of CDK5 plus p25 (negative control, Fig7C and E). NDE1T191I showed the same pattern in cells transfected with activated CDK5 (Fig7B and E). However, wild-type NDE1 reached half of its starting concentration within ˜4.5 h after the addition of emetine in the presence of activated CDK5 (Fig7A and E), indicating that the stability of wild-type but not mutant NDE1 protein was reduced in the presence of activated CDK5.
Figure 7.
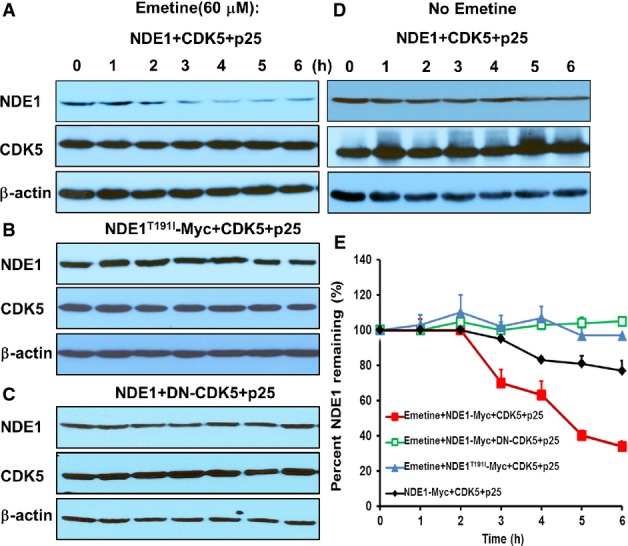
- A–D HEK293T cells were transiently transfected with indicated plasmids. Twenty-four hours after transfection, cells were treated with emetine (60 μM) at indicated time point (A–C) or left untreated (D). Cells were lysed at 0–6 h following emetine treatment, and expression levels of NDE1, CDK5, and β-actin were determined by immunoblotting.
- E Kinetics of percent NDE1 remaining in whole cell lysates after emetine treatment. Data represent mean ± SEM from three independent transfections.
CDK5-mediated downregulation of NDE1 increases ciliary length
Next, we examined whether endogenous CDK5 could regulate overall NDE1 levels in RPE1-hTERT cells. RPE1-hTERT cells depleted of CDK5 showed increased expression of NDE1 without a significant change in FBW7 (Fig8A). An accumulation of NDE1 in the centrosome/basal body in cells depleted of CDK5 compared to wild type cells was also seen (Appendix Fig S6). These data show that NDE1 is negatively regulated by CDK5 when cells are induced to enter G1 or exit the cell cycle by serum starvation. Given the negative effect of NDE1 on ciliary length, we reasoned that cells depleted of CDK5 should have abnormally short cilia, which should be rescued in cells depleted of both CDK5 and NDE1. Indeed, cells depleted of NDE1 showed increased average ciliary length from 2.3 to 6.8 μm, as expected (Kim et al, 2011). Cells depleted of CDK5 had an average ciliary length of 1.8 μm, which was increased to 4.6 μm in cells depleted of both proteins (Fig8B and C). The percentage of ciliated cells at 24 h following serum starvation did not change in cells with single and double knockdowns (Fig8C), suggesting that the effect of CDK5 on ciliary length was not as severe as that of FBW7, but within the same pathway as NDE1.
Figure 8.
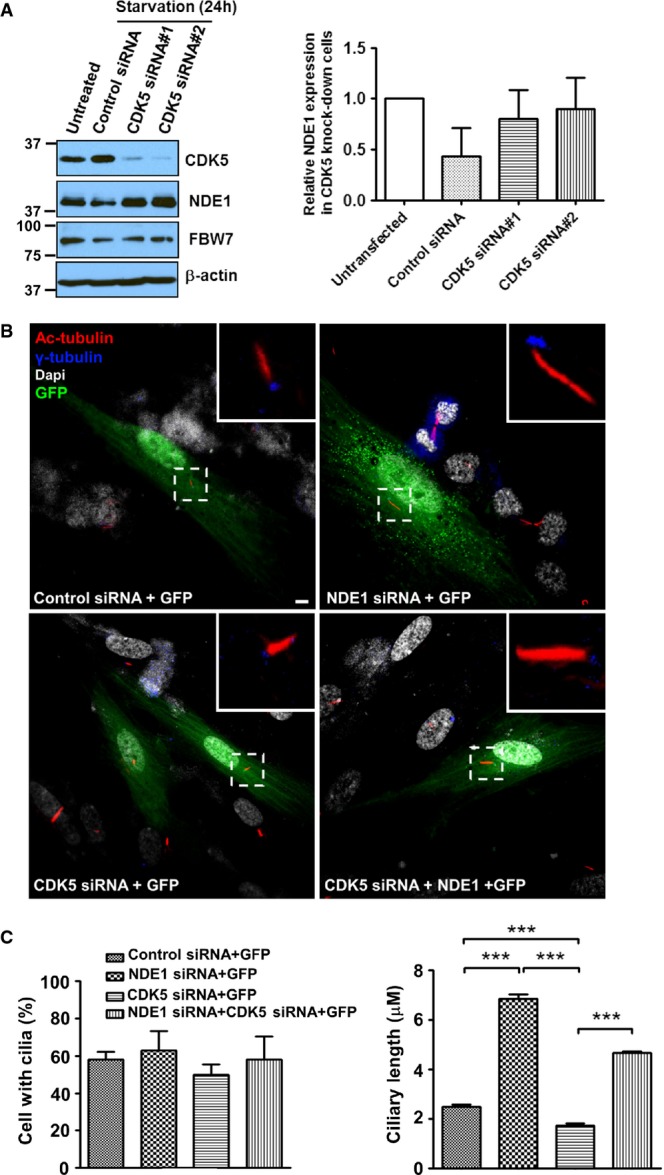
- A RPE1-hTERT cells were transiently transfected with a scrambled siRNA (Control siRNA) or two human CDK5-specific siRNAs (CDK5 siRNA#1 or CDK5 siRNA#2) separately, and serum starved for 24 h. Expression levels of CDK5, NDE1, FBW7, or β-actin in serum-starved cells (lanes 2–4) and untransfected, exponentially growing cells (lane 1) were determined by immunoblotting. Quantification of changes in NDE1 levels in mock-transfected or CDK5-depleted serum-starved cells relatively to NDE1 levels in exponentially growing cells are shown in the bar graph. Data represent mean ± SEM from three independent transfections.
- B RPE1-hTERT cells were co-transfected with indicated siRNAs and GFP and serum starved for 24 h following transfection to allow cilia formation. Cilia were visualized using a mouse monoclonal antibody (611B) against acetylated tubulin (shown in red), whereas basal bodies were stained with a rabbit polyclonal antibody against γ-tubulin (shown in yellow). Scale bar: 5 μm.
- C Quantification of percent of ciliated cells and ciliary length in mock-transfected cells (Control siRNA + GFP), cells transfected with an NDE1-specific siRNA (NDE1 siRNA + GFP), a CDK5-specific siRNA (CDK5 siRNA + GFP), or double-transfected with NDE1 and CDK5 siRNAs (NDE1 siRNA + CDK5 siRNA + GFP). Percentages of cells with cilia in all four groups were determined from GFP-positive cells from three independent experiments. Ciliary length was determined from a total of 190 ciliated cells in mock-transfected group, 189 ciliated cells in NDE1 siRNA group, 194 ciliated cells in CDK5 siRNA group, and 197 ciliated cells in NDE1 + CDK5 siRNA group pooled from three independent transfections. Data represent mean ± SEM. Significance was determined by one-way ANOVA followed by Newman–Keuls post-test, ***P < 0.005.
Discussion
In this study, we identify the CDK5-FBW7-NDE1 module as a critical regulator of ciliary length in a cell cycle-dependent manner. This conclusion is based on the following lines of evidence: first, NDE1 can be phosphorylated by p35-/p25-activated CDK5 at a site (T191) located within an FBW7-specific phosphodegron. Second, phosphorylated NDE1 physically interacts with FBW7 and cells depleted of FBW7 show increased levels of NDE1. Third, activation of CDK5 destabilizes NDE1. Fourth, the mutation of T191 to isoleucine in NDE1 diminishes the physical interaction with FBW7 and renders the protein insensitive to CDK5-mediated destabilization. Fifth, depletion of CDK5 or FBW7 results in the accumulation of NDE1 and a reduction in ciliary length, which is corrected in cells depleted of CDK5 and NDE1 or FBW7 and NDE1. Sixth and final, the effects of single and double depletions on ciliary length correlated with effects on the activation of the Hedgehog pathway, which is dependent on primary cilia. These data lead us to propose a model in which, upon entry into G1, levels and/or activity of CDK5 rise inducing the phosphorylation of NDE1. Phospho-NDE1 is recognized by FBW7 and targeted for UPS-mediated degradation, allowing cilium assembly to proceed and cilia to function properly (Fig9). Accordingly, a low level of NDE1 is maintained throughout G1 and G0. During cell cycle re-entry, levels of CDK5 are reduced (Zhang et al, 2012) and remaining activity is further suppressed through known interactions with cyclin D1 (Xiong et al, 1992; Modi et al, 2012) and cyclin E (Odajima et al, 2011), allowing NDE1 levels to accumulate and promote ciliary resorption.
Figure 9.
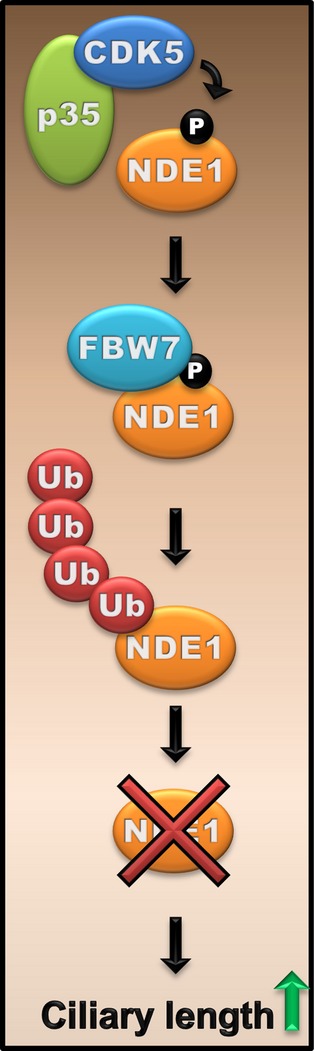
Schematic representation of the CDK5-FBW7-NDE1 pathway in the regulation of ciliary length
NDE1 is phosphorylated by CDK5 at T191. Phosphorylated NDE1 is recognized by FBW7 and targeted for degradation via the UPS. Degradation of NDE1 allows cilia to reach their normal length..
Our studies provide evidence for an essential role of FBW7 in the regulation of ciliary length. To our knowledge, this is the first time that FBW7 has been implicated in cilium formation and function. We show that depletion of FBW7 results in a dramatic decrease in the number of ciliated cells accompanied by a robust reduction in the expression levels of core components of the Hedgehog pathway. Effects on ciliary length and Hedgehog pathway are partially rescued in cells lacking both FBW7 and NDE1, suggesting that NDE1 functions downstream of and antagonistically to FBW7 in cilium formation and function. The FBW7-mediated recognition of NDE1 and degradation by the SCFFBW7 enzyme complex are likely to occur at the centrosome, because NDE1 and FBW7 co-localize there and MG132 treatment increases the amount of NDE1 at the centrosome. This is consistent with previous work in which Skp1 and Cul1 were identified as core components of the centrosome (Freed et al, 1999) and proteasomal subunits co-localize with γ-tubulin in HeLa cells (Wigley et al, 1999). Further, Polo-like kinase 2 co-immunoprecipitates and phosphorylates FBW7 in centrosomal preparatio n in HeLa cells (Cizmecioglu et al, 2012).
Depletion of FBW7 reduced the expression levels of GLI2, which were partially restored in cells depleted of both FBW7 and NDE1. This effect mirrored the effect of single and double deletions on ciliary length. We propose that full-length GLI2 is stabilized in the presence of cilia and this stabilization depends on ciliary length. The depletion of NDE1 seems to affect the Hedgehog pathway in a mechanism similar to a weak allele of Ift144, which caused ectopic activation of the pathway, and activation was not induced further by SAG (Liem et al, 2012). IFT144 is part of the IFT-A complex mediating anterograde transport. We had shown earlier that NDE1 affected ciliary length by tethering the dynein light chain, DYNLL1 (or LC8), also an IFT-A component, at the basal body (Kim et al, 2011). Thus, the stabilization of GLI2 in the presence of cilia could involve the IFT-A complex.
Our results show that FBW7 affects ciliary length and perhaps ciliogenesis in general, not only by suppressing the expression of NDE1, but also through an effect on other proteins. At the present time, we do not know all stage(s) and ciliogenesis-relevant pathways affected by FBW7. The fact that some of these effects were rescued by the depletion of NDE1 indicates that FBW7 may have affected later stages of ciliogenesis/maintenance modulating the IFT machinery through NDE1. We believe that NDE1 suppresses ciliogenesis at the stage of cilium assembly and maintenance because massive overexpression of NDE1 still allowed the formation of stumpy cilia (Kim et al, 2011), suggesting that early stages of ciliogenesis, such as centriole docking, must be intact. It is tempting to speculate on a regulatory role of cyclin E in FBW7-mediated destruction of NDE1. Cyclin E is expressed in high levels in post-mitotic neurons where it negatively regulates CDK5 by sequestering it away from its activators (Odajima et al, 2011). Although the accumulation of cyclin E in G0 might be limited to neurons, it is conceivable that at the onset of S phase, elevated levels of cyclin E could inhibit CDK5, terminating its priming effect on NDE1 and subsequent destruction of NDE1 by the UPS. Accumulated NDE1 could contribute to ciliary disassembly.
Our data show that activation of CDK5 via p35 or p25 can induce T191 phosphorylation and destabilization of NDE1. NDE1 was also found to associate with p35 or p25, but not with CDK5 directly. Activation of CDK5 via p39 did not have an effect on NDE1, and p39 did not associate with NDE1. These data suggest that CDK5 is recruited to NDE1 through a physical interaction with its activators. The subcellular localization of p35 and p25 are not completely overlapping with p35 being predominantly localized in the plasma membrane, whereas p25 being localized in the cytosol (Shah & Lahiri, 2014). p35 was shown to be present in the centrosome (Rosales et al, 2010b). We confirmed its localization there. A splice variant of CDK5 has been shown to be present in the centrosome (Kim et al, 2010). Furthermore, CDK5 has an indispensable role in cilia formation (Rosales et al, 2010a). Although, we could not detect endogenous CDK5 in the centrosome in RPE1-hTERT cells, it is possible that p35 transiently recruits CDK5 to the centrosome where it can phosphorylate NDE1 and possibly other substrates. Thus, priming and recognition of NDE1 by CDK5 and FBW7, respectively, may likely occur at the centrosome.
The effect of CDK5 on ciliary length seems to be in agreement with biochemical experiments showing that CDK5 primes NDE1 for FBW7-mediated destruction, since depletion of both proteins, CDK5 and NDE1, partially restored ciliary length. These data lead us to propose the following suggestions: first, the effect of CDK5 on ciliary length is less severe than that of FBW7. This idea is due to the fact that cells depleted of FBW7 had a reduced number of ciliated cells, most likely due to a strong effect on ciliary length that would make cilia invisible for counting, whereas that was not the case in cells depleted of CDK5 where reduction in ciliary length was less dramatic (from 2.3 to 1.8 μm). This observation suggests that CDK5 could only modify a subset of FBW7 targets with a negative role in ciliary length regulation. Other kinases could prime these putative FBW7 targets. Second, CDK5 seemed to have affected other substrates, in addition to NDE1. These other substrates may have positive or negative roles in ciliary assembly. Identification of these targets may provide new information on mechanisms controlling ciliary length.
Our study shows that CDK5 and FBW7 can function in a linear pathway to maintain low levels of NDE1 and ensure proper cilia formation and function. Because CDK5 activity is high in G1/G0, it is likely that the CDK5/FBW7 module contributes to the regulation of NDE1 in the G1/G0 phases of the cell cycle. The functionality of CDK5 and FBW7 in a G1/G0-specific function, such as cilium formation and function through NDE1, is shown here. However, it is possible that FBW7 may regulate the abundance of NDE1 in other phases of the cell cycle, where priming could occur by another kinase. NDE1 has important functions in mitosis (Feng & Walsh, 2004; Vergnolle & Taylor, 2007) and S phase (Houlihan & Feng, 2014), and naturally occurring inactivating mutations in the NDE1 gene in humans result in microcephaly (Alkuraya et al, 2011; Bakircioglu et al, 2011), a condition associated with the reduction the neuronal progenitor pool through effects on cell cycle progression and apoptosis. Given the well-known role of FBW7 as a tumor suppressor (Crusio et al, 2010; Wang et al, 2012; Davis et al, 2014), it is tempting to speculate that an excessive accumulation of NDE1 throughout the cell cycle could contribute to tumorigenesis induced by inactivating mutations in FBXW7. In addition to its tumor suppressor activity, FBW7 can affect differentiation, survival, and reprogramming of stem cells and progenitors in tissues that are ciliated (Hoeck et al, 2010; Sancho et al, 2014). Based on our data, it is conceivable that some of these effects could be mediated through the FBW7-mediated suppression of important regulators of cilium formation and function, such as the Hedgehog pathway.
Here, we have identified a pathway that negatively regulates the abundance of NDE1 in quiescent cells providing mechanistic leads into the connection of the cell cycle with cilium formation, assembly, and function. These findings can spur future studies to identify regulators of cilium biogenesis, assembly, and/or maintenance. They also have important implications in clinical conditions associated not only with well-known ciliopathies, but with cancer and microcephaly.
Materials and Methods
Cell culture
HEK293T, BALB/C 3T3, and RPE1-hTERT cells were obtained from ATCC, and cell cultures were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) plus 10% fetal bovine serum. DLD-1 and DLD-1-FBXW7−/− cells were obtained from Dr. Wei (Harvard).
Primary hippocampal neuronal cultures and antibody staining were prepared as follows: Hippocampi from newborn mice were dissected away from the cortex and digested with Papain and DNase-I for 25 min. Hippocampi were then triturated with a glass Pasteur pipette, and cell suspensions were plated on poly-L-lysine-coated coverslips. Primary hippocampal cultures were maintained for 3 weeks in Neuralbasal media supplemented with B27 and Glutamax-I with treatment of the mitotic inhibitor 5′-fluoro-2′-deoxyuridine on day 3 in culture. Cells were fixed in 4% paraformaldehyde and stained for designated primary antibodies for 1 h at 37°C and complementary secondary antibodies for 30 min at 37°C. Coverslips were mounted on slides with ProLong Gold, and nuclei were labeled with DAPI. Tau+ neurons were imaged the following day on a Leica SP2 scanning confocal microscope with an oil immersion 63× objective.
Reagents
Nocodazole, emetine, lactacystin, and MG132 were purchased from Sigma. Phos-tag reagent was purchased from Wako Chemicals. SAG was purchased from Millipore.
Plasmids
Mouse and human NDE1 were obtained from Open Biosystems and were subcloned into myc-pFlag-CMV-2 vector (Kim et al, 2011). GFP-CDK5, GFP-DN-CDK5, MYC-p35, GFP-p25, FLAG-β-TrCP1, HA-CDK1, HA-CDK2, HA-GSK3β, cyclin A, cyclin E, MYC-FBW7, and MYC-FBW7R465C were obtained from Addgene. tGFP-p39 was obtained from Origene. MYC-NDE1T191I and FLAG-NDE1T191I were generated using QuikChange mutagenesis kit according to the manufacturer’s instructions (Stratagene).
Transient transfection
siRNAs were transfected into RPE1-hTERT cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Plasmids were transfected into 293T cells using calcium phosphate method, as described (Kim et al, 2011).
Immunoblotting
HEK293T, BALB/C 3T3, or RPE1-hTERT cells were lysed in 1% Triton X-100, 150 mM NaCl, 10 mM Tris–HCl at pH 7.5, 1 mM EGTA, 1 mM EDTA, 10% sucrose and protease inhibitor cocktail (Roche Applied Science), and phosphatase inhibitors 0.2 mM Na3VO4 and 1 mM NaF at 4°C for 30 min. Antibodies against NDE1, (Proteintech), c-Myc, Flag (Sigma-Aldrich), CDK5 (Cell Signaling Technology), p35 (used to detect both p35 and p25) (C-19:sc-820, Santa Cruz Biotechnology), β-actin (Santa Cruz Biotechnology), mouse-FBW7 (Abnova), rabbit-FBW7 (A301-720A; A301-721A) (Bethyl Laboratories), rabbit-GLI2 (Cell Signaling), rabbit-Aurora A Kinase (Cell signaling), and α-tubulin (Sigma Aldrich) were used at a 1:1,000 dilution. Densitometric quantification of autoradiograms was analyzed by ImageJ software.
Lambda (λ-) phosphatase treatment
RPE1-hTERT cells were lysed in 1% Triton X-100, 150 mM NaCl, 10 mM Tris–HCl at pH 7.5, 1 mM EGTA, 1 mM EDTA, 10% sucrose and protease inhibitor cocktail (Roche Applied Science), and phosphatase inhibitors 0.2 mM Na3VO4 and 1 mM NaF at 4°C for 30 min. NDE1 was immunoprecipitated with rabbit α-NDE1, and complexes were washed three times with lysis buffer followed by a wash with phosphate-buffered saline (PBS). A second wash was performed with λ-phosphatase buffer containing MnCl2 (1 mM). Half of immunoprecipitated NDE1 was treated with 1.25 units of λ-phosphatase (Biolabs, New England) for 1 h at 30°C followed by boiling for 5 min at 96°C.
Indirect immunofluorescence
RPE1-hTERT cells were grown on glass coverslips and fixed in 1:1 methanol/acetone or 4% paraformaldehyde, permeabilized in 0.2% Triton X-100 in PBS, blocked in 2% heat-activated goat serum/0.2% Triton X-100 in PBS (blocking buffer), and incubated overnight with primary antibodies diluted in blocking buffer at 4°C. Primary antibodies were used against mouse acetylated α-tubulin, at 1:1,000 (Sigma Aldrich), rabbit γ-tubulin 1:1,000 (Sigma-Aldrich), mouse γ-tubulin 1:1,000 (Abcam), rabbit Flag 1:1,000, rabbit CEP164, 1:1,000 (Novus Biologicals), mouse ninein 1:500, mouse centrin-2 1:500 (Millipore), rabbit ARL13B at 1:500, rabbit NDE1 at 1:1,000 (Proteintech), mouse FBW7 1:1,000 (Abnova), and rabbit FBW7 (A301-720A; A301-721A) (Bethyl Laboratories). Cells were washed three times with PBS and incubated for 2 h at 4°C with appropriate combinations of fluorescence-labeled secondary antibodies at 1:2,000 dilution. Secondary antibodies were donkey anti-rabbit Alexa 488, donkey anti-mouse Alexa 568, goat anti-mouse Alexa 488, donkey anti-mouse Alexa 568, or donkey anti-rabbit Alexa 647 (Molecular Probes). Excess of secondary antibodies were removed by four washes in PBS. Coverslips were mounted with ProLong Gold antifade reagent with DAPI (Molecular Probes). Images were acquired with Leica SP2MP confocal microscope and processed with ImageJ software and Adobe Photoshop 6.0. Quantification of fluorescence intensity ratio of NDE1/γ-tubulin (red/green) at the centrosome (Fig2B; Appendix Fig S6) was done as described earlier (Kim et al, 2011).
Cell cycle analysis
BALB/C 3T3 and RPE1-hTERT cells were fixed with 70% ethanol and stored at −20°C for no longer than 24 h. After fixation, cells were stained with 40 μg/ml propidium iodide (Sigma Aldrich) plus 200 μg/ml RNase A (Sigma Aldrich) in PBS and subjected to flow cytometry analysis. At least 15,000 cells were analyzed per sample using a FACSCalibur™ flow cytometer.
siRNA sequences
The human non-specific siRNAs 5′-UUCUCCGAACGUGUCACGU-3′ and 5′-ACGUGACACGUUCGGAGAA-3′ were obtained from QIAGEN. Human NDE1-specific smart pool siRNA (5′-GGACCCAGCUCAAGUUUAA-3′, 5′-GCGCAGACCAAAGCCAUUA-3′, 5′-GCUGAAGCCUGUUCUUGGU-3′, 5′-GCAGCACUCUGAAGGCUAC-3′), human p35 (CDK5R1)-specific smart pool siRNA (5′-GCAGAUAAAUGCCGACCCA-3′, 5′-UCACGCACCUCAACAAUGA-3′, 5′-GGAAGGCCACGCUGUUUGA-3′, 5′-UCACACAGGUCUUCUCCGA-3′), and human CDK5-1 specific smart pool siRNA (5′-UAUAAGCCCUAUCCGAUGU-3′, 5′-CCGGGAGACUCAUGAGAUC-3′, 5′-GGGCUGGGAUUCUGUCAUA-3′, 5′-GGAUUCCCGUCCGCUGUUA-3′) were obtained from Dharmacon (Thermo scientific) and human-specific CDK5-2 5′-UAUGACAGAAUCCCAGCC-3′, 5′-GGGCUGGGAUUCUGUCAUA-3′. Human FBW7-1-specific siRNAs 5-GCAUUUCUCUCUCCAGAGAAGGU-3′ and 5′-ACCUUCUCUGGAGAGAGAAAUGC-3′ and FBW7-2-specific siRNAs 5-UCUCCAGUCUCUGCAUUCCACAC-3′ and 5′-GUGUGGAAUGCAGAGACUGGAGA-3′ were initially obtained from Dharmacon (Thermo Fisher) and Integrated DNA Technology.
Ubiquitylation assay
In vivo ubiquitylation assay was done according to Li et al (2013). Briefly, HEK293T cells were transiently co-transfected with FLAG-tagged NDE1 or FLAG-tagged Nde1T191I and pcB6-His ubiquitin (gift from R. Baer). After 24 h, cells were treated with 10 μM MG132 (Peptides international) for 5 h, and cells were lysed and briefly sonicated in buffer A (100 mM Na2PO4, 10 mM Tris–HCl, 6 M guanidine-HCl, 10 mM imidazole pH 8.0). Cell lysates were incubated with nickel-NTA resin (Qiagen) to precipitate His-tagged ubiquitylated proteins for 2–3 h at room temperature (24°C). Ni-NTA beads were washed three times with buffer A, two times with washes A/TI (1 volume buffer A and 1 volume TI buffer) (TI buffer, 25 mM Tris–Cl pH 6.8 and 20 mM imidazole), and then wash with TI buffer. Finally, Ni-NTA beads were eluted in SDS loading buffer containing 200 mM imidazole, separated by SDS–PAGE, and detected by immunoblotting.
Statistics
Data are presented as mean ± SEM. One-way ANOVA followed by Newman–Keuls post-test was used to determine statistical significance among multiple measurements. *P < 0.05, **P < 0.01, ***P < 0.005, nsP > 0.05.
Acknowledgments
We would like to thank Dr. Axel Behrens for helpful suggestions and Drs. Wei for DLD-1 cells, R. Baer for pcB6-His ubiquitin plasmid, S. Rankin for help with the APC/C assay using Xenopus egg extracts, and M. Ahmad for help with the preparation of hippocampal cultures. This work was supported by PBBEP3-141439 from the Swiss National Science Foundation (DM); GM074692 from NIH (GL); DK59599 from NIH, Oklahoma Center for the Advancement of Science and Technology; and the John S. Gammill Endowed Chair in Polycystic Kidney Disease (LT).
Author contributions
DM performed biochemical and imaging experiments in HEK293T, RPE1-hTERT, and DLD-1 cells; MCM and GL performed imaging experiments and analyzed data on primary hippocampal neurons; SeoK performed biochemical experiments; SehK performed APC/C assays; ECO performed initial mutagenesis experiments; and LT supervised the project, performed biochemical experiments, analyzed data, and wrote the paper together with DM.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Appendix
Review Process File
References
- Alkuraya FS, Cai X, Emery C, Mochida GH, Al-Dosari MS, Felie JM, Hill RS, Barry BJ, Partlow JN, Gascon GG, Kentab A, Jan M, Shaheen R, Feng Y, Walsh CA. Human mutations in NDE1 cause extreme microcephaly with lissencephaly. Am J Hum Genet. 2011;88:536–547. doi: 10.1016/j.ajhg.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arquint C, Nigg EA. STIL microcephaly mutations interfere with APC/C-mediated degradation and cause centriole amplification. Curr Biol. 2014;24:351–360. doi: 10.1016/j.cub.2013.12.016. [DOI] [PubMed] [Google Scholar]
- Bakircioglu M, Carvalho OP, Khurshid M, Cox JJ, Tuysuz B, Barak T, Yilmaz S, Caglayan O, Dincer A, Nicholas AK, Quarrell O, Springell K, Karbani G, Malik S, Gannon C, Sheridan E, Crosier M, Lisgo SN, Lindsay S, Bilguvar K, et al. The essential role of centrosomal NDE1 in human cerebral cortex neurogenesis. Am J Hum Genet. 2011;88:523–535. doi: 10.1016/j.ajhg.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskaran N, van Drogen F, Ng HF, Kumar R, Ekholm-Reed S, Peter M, Sangfelt O, Reed SI. Fbw7alpha and Fbw7gamma collaborate to shuttle cyclin E1 into the nucleolus for multiubiquitylation. Mol Cell Biol. 2013;33:85–97. doi: 10.1128/MCB.00288-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busino L, Millman SE, Scotto L, Kyratsous CA, Basrur V, O’Connor O, Hoffmann A, Elenitoba-Johnson KS, Pagano M. Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat Cell Biol. 2012;14:375–385. doi: 10.1038/ncb2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow HM, Guo D, Zhou JC, Zhang GY, Li HF, Herrup K, Zhang J. CDK5 activator protein p25 preferentially binds and activates GSK3beta. Proc Natl Acad Sci USA. 2014;111:E4887–E4895. doi: 10.1073/pnas.1402627111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen ST, Clement CA, Satir P, Pedersen LB. Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. J Pathol. 2012;226:172–184. doi: 10.1002/path.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizmecioglu O, Krause A, Bahtz R, Ehret L, Malek N, Hoffmann I. Plk2 regulates centriole duplication through phosphorylation-mediated degradation of Fbxw7 (human Cdc4) J Cell Sci. 2012;125:981–992. doi: 10.1242/jcs.095075. [DOI] [PubMed] [Google Scholar]
- Crusio KM, King B, Reavie LB, Aifantis I. The ubiquitous nature of cancer: the role of the SCF(Fbw7) complex in development and transformation. Oncogene. 2010;29:4865–4873. doi: 10.1038/onc.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell. 2014;26:455–464. doi: 10.1016/j.ccell.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Olson EC, Stukenberg PT, Flanagan LA, Kirschner MW, Walsh CA. LIS1 regulates CNS lamination by interacting with mNudE, a central component of the centrosome. Neuron. 2000;28:665–679. doi: 10.1016/s0896-6273(00)00145-8. [DOI] [PubMed] [Google Scholar]
- Feng Y, Walsh CA. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron. 2004;44:279–293. doi: 10.1016/j.neuron.2004.09.023. [DOI] [PubMed] [Google Scholar]
- Freed E, Lacey KR, Huie P, Lyapina SA, Deshaies RJ, Stearns T, Jackson PK. Components of an SCF ubiquitin ligase localize to the centrosome and regulate the centrosome duplication cycle. Genes Dev. 1999;13:2242–2257. doi: 10.1101/gad.13.17.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Liem KF, Jr, Anderson KV. The spinocerebellar ataxia-associated gene Tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell. 2012;151:847–858. doi: 10.1016/j.cell.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell. 2007;26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Hellmich MR, Pant HC, Wada E, Battey JF. Neuronal cdc2-like kinase: a cdc2-related protein kinase with predominantly neuronal expression. Proc Natl Acad Sci USA. 1992;89:10867–10871. doi: 10.1073/pnas.89.22.10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeck JD, Jandke A, Blake SM, Nye E, Spencer-Dene B, Brandner S, Behrens A. Fbw7 controls neural stem cell differentiation and progenitor apoptosis via Notch and c-Jun. Nat Neurosci. 2010;13:1365–1372. doi: 10.1038/nn.2644. [DOI] [PubMed] [Google Scholar]
- Houlihan SL, Feng Y. The scaffold protein Nde1 safeguards the brain genome during S phase of early neural progenitor differentiation. eLife. 2014;3:e03297. doi: 10.7554/eLife.03297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Marshall WF. Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol. 2011;12:222–234. doi: 10.1038/nrm3085. [DOI] [PubMed] [Google Scholar]
- Kasahara K, Kawakami Y, Kiyono T, Yonemura S, Kawamura Y, Era S, Matsuzaki F, Goshima N, Inagaki M. Ubiquitin-proteasome system controls ciliogenesis at the initial step of axoneme extension. Nat Commun. 2014;5:5081. doi: 10.1038/ncomms6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Law V, Rosales JL, Lee KY. Cdk5 variant 1 (cdk5-v1), but not full-length cdk5, is a centrosomal protein. Cell Cycle. 2010;9:2251–2253. doi: 10.4161/cc.9.11.11652. [DOI] [PubMed] [Google Scholar]
- Kim S, Tsiokas L. Cilia and cell cycle re-entry: more than a coincidence. Cell Cycle. 2011;10:2683–2690. doi: 10.4161/cc.10.16.17009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Zaghloul NA, Bubenshchikova E, Oh EC, Rankin S, Katsanis N, Obara T, Tsiokas L. Nde1-mediated suppression of ciliogenesis affects cell cycle re-entry. Nat Cell Biol. 2011;13:351–360. doi: 10.1038/ncb2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Dynlacht BD. Assembling a primary cilium. Curr Opin Cell Biol. 2013;25:506–511. doi: 10.1016/j.ceb.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Dynlacht BD. Regulating the transition from centriole to basal body. J Cell Biol. 2011;193:435–444. doi: 10.1083/jcb.201101005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294:173–177. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- Lew J, Huang QQ, Qi Z, Winkfein RJ, Aebersold R, Hunt T, Wang JH. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- Li J, D’Angiolella V, Seeley ES, Kim S, Kobayashi T, Fu W, Campos EI, Pagano M, Dynlacht BD. USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature. 2013;495:255–259. doi: 10.1038/nature11941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liem KF, Jr, Ashe A, He M, Satir P, Moran J, Beier D, Wicking C, Anderson KV. The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J Cell Biol. 2012;197:789–800. doi: 10.1083/jcb.201110049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlepage LE, Ruderman JV. Identification of a new APC/C recognition domain, the A box, which is required for the Cdh1-dependent destruction of the kinase Aurora-A during mitotic exit. Genes Dev. 2002;16:2274–2285. doi: 10.1101/gad.1007302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, Balmain A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321:1499–1502. doi: 10.1126/science.1162981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432:775–779. doi: 10.1038/nature03155. [DOI] [PubMed] [Google Scholar]
- Modi PK, Komaravelli N, Singh N, Sharma P. Interplay between MEK-ERK signaling, cyclin D1, and cyclin-dependent kinase 5 regulates cell cycle reentry and apoptosis of neurons. Mol Biol Cell. 2012;23:3722–3730. doi: 10.1091/mbc.E12-02-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS, Morabito M, Tsai LH. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- Nigg EA, Stearns T. The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol. 2011;13:1154–1160. doi: 10.1038/ncb2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;10:816–825. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- Odajima J, Wills ZP, Ndassa YM, Terunuma M, Kretschmannova K, Deeb TZ, Geng Y, Gawrzak S, Quadros IM, Newman J, Das M, Jecrois ME, Yu Q, Li N, Bienvenu F, Moss SJ, Greenberg ME, Marto JA, Sicinski P. Cyclin E constrains Cdk5 activity to regulate synaptic plasticity and memory formation. Dev Cell. 2011;21:655–668. doi: 10.1016/j.devcel.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R, Christiansen H, Berwanger B, Eilers M. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15:67–78. doi: 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Pan J, Snell W. The primary cilium: keeper of the key to cell division. Cell. 2007;129:1255–1257. doi: 10.1016/j.cell.2007.06.018. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Pei Z, Lang B, Fragoso YD, Shearer KD, Zhao L, McCaffery PJ, Shen S, Ding YQ, McCaig CD, Collinson JM. The expression and roles of Nde1 and Ndel1 in the adult mammalian central nervous system. Neuroscience. 2014;271:119–136. doi: 10.1016/j.neuroscience.2014.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikova OV, Golemis EA, Pugacheva EN. Cell cycle-dependent ciliogenesis and cancer. Cancer Res. 2008;68:2058–2061. doi: 10.1158/0008-5472.CAN-07-5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129:1351–1363. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarmby LM, Parker JD. Cilia and the cell cycle? J Cell Biol. 2005;169:707–710. doi: 10.1083/jcb.200503053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- Rosales JL, Rattner JB, Lee KY. Cdk5 in the centriolar appendages mediates cenexin1 localization and primary cilia formation. Cell Cycle. 2010a;9:2037–2039. doi: 10.4161/cc.9.10.11600. [DOI] [PubMed] [Google Scholar]
- Rosales JL, Rattner JB, Lee KY. The primary microcephaly 3 (MCPH3) interacting protein, p35 and its catalytic subunit, Cdk5, are centrosomal proteins. Cell Cycle. 2010b;9:618–620. doi: 10.4161/cc.9.3.10597. [DOI] [PubMed] [Google Scholar]
- Sancho R, Gruber R, Gu G, Behrens A. Loss of Fbw7 reprograms adult pancreatic ductal cells into alpha, delta, and beta cells. Cell Stem Cell. 2014;15:139–153. doi: 10.1016/j.stem.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Mori D, Toyo-oka K, Chen A, Garrett-Beal L, Muramatsu M, Miyagawa S, Hiraiwa N, Yoshiki A, Wynshaw-Boris A, Hirotsune S. Complete loss of Ndel1 results in neuronal migration defects and early embryonic lethality. Mol Cell Biol. 2005;25:7812–7827. doi: 10.1128/MCB.25.17.7812-7827.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley ES, Nachury MV. The perennial organelle: assembly and disassembly of the primary cilium. J Cell Sci. 2010;123:511–518. doi: 10.1242/jcs.061093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Lahiri DK. Cdk5 activity in the brain - multiple paths of regulation. J Cell Sci. 2014;127:2391–2400. doi: 10.1242/jcs.147553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spektor A, Tsang WY, Khoo D, Dynlacht BD. Cep97 and CP110 suppress a cilia assembly program. Cell. 2007;130:678–690. doi: 10.1016/j.cell.2007.06.027. [DOI] [PubMed] [Google Scholar]
- Su SC, Tsai LH. Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol. 2011;27:465–491. doi: 10.1146/annurev-cellbio-092910-154023. [DOI] [PubMed] [Google Scholar]
- Sung CH, Leroux MR. The roles of evolutionarily conserved functional modules in cilia-related trafficking. Nat Cell Biol. 2013;15:1387–1397. doi: 10.1038/ncb2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Li A. Ciliary resorption modulates G1 length and cell cycle progression. Cell Cycle. 2011;10:2825–2826. doi: 10.4161/cc.10.17.16943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature. 2013;502:254–257. doi: 10.1038/nature12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- Tsang WY, Dynlacht BD. CP110 and its network of partners coordinately regulate cilia assembly. Cilia. 2013;2:9. doi: 10.1186/2046-2530-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnolle MA, Taylor SS. Cenp-F links kinetochores to Ndel1/Nde1/Lis1/dynein microtubule motor complexes. Curr Biol. 2007;17:1173–1179. doi: 10.1016/j.cub.2007.05.077. [DOI] [PubMed] [Google Scholar]
- Wang Z, Inuzuka H, Fukushima H, Wan L, Gao D, Shaik S, Sarkar FH, Wei W. Emerging roles of the FBW7 tumour suppressor in stem cell differentiation. EMBO Rep. 2012;13:36–43. doi: 10.1038/embor.2011.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wu T, Kirschner MW. The master cell cycle regulator APC-Cdc20 regulates ciliary length and disassembly of the primary cilium. eLife. 2014;3:e03083. doi: 10.7554/eLife.03083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr Biol. 2004;14:1852–1857. doi: 10.1016/j.cub.2004.09.083. [DOI] [PubMed] [Google Scholar]
- Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- Wigley WC, Fabunmi RP, Lee MG, Marino CR, Muallem S, DeMartino GN, Thomas PJ. Dynamic association of proteasomal machinery with the centrosome. J Cell Biol. 1999;145:481–490. doi: 10.1083/jcb.145.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Zhang H, Beach D. D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell. 1992;71:505–514. doi: 10.1016/0092-8674(92)90518-h. [DOI] [PubMed] [Google Scholar]
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li H, Zhou T, Zhou J, Herrup K. Cdk5 levels oscillate during the neuronal cell cycle: Cdh1 ubiquitination triggers proteosome-dependent degradation during S-phase. J Biol Chem. 2012;287:25985–25994. doi: 10.1074/jbc.M112.343152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File