

Abstract
Background. Staphylococcus epidermidis causes late-onset sepsis in preterm infants. Staphylococcus epidermidis activates host responses in part via Toll-like receptor 2 (TLR2). Epidemiologic studies link bacteremia and neonatal brain injury, but direct evidence is lacking.
Methods. Wild-type and TLR2-deficient (TLR2−/−) mice were injected intravenously with S. epidermidis at postnatal day 1 prior to measuring plasma and brain cytokine and chemokine levels, bacterial clearance, brain caspase-3 activation, white/gray matter volume, and innate transcriptome.
Results. Staphylococcus epidermidis bacteremia spontaneously resolved over 24 hours without detectable bacteria in the cerebrospinal fluid (CSF). TLR2−/− mice demonstrated delayed S. epidermidis clearance from blood, spleen, and liver. Staphylococcus epidermidis increased the white blood cell count in the CSF, increased interleukin 6, interleukin 12p40, CCL2, and CXCL1 concentrations in plasma; increased the CCL2 concentration in the brain; and caused rapid (within 6 hours) TLR2-dependent brain activation of caspase-3 and TLR2-independent white matter injury.
Conclusions. Staphylococcus epidermidis bacteremia, in the absence of bacterial entry into the CSF, impairs neonatal brain development. Staphylococcus epidermidis bacteremia induced both TLR2-dependent and –independent brain injury, with the latter occurring in the absence of TLR2, a condition associated with an increased bacterial burden. Our study indicates that the consequences of transient bacteremia in early life may be more severe than commonly appreciated, and our findings may inform novel approaches to reduce bacteremia-associated brain injury.
Keywords: Staphylococcus epidermidis, brain injury, preterm, Toll-like receptor
Preterm birth is associated with a high risk of brain injury that can contribute to neurological disorders such as cerebral palsy, autism, and possibly schizophrenia [1, 2]. Cerebral white-matter injury is the most common form of injury to the preterm brain and is associated with a high risk of neurodevelopmental impairment. Indeed, preterm infants exposed to chorioamnionitis, fetal vasculitis, and postnatal sepsis are particularly susceptible to cerebral white-matter injury and cerebral palsy [3–6]. In animal models, inflammation, such as that caused by systemic administration of lipopolysaccharide, induces cerebral white-matter injury similar to that seen in preterm human infants [3, 7, 8]. Fewer studies have addressed the ability of live bacteria to induce brain injury [9], and to our knowledge none of these have modeled this in newborn animals that express distinct immune function and susceptibility to brain injury [2, 10].
Improvements in perinatal and neonatal care have changed the pattern of prenatal/neonatal infectious pathogens from high-virulence bacteria, such as group B Streptococcus and Escherichia coli, to predominantly low-virulence bacteria that cause nosocomial, late-onset infections [11]. Coagulase-negative staphylococci such as S. epidermidis are the most common causes of bacteremia in preterm infants, especially those of low gestational age [12, 13]. Staphylococcus epidermidis is a ubiquitous skin commensal responsible for approximately 50% of all cases of late-onset neonatal septicemia [14]. Although S. epidermidis infection results in relatively low mortality, it is associated with an increased risk of common adverse outcomes, prolonged hospital stays, and increased costs [15, 16]. Staphylococcus epidermidis, via expression of soluble factors, including lipopeptides, activates host responses in part via Toll-like receptor 2 (TLR2) [17], which mediates recognition and clearance of S. epidermidis bacteremia in adult mice in vivo [18]. Moreover, repeated systemic (intraperitoneal) administration of the bacterial lipopeptide TLR2 agonist Pam3CSK4 adversely influences brain development in newborn mice [19], raising the possibility that bacteremia due to pathogens that activate TLR2, such as S. epidermidis, may trigger neonatal brain injury even in the absence of entry of bacteria into the central nervous system (CNS).
To assess the possible link between S. epidermidis bacteremia and early life brain injury, we characterized the impact of S. epidermidis bacteremia and the potential role of TLR2 using an established neonatal mouse model [20]. We found that S. epidermidis bacteremia at postnatal day 1 (PND1), even in the absence of bacteria in the cerebrospinal fluid (CSF), induces both systemic and brain inflammation and associated TLR2-dependent early brain caspase-3 activation (6 hours after injection on PND1) and TLR2-independent late white matter injury (PND14) providing fresh insights into the potential role of bacteremia in preterm brain injury.
MATERIALS AND METHODS
Animals
Control C57BL/6J wild-type (WT) and matched TLR2-deficient B6.129-Tlr2tm1Kir/J mice (hereafter, “TLR2−/− mice”) were obtained from the Jackson Laboratory and continuously bred in an accredited animal facility (Experimental Biomedicine, University of Gothenburg, Sweden). Animal protocols were approved by Gothenburg Animal Ethics Committee (protocol 21/2011).
Bacteria and Preparation of Inoculum
WT S. epidermidis 1457, a clinical strain obtained from an adult patient with a central venous catheter infection [21], was a kind gift from Dr Michael Otto's laboratory. For the preparation of inocula, 500 mL of bacterial stock was added to 30 mL of trypticase soy broth in a 125 mL baffled flask. Bacteria were grown for 16–20 hours in an incubator/shaker at 37°C and 240 rpm (MaxQ™ 4450 Benchtop Orbital Shakers - Thermo Scientific, Stockholm, Sweden), as previously described [20]. For murine injections, 1 × 108 S. epidermidis were resuspended in 1 mL of sterile, pyrogen-free saline, and mice were injected with 5 × 106 S. epidermidis in 50 μL saline.
Bacteremia Model
PND1 mice (≤24 hours of age), similar to preterm human infants with respect to CNS development [22], were injected via the intrajugular vein with 50 μL of either saline (control) or 5 × 106 S. epidermidis as previously described [20].
Sample Collection, Organ Extraction, and Bacterial Culture
At different time points after S. epidermidis injection, approximately 20 μL of blood was collected via cardiac puncture mixed with 5 μL pyrogen-free heparin sodium (1000 USP units/mL; Sagent Pharmaceutical, Schaumburg, Illinois). After a brief intracardial perfusion with saline, the brain, spleen, and liver were collected as described previously [20]. Tissue specimens were homogenized and were spread onto trypticase soy agar plates (TSA II; BD Diagnostics; Franklin Lakes, New Jersey) with 5% sheep blood and incubated at 37°C for 24 hours to enumerate colony-forming units (CFUs), as described previously [20].
CSF Sample Collection and White Blood Cell (WBC) Counting
To assess whether intrajugular administration of S. epidermidis resulted in entry of bacteria into the CSF, approximately 3–4 μL of CSF was collected from the cisterna magna of saline-injected and S. epidermidis–injected animals as previously described [23]. CSF sampling was assisted by a surgical microscope and CSF samples with any suspicion of blood contamination were discarded. Samples were mixed with methyl violet, and the number of WBCs was determined in a Bürker chamber (Thermo Scientific, Braunschweig, Germany).
Immunohistochemical Staining and Brain Injury Evaluation
PND14 mice were deeply anesthetized with 50 mg/mL pentothal and perfused intracardially with saline followed by 5% buffered formaldehyde (Histofix; Histolab, Gothenburg). Brains were cut into 10-µm coronal sections, collected at 50-section intervals, and serial sections were used for histologic staining as previously described [19, 24]. The following primary antibodies were used: anti-microtubule associated protein-2 (MAP-2; clone HM-2; Sigma), mouse anti-myelin basic protein (MBP; SMI 94; Sternberger Monoclonal, Lutherville, Massachusetts) and anti-ionized calcium-binding adapter molecule 1 (Iba-1; 019-19741 Wako Chemicals, Richmond, Virginia). Volumes of gray matter (MAP-2) and white matter (MBP) were measured and calculated as described previously [25]. The number of Iba-1–positive cells was counted as previously described [19].
Quantitative Reverse Transcription–Polymerase Chain Reaction (qRT-PCR)
Brain tissue RNA was extracted using the RNeasy Lipid Tissue Mini Kit (Qiagen) according to the manufacturer's instructions. Total RNA was measured in a spectrophotometer at 260 nm absorbance. Messenger RNA (mRNA; 500 ng) was reverse transcribed to complementary DNA in a Bio-Rad T100 ThermoCycler using the protocol provided in the RT2 First Strand Kit (Qiagen).
Gene expression levels were assessed by qRT-PCR analysis using an ABI 7300 real-time PCR system (Applied Biosystems, Foster City, California). A Mouse TLR Signaling Pathway PCR Array (PAMM-018A; Qiagen) containing primers for 84 genes of interest and 12 controls was used as described elsewhere [20].
Multiplex Cytokine and Chemokine Assay
Cytokine and chemokine levels in plasma and supernatants of brain homogenate were measured 6 hours after S. epidermidis or saline injection. Snap-frozen brains were homogenized by sonication in ice-cold homogenization buffer containing PMSF (P7626 Sigma), dimethyl sulfoxide (D2650, Sigma), and cell lysis buffer factor 1 and factor 2 (Bio-Plex Cell Lysis Kit; Life Science Research, Hercules, California). Mixtures were centrifuged at 4°C at 4500g for 20 minutes, and supernatants were collected and stored at −80°C until use. A panel of mouse cytokines and chemokines (interleukin 1β [IL-1β], interleukin 6 [IL-6], CXCL1, CCL2, tumor necrosis factor α [TNF-α], interleukin 12p40 [IL-12p40], interleukin 12p70 [IL-12p70], interleukin 10 [IL-10], interleukin 17 [IL-17], and interferon γ [IFN-γ]) was measured with the Bio-Plex Multiplex Cytokine Assay (Bio-Rad). Data were acquired on a Luminex 200 system (Bio-Rad). Brain homogenate cytokine concentrations were normalized to the amount of protein per sample as determined with the Bio-Rad DC detergent-compatible protein assay (Bio-Rad).
Caspase-3 Activity Measurement
Caspase-3 activity was measured at 6 hours after S. epidermidis or saline injection. PND1 mouse brains were rapidly dissected out and immediately frozen in isopentane on dry ice. Each hemisphere (approximately 50 mg) was sonicated in ice-cold homogenization buffer (50 mM Tris, pH 7.3, 2 mM ethylenediaminetetraacetic acid, 1% protease inhibitor cocktail [P8340; Sigma]). The homogenate was centrifuged at 10 000g for 10 minutes at 4°C, and the supernatant (S2; cytosolic fraction) was used in the determination of caspase-3 activity as described previously [26, 27].
Statistical Analyses
Statistical Package for the Social Sciences, version 19.0 (IBM, Armonk, New York), and GraphPad Prism, version 6.0 for Windows (San Diego, California), were used for all analyses. One-way analysis of variance followed by a least significant difference post hoc test was used for comparison of data from >2 groups. For all other comparisons, Student's unpaired t tests were used. Data groups with unequal variances were analyzed using Mann–Whitney U tests. A lower limit of detection of 1 CFU/mL was determined and used for statistical analyses. P values of <.05 were considered statistically significant.
RESULTS
TLR2 Accelerates Clearance of S. epidermidis Bacteremia in Neonatal Mice
As a first step in characterizing the potential impact of S. epidermidis bacteremia on the neonatal CNS, we assessed clearance of S. epidermidis from peripheral blood, solid organs, and CSF in WT and TLR2−/− neonatal mice. To this end, we measured bacterial CFUs in blood (Figure 1A), spleen (Figure 1B), liver (Figure 1C), and CSF (Figure 1D) 4, 24, 48, and 72 hours after S. epidermidis injection in WT and TLR2−/− mice. After peaking 4 hours after injection, there was a significant decrease in the numbers of CFUs in the blood and organs from 4 to 48 hours in WT mice, confirming that neonatal mice clear S. epidermidis infection [20]. However, TLR2−/− mice demonstrated impaired bacterial clearance compared with WT mice, and they had significantly higher concentrations of S. epidermidis in blood, spleen, and liver 24 hours after S. epidermidis injection and clearance was delayed until 72 hours after injection (Figure 1). Of note, no viable S. epidermidis (ie, CFUs) were detected in CSF at any of the time points examined, suggesting that S. epidermidis infection remained outside the CNS.
Figure 1.

Neonatal mice clear Staphylococcus epidermidis bacteremia within 48 hours without entry of bacteria into the cerebrospinal fluid (CSF). Wild-type (WT) and Toll-like receptor 2 (TLR2)–deficient (TLR2−/−) mice were injected intravenously with 5 × 106 S. epidermidis at postnatal day 1 (PND1) and euthanized 4, 24, 48, or 72 hours after injection prior to measuring bacterial colony-forming units (CFU) in blood (A), spleen (B), liver (C), and CSF (D). Each bar represents the mean value and standard error for 6–15 animals/group. Comparisons were made using the Mann–Whitney U test.
S. epidermidis Bacteremia Induces Blood and Brain Cytokines and Chemokines and Triggers TLR2-Dependent Brain Caspase-3 Activation
To characterize S. epidermidis–induced systemic inflammation and assess whether inflammation may extend to the brain, we measured cytokine and chemokine concentrations in plasma and brain homogenates after injection of saline or S. epidermidis into the intrajugular vein of PND1 mice (Figure 2). Cytokine production in blood plasma specimens and homogenates of brain collected 6 hours after S. epidermidis injection on PND1 from WT and TLR2−/− mice was measured using a multiplex cytokine/chemokine assay. Staphylococcus epidermidis injection induced increases of plasma, IL-6, CCL2, CXCL1, and IL-12p40 concentrations in both WT and TLR2−/− mice, while CCL2 and IL-12p40 concentrations were significantly higher in TLR2−/− mice, compared with WT mice, corresponding with the higher bacterial burden and delayed bacterial clearance in TLR2−/− mice (Figure 1A–C). In brain homogenate, CCL2 was selectively and significantly increased at 6 hours after S. epidermidis injection in WT and TLR2−/− mice. In contrast to the levels of aforementioned cytokines that were higher in S. epidermidis-injected mice, S. epidermidis injection did not induce detectable increases in blood or brain concentrations of the other cytokines measured, including IL-1β, TNF-α, IL-10, IL-17, IL12p70, and IFN-γ, in either WT or TLR2−/− mice (Supplementary Tables 1 and 2).
Figure 2.

Staphylococcus epidermidis bacteremia induces systemic cytokine and brain CCL2 chemokine production 6 hours after injection. Newborn mice were injected intravenously with 5 × 106 S. epidermidis on postnatal day 1 (PND1) and then euthanized at 6 hours after injection for collection of plasma specimens and brain for homogenization. Levels of cytokines and chemokines were then measured. Each bar represents the mean value and standard error for 6–8 animals. *P < .05, **P < .01, and ***P < .01, by unpaired t tests, for comparisons between S. epidermidis and saline recipients. #P < .05 and ##P < .01, by unpaired t tests, for comparisons between WT and Toll-like receptor 2–deficient (TLR2−/−) mice. Abbreviations: IL-6, interleukin 6; IL-12p40, interleukin 12p40; IL-12p70, interleukin 12p70.
To assess potential recruitment of leukocytes into the systemic circulation and CNS, we determined the number of WBCs in blood (Figure 3A and 3C) and CSF (Figure 3B and 3D) specimens collection 6 hours (Figure 3A and 3B) and 24 hours (Figure 3C and 3D) after S. epidermidis injection in WT and TLR2−/− mice. Six hours after S. epidermidis injection, WBC counts in blood (Figure 3A) and CSF (Figure 3B) specimens were significantly increased in WT and TLR2−/− mice. By 24 hours after S. epidermidis injection, there were no significant differences in the number of WBCs in blood (Figure 3C) or CSF (Figure 3C) specimens from WT or TLR2−/− mice.
Figure 3.

Staphylococcus epidermidis injection at postnatal day 1 (PND1) increased peripheral blood and cerebrospinal fluid (CSF) white blood cell (WBC) counts. WBC counts in blood (A and C) and CSF (B and D) specimens from wild-type (WT) and Toll-like receptor 2–deficient (TLR2−/−) mice 6 (A and B) and 24 (C and D) hours after S. epidermidis injection on PND1. Each bar represents the mean value ± standard error for 5–8 animals. *P < .05 and **P < .01, by unpaired t tests, for comparisons between WT and TLR2−/− mice.
To assess whether S. epidermidis triggers brain apoptosis, we measured caspase-3 activation in brain homogenates (Figure 4). Six hours after S. epidermidis injection, there was a significant increase in caspase-3 activation in brain homogenates of WT but not TLR2−/− neonatal mice.
Figure 4.

Staphylococcus epidermidis bacteremia increases Toll-like receptor 2 (TLR2)–mediated brain caspase-3 activation 6 hours after injection. Caspase-3 activation was measured in brain homogenates as described in “Methods” section. Each bar represents the mean value and standard error for 5–6 animals. Significant changes within the WT groups are shown. *P < .05, by the unpaired t test, for comparison between the wild-type (WT) groups. Abbreviation: TLR2−/−, TLR2 deficient.
S. epidermidis Bacteremia Induces Early Selective Upregulation of Brain TLR2 and Downstream Target Genes
Having observed that S. epidermidis bacteremia induces increased brain CCL2 concentrations (Figure 2), we sought to further characterize S. epidermidis bacteremia–induced innate immune activation in the brain. To this end, we harvested brains from WT mice 6 and 24 hours after S. epidermidis injection and analyzed total RNA by targeted qRT-PCR TLR signaling pathway gene arrays. Compared with saline-injected animals, S. epidermidis–injected animals demonstrated selective and significant upregulation at 6 hours of mRNAs encoding TLR2 (but not the other TLRs tested), IFN regulatory factor-1 (IRF-1; a transcription factor), and prostaglandin-endoperoxide synthase 2 (PTGS2; also known as cyclooxygenase-2; Figure 5A), and at 24 hours of mRNAs encoding IRF-1 and nuclear factor-κ light polypeptide gene enhancer in B-cells inhibitor-β, an inhibitor of nuclear factor–κB (Figure 5B). A complete list of the genes for which mRNA expression was measured and their fold-changes appears in Supplementary Tables 3 and 4. Overall, these results indicate that S. epidermidis bacteremia, in the absence of detectable entry of live S. epidermidis to the CSF, activates innate, including TLR2-based, inflammatory signaling pathways in the brain.
Figure 5.
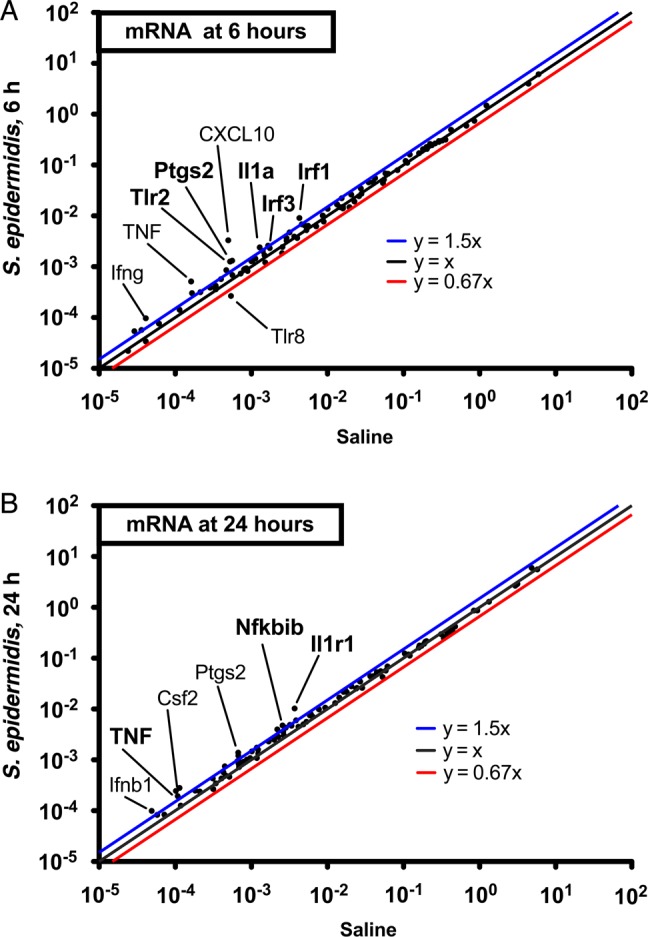
Staphylococcus epidermidis bacteremia induces expression of a selective innate transcriptome, including Toll-like receptor 2 (TLR2), in the brain. Mice were injected with 5 × 106 S. epidermidis intravenously at postnatal day 1 (PND1) prior to collection of brains 6 or 24 hours after infection for total brain RNA purification. Expression of TLR signaling pathway gene messenger RNA (mRNA) transcripts was analyzed by real-time polymerase chain reaction in brains collected at 6 hours (A) and 24 hours (B). Log10-normalized brain gene expression levels are plotted for both saline-injected mice (x-axis) and S. epidermidis–injected mice (y-axis). The asterisks represent mRNA transcripts that are significantly upregulated as calculated by the Mann–Whitney U test. *P < .05. Abbreviation: TNF, tumor necrosis factor.
S. epidermidis Bacteremia Does Not Affect Neonatal Mouse Survival
As S. epidermidis bacteremia, in the absence of meningitis, induced both systemic and brain inflammation, we next sought to assess whether these inflammatory changes were accompanied by signs of brain injury. Prior studies of S. epidermidis infection in newborn mice focused on the first 48 hours after injection [20], but to determine any potential impact of S. epidermidis bacteremia on brain injury, longer follow-up was desirable. Accordingly, we next characterized both short-term (≤3 days) and long-term (4–13 days) survival of WT and TLR2−/− mice after injection with saline or S. epidermidis (Supplementary Table 5). As there was no significant survival difference between the WT and TLR2−/− mice, the data from these two groups of mice were pooled. Consistent with the low rate of mortality in S. epidermidis–infected human newborns and the relatively low virulence of S. epidermidis [28], there were no significant differences in either short-term or long-term survival rates between saline-injected and S. epidermidis–injected newborn mice, indicating that S. epidermidis bacteremia did not significantly affect survival. Mice infected with S. epidermidis at PND1 demonstrated >80% long-term survival (4–13 days after infection; Supplementary Table 5).
S. epidermidis Bacteremia Impairs Development of White and Gray Matter in WT and TLR2−/− Mice
The early (within 24 hours) induction of systemic and brain inflammation after injection of S. epidermidis suggested that bacteremia may induce brain injury. Immunohistochemical staining for Iba-1, a protein expressed in microglia, indicated that S. epidermidis injection was not accompanied by an increase in the number of brain microglia (Supplementary Figure 1A and 1B). To assess whether S. epidermidis bacteremia in neonatal mice may influence brain development, changes in brain white and gray matter after S. epidermidis injection were measured by immunohistochemical staining at PND14, an age when brain myelin is formed and is measurable in mice [29]. The subcortical white-matter volume was measured using the marker MBP (Figure 6A and 6C), and the volume of cerebral gray matter was measured using the neuronal marker MAP-2 (Figure 6B). Compared with saline-injected mice, S. epidermidis–injected WT and TLR2−/− mice had significantly decreased volumes of cerebral white and gray matter at PND14 (Figure 6). Although neither tissue infarction, hemorrhage, nor any other forms of focal insult were observed, disruption of myelin processes was noted in the cortical region of the S. epidermidis–injected mice in both WT and TLR2−/− groups (Figure 6C).
Figure 6.

Staphylococcus epidermidis bacteremia initiated at postnatal day (PND) 1 impairs development of white and gray matter by PND14. Neonatal wild-type (WT) and Toll-like receptor 2–deficient (TLR2−/−) mice were injected with S. epidermidis on PND1 and euthanized for collection of brains at PND14. A, Quantitative analysis of subcortical white matter volume. B, Quantitative analysis of cerebral gray matter volume. C, Representative microphotographs of mouse anti-myelin basic protein (MBP) staining in the subcortical area. Bars represent means ± standard error of the mean (n = 6–14 animals/group). *P < .05 and ***P < .001, by unpaired t tests, for comparisons between saline-injected and S. epidermidis–injected WT or TLR2−/− animals. Abbreviation: MAP-2, microtubule-associated protein-2.
DISCUSSION
Prompted by the growing epidemiologic and clinical evidence that bacteremia in human newborns, even in the absence of positive CSF cultures, is associated with subsequent CNS injury [2–5, 30, 31], we tested the hypothesis that S. epidermidis bacteremia can induce brain injury and we characterized the potential roles of TLR2 in this process. To this end, we used an established neonatal S. epidermidis sepsis model [20] to demonstrate for the first time that (1) S. epidermidis bacteremia, in the absence of viable bacteria in the CSF, has a detrimental impact on the development of the neonatal brain via both rapid TLR2-dependent caspase-3 activation, as well as longer-term TLR2-independent pathways, culminating in reduced white and gray matter volumes; and (2) neonatal TLR2 is functional in host defense against S. epidermidis and serves to accelerate bacterial clearance. Our results are thus in agreement with the observations that human preterm infants with bacteremia, even in the absence of positive CSF cultures, experience CNS injury [31].
Staphylococcus epidermidis injection triggered systemic inflammation, including elevations in the peripheral WBC count and increases in concentrations of systemic plasma cytokines and chemokines, including IL-6, IL-12p40, CCL2, and CXCL1. However, with respect to cytokines and chemokines in brain homogenates, S. epidermidis injection increased concentrations of CCL2 but not the other chemokines/cytokines examined. CCL2 is rapidly upregulated in response to a variety of acute and chronic CNS disorders [32], including neonatal brain injury in rodent models [33] and human infants [34]. CCL2 is released across the apical and basolateral membranes of the choroid epithelium and can mediate leukocyte recruitment to the choroid plexus [35, 36]. Of note, functional inhibition of CCL2 by using a neutralizing antibody protects the neonatal rat brain from acute excitotoxic injury [37]. As there was a significant increase in leukocyte numbers in the CSF 6 hours after S. epidermidis, similar in magnitude to that observed after intraperitoneal injection of lipopolysaccharide (0.3 mg/kg) [38], there is apparently recruitment of cells across the choroid plexus early after S. epidermidis injection, which might contribute to CCL2 production in the brain and subsequent impaired brain development.
Apoptosis represents a common pathway for brain injury [39]. Indeed, activated caspase-3 plays a key role in the regulation of microglia activation, and TLR-mediated caspase-3 activation correlates positively with neurotoxicity [40]. Priming of microglia with the neuronal tissue protein α-synuclein enhances TLR2-mediated responses to bacterial lipopeptides (Pam3Cys) including caspase-3 activation and CCL2 secretion [41]. Our study demonstrates for the first time that S. epidermidis induces rapid and TLR2-dependent brain caspase-3 activation, suggesting a prominent role for TLR2 in early inflammatory responses to S. epidermidis that is consistent with our prior studies [18].
As a pattern-recognition receptor that mediates responses to gram-positive bacteria, TLR2 plays multiple roles in responses to infection. TLR2 is important to host defense and mediates clearance of S. epidermidis bacteremia in vivo in adult mice [18]. Indeed, in our current study TLR2−/− neonatal mice demonstrated delayed clearance of S. epidermidis from the bloodstream, spleen, and liver. In addition to its important roles in host defense in clearing bacteria, TLR2 may mediate some forms of inflammatory brain injury [42], including middle cerebral artery occlusion [43] and neonatal hypoxia-ischemia–induced brain injury [44]. Indeed, consistent with the known role of TLR2 in mediating neuroinflammation and neuronal damage [45], we have previously demonstrated that a single intraperitoneal injection of the TLR2 agonist Pam3CSK4 revealed a robust increase in the WBC count in the CSF in mice on PND8 [38], and repeated administration of Pam3CSK4 induced brain injury in newborn animals [19]. S. epidermidis bacteremia induced TLR2 expression in the brain in our current study (Figure 5). However, the effects of ablating TLR2 on inflammatory events after S. epidermidis injection are complex because the consequently greater bacterial load might enhance inflammation via TLR-independent pathways. Consistent with this view, we noted greater S. epidermidis–induced systemic cytokine and chemokine production in TLR2−/− mice, compared with WT mice, likely reflecting the greater exposure over time to gram-positive bacteria in the absence of TLR2-mediated bacterial clearance.
Despite evidence of the relevance of TLR2 to host defense and inflammatory responses to S. epidermidis, we also demonstrate that S. epidermidis can activate systemic inflammation and induce brain injury independently of TLR2. In the presence of high bacterial concentrations and/or prolonged bacteremia, TLR2-independent pathways can contribute to systemic inflammation and brain injury. Our prior studies have indicated that S. epidermidis–triggered inflammatory responses are concentration dependent and, at high concentrations of bacteria and/or later time points, can proceed via TLR2-independent pathways both in vitro (cultured macrophages) and in vivo (cytokinemia) [18]. Accordingly, engagement of additional TLR2-independent pattern-recognition receptor pathways, such as NOD-like receptors, β-integrins, C-type lectins, inflammasomes, or other innate pathways [46–49], may contribute to S. epidermidis bacteremia–induced brain injury, especially at high concentrations of bacteria, and should be characterized in future studies. Consistent with these observations, Streptococcus pneumoniae induced neuronal injury within hours of bacteremia in adult mice, preceding detectable CNS penetration that was only partially mitigated in TLR2-deficient animals [9].
As outlined in a working model depicted in Figure 7, our study is the first to demonstrate that S. epidermidis bacteremia in the absence of entry of live S. epidermidis to the CSF has a detrimental impact on the development of both white and gray matter in neonatal mice. However, several steps in this process remain to be further characterized in future studies, including the cellular source of inflammatory mediators in the brain, such as CCL2, and whether dead bacteria or bacterial products, such as the TLR2 agonist bacterial lipopeptides, spill into the CNS. Notably, it has been previously shown that S. pneumoniae bacterial cell wall debris can trigger preterm brain injury [9].
Figure 7.

Schematic diagram of possible steps leading to brain injury after initiation of Staphylococcus epidermidis bacteremia in mice on postnatal day 1 (PND1). The current study, interpreted in the context of published literature, suggests that S. epidermidis bacteremia at PND1 can trigger inflammation in both the bloodstream and central nervous system (CNS) that culminates in brain injury without entry of bacteria into the cerebrospinal fluid (CSF). Injection of S. epidermidis results in early (within 6 hours) generation of systemic cytokines in the bloodstream via both Toll-like receptor 2 (TLR2; yellow dimers) and other pattern-recognition receptors (PRRs; generically depicted as black/square receptors; a), associated entry into the CNS of white blood cells (WBCs; b), potential entry of dead bacteria and/or bacterial products such as bacterial lipoproteins (BLPs) into the CNS (this is under question, given the lack of direct evidence in the literature; c), potential entry of endothelium-derived cytokines (d) and/or systemic cytokines (e) into the CNS, and potential local (brain) generation of inflammatory mediators (eg, CCL2 detected in blood and brain and prostaglandins synthesized by the enzyme PTGS2, which was upregulated in the brain at the RNA level; f), culminating in early (within 6 hours) TLR2-dependent caspase-3 activation and late (PND14) brain injury (g), which can occur in a TLR2-independent manner that manifests as reduced gray and white matter volume.
Although early bacterial clearance and caspase-3 activation were TLR2 dependent, the late PND14 brain injury occurred in WT and TLR2−/− mice. In interpreting these results, it is important to bear in mind that TLR2−/− mice experience prolonged bacteremia and a higher overall exposure to S. epidermidis that might signal via TLR2-independent pathways [18]. Thus, it may be that low concentrations of S. epidermidis induce TLR2-dependent, including caspase-3–dependent, brain injury, whereas higher numbers of S. epidermidis (like those noted in the absence of TLR2) may engage additional TLR2-independent inflammatory pathways to induce brain injury. Indeed, there is precedent for different bacterial inocula inducing distinct beneficial or harmful TLR-mediated effects [50]. Future work will focus on dissection of the mechanisms by which the peripheral inflammatory signals are transmitted to the CNS and result in brain injury, including probing the potential role of CCL2, prostaglandins, and/or other inflammatory mediators.
In summary, our study provides the first direct evidence that the consequences of transient bacteremia due to S. epidermidis, and possibly other bacterial pathogens [9], in early life may be substantially more severe than commonly appreciated and may include both TLR2-dependent and TLR2-independent brain inflammation and injury. These observations may indicate novel approaches to reduce bacteremia-associated brain injury in early life, including development of targeted antiinfective/antiinflammatory agents designed to accompany conventional antibiotics, thereby potentially enhancing bacterial clearance while reducing brain injury.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgment. We thank Ms Kristin Johnson, medical graphics specialist at Boston Children's Hospital, for expert assistance with creating Figure 7.
Financial support. This work was supported by Swedish Medical Society (SLS-328431, to X. W.), Signihild Engkvists Stiftelse (to X. W.), the Wilhelm and Martina Lundgren Foundation (vet2-83/2011 to X. W. and vet2-32/3013 and vet2-21/2014 to C. J. E.), the Swedish Medical Research Council (2012–2992 to C. M., VR 522-2008-2286 and VR 2013-2475 to X. W., and VR 2012-3500 to H. H.), the Public Health Service at the Sahlgrenska University Hospital (ALFGBG-432291 to C. M., ALFGBG-429801 to X. W., and ALFGBG-2863 to H. H.), the VINNMER programme (VINNOVA 2011-03458 to X. W.), Grand Challenge Explorations and Global Health (OPP1036135, via the Bill and Melinda Gates Foundation, to X. W.), the Leducq Foundation (DSRR_P34404 to C. M.), the Åhlens Foundation (to C. M.), the Swedish Brain Foundation (FO2014-0080 to C. M.), the Bill and Melinda Gates Foundation (OPPGH5284 and OPP1035192 to O. L.), the National Institutes of Health National Institute of Allergy and Infectious Diseases (1R01AI100135‐01 and RO1 supplement PA-14-027 to O. L.), MedImmune (to O. L.), Crucell (to O. L.), the Åke Wiberg Foundation (726300470 to C. J. E.), and the Magnus Bergvall Foundation (to C. J. E.).
Potential conflicts of interest. O. L.'s laboratory is supported in part by sponsored research support from Crucell and MedImmune, companies that develop vaccines, as well as by Shire, a company that develops antiinfective biologics. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Boksa P. Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav Immun 2010; 24:881–97. [DOI] [PubMed] [Google Scholar]
- 2.Strunk T, Inder T, Wang X, Burgner D, Mallard C, Levy O. Infection-induced inflammation and cerebral injury in preterm infants. Lancet Infect Dis 2014; 14:751–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dammann O, Leviton A. Inflammatory brain damage in preterm newborns--dry numbers, wet lab, and causal inferences. Early Hum Dev 2004; 79:1–15. [DOI] [PubMed] [Google Scholar]
- 4.Babcock MA, Kostova FV, Ferriero DM et al. Injury to the preterm brain and cerebral palsy: clinical aspects, molecular mechanisms, unanswered questions, and future research directions. J Child Neurol 2009; 24:1064–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas W, Speer CP. Chorioamnionitis: important risk factor or innocent bystander for neonatal outcome? Neonatology 2011; 99:177–87. [DOI] [PubMed] [Google Scholar]
- 6.Mitha A, Foix-L'Helias L, Arnaud C et al. Neonatal infection and 5-year neurodevelopmental outcome of very preterm infants. Pediatrics 2013; 132:e372–80. [DOI] [PubMed] [Google Scholar]
- 7.Mallard C, Welin AK, Peebles D, Hagberg H, Kjellmer I. White matter injury following systemic endotoxemia or asphyxia in the fetal sheep. Neurochem Res 2003; 28:215–23. [DOI] [PubMed] [Google Scholar]
- 8.Mallard C, Wang X. Infection-induced vulnerability of perinatal brain injury. Neurol Res Int 2012; 2012:102153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orihuela CJ, Fillon S, Smith-Sielicki SH et al. Cell wall-mediated neuronal damage in early sepsis. Infect Immun 2006; 74:3783–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Mallard C, Levy O. Potential Role of Coagulase-negative Staphylococcus Infection in Preterm Brain Injury. Adv Neuroimmune Biol 2012; 3:7. [Google Scholar]
- 11.Hartel C, Osthues I, Rupp J et al. Characterisation of the host inflammatory response to Staphylococcus epidermidis in neonatal whole blood. Arch Dis Child Fetal Neonatal Ed 2008; 93:F140–5. [DOI] [PubMed] [Google Scholar]
- 12.Stoll BJ, Hansen N, Fanaroff AA et al. Late-onset sepsis in very low birth weight neonates: the experience of the NICHD Neonatal Research Network. Pediatrics 2002; 110:285–91. [DOI] [PubMed] [Google Scholar]
- 13.Power Coombs MR, Kronforst K, Levy O. Neonatal host defense against Staphylococcal infections. Clin Dev Immunol 2013; 2013:826303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vergnano S, Menson E, Kennea N et al. Neonatal infections in England: the NeonIN surveillance network. Arch Dis Child Fetal Neonatal Ed 2011; 96:F9–14. [DOI] [PubMed] [Google Scholar]
- 15.Payne NR, Carpenter JH, Badger GJ, Horbar JD, Rogowski J. Marginal increase in cost and excess length of stay associated with nosocomial bloodstream infections in surviving very low birth weight infants. Pediatrics 2004; 114:348–55. [DOI] [PubMed] [Google Scholar]
- 16.Cheung GY, Otto M. Understanding the significance of Staphylococcus epidermidis bacteremia in babies and children. Curr Opin Infect Dis 2010; 23:208–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zahringer U, Lindner B, Inamura S, Heine H, Alexander C. TLR2 - promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 2008; 213:205–24. [DOI] [PubMed] [Google Scholar]
- 18.Strunk T, Power Coombs MR, Currie AJ et al. TLR2 mediates recognition of live Staphylococcus epidermidis and clearance of bacteremia. PLoS One 2010; 5:e10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du X, Fleiss B, Li H et al. Systemic stimulation of TLR2 impairs neonatal mouse brain development. PLoS One 2011; 6:e19583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kronforst KD, Mancuso CJ, Pettengill M et al. A neonatal model of intravenous Staphylococcus epidermidis infection in mice <24 h old enables characterization of early innate immune responses. PLoS One 2012; 7:e43897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mack D, Fischer W, Krokotsch A et al. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis. J Bacteriol 1996; 178:175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Craig A, Ling Luo N, Beardsley DJ et al. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp Neurol 2003; 181:231–40. [DOI] [PubMed] [Google Scholar]
- 23.Ek CJ, Habgood MD, Dziegielewska KM, Potter A, Saunders NR. Permeability and route of entry for lipid-insoluble molecules across brain barriers in developing Monodelphis domestica. J Physiol 2001; 536:841–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Hagberg H, Zhu C, Jacobsson B, Mallard C. Effects of intrauterine inflammation on the developing mouse brain. Brain Res 2007; 1144:180–5. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Carlsson Y, Basso E et al. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J Neurosci 2009; 29:2588–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Karlsson JO, Zhu C, Bahr BA, Hagberg H, Blomgren K. Caspase-3 activation after neonatal rat cerebral hypoxia-ischemia. Biol Neonate 2001; 79:172–9. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Stridh L, Li W et al. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J Immunol 2009; 183:7471–7. [DOI] [PubMed] [Google Scholar]
- 28.Isaacs D; Australasian Study Group For Neonatal I. A ten year, multicentre study of coagulase negative staphylococcal infections in Australasian neonatal units. Arch Dis Child Fetal Neonatal Ed 2003; 88:F89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vincze A, Mazlo M, Seress L, Komoly S, Abraham H. A correlative light and electron microscopic study of postnatal myelination in the murine corpus callosum. Int J Dev Neurosci 2008; 26:575–84. [DOI] [PubMed] [Google Scholar]
- 30.Mittendorf R, Roizen N, Moawad A, Khoshnood B, Lee KS. Association between cerebral palsy and coagulase-negative staphylococci. Lancet 1999; 354:1875–6. [DOI] [PubMed] [Google Scholar]
- 31.Chau V, Brant R, Poskitt KJ, Tam EW, Synnes A, Miller SP. Postnatal infection is associated with widespread abnormalities of brain development in premature newborns. Pediatr Res 2012; 71:274–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Semple BD, Kossmann T, Morganti-Kossmann MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab 2010; 30:459–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ivacko J, Szaflarski J, Malinak C, Flory C, Warren JS, Silverstein FS. Hypoxic-ischemic injury induces monocyte chemoattractant protein-1 expression in neonatal rat brain. J Cereb Blood Flow Metab 1997; 17:759–70. [DOI] [PubMed] [Google Scholar]
- 34.Jenkins DD, Rollins LG, Perkel JK et al. Serum cytokines in a clinical trial of hypothermia for neonatal hypoxic-ischemic encephalopathy. J Cereb Blood Flow Metab 2012; 32:1888–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitchell K, Yang HY, Berk JD, Tran JH, Iadarola MJ. Monocyte chemoattractant protein-1 in the choroid plexus: a potential link between vascular pro-inflammatory mediators and the CNS during peripheral tissue inflammation. Neuroscience 2009; 158:885–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szmydynger-Chodobska J, Strazielle N, Gandy JR et al. Posttraumatic invasion of monocytes across the blood-cerebrospinal fluid barrier. J Cereb Blood Flow Metab 2012; 32:93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galasso JM, Liu Y, Szaflarski J, Warren JS, Silverstein FS. Monocyte chemoattractant protein-1 is a mediator of acute excitotoxic injury in neonatal rat brain. Neuroscience 2000; 101:737–44. [DOI] [PubMed] [Google Scholar]
- 38.Stridh L, Ek CJ, Wang X, Nilsson H, Mallard C. Regulation of Toll-like receptors in the choroid plexus in the immature brain after systemic inflammatory stimuli. Transl Stroke Res 2013; 4:220–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35:495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burguillos MA, Deierborg T, Kavanagh E et al. Caspase signalling controls microglia activation and neurotoxicity. Nature 2011; 472:319–24. [DOI] [PubMed] [Google Scholar]
- 41.Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E et al. Preconditioning of microglia by alpha-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS One 2013; 8:e79160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lehnardt S, Henneke P, Lien E et al. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol 2006; 177:583–92. [DOI] [PubMed] [Google Scholar]
- 43.Lehnardt S, Lehmann S, Kaul D et al. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol 2007; 190:28–33. [DOI] [PubMed] [Google Scholar]
- 44.Stridh L, Smith PL, Naylor AS, Wang X, Mallard C. Regulation of toll-like receptor 1 and -2 in neonatal mice brains after hypoxia-ischemia. J Neuroinflammation 2011; 8:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffmann O, Braun JS, Becker D et al. TLR2 mediates neuroinflammation and neuronal damage. J Immunol 2007; 178:6476–81. [DOI] [PubMed] [Google Scholar]
- 46.Cuzzola M, Mancuso G, Beninati C et al. Beta 2 integrins are involved in cytokine responses to whole Gram-positive bacteria. J Immunol 2000; 164:5871–6. [DOI] [PubMed] [Google Scholar]
- 47.Albanyan EA, Edwards MS. Lectin site interaction with capsular polysaccharide mediates nonimmune phagocytosis of type III group B streptococci. Infect Immun 2000; 68:5794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shaw MH, Reimer T, Kim YG, Nunez G. NOD-like receptors (NLRs): bona fide intracellular microbial sensors. Curr Opin Immunol 2008; 20:377–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strunk T, Richmond P, Prosser A et al. Method of bacterial killing differentially affects the human innate immune response to Staphylococcus epidermidis. Innate Immun 2011; 17:508–16. [DOI] [PubMed] [Google Scholar]
- 50.Mancuso G, Midiri A, Beninati C et al. Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J Immunol 2004; 172:6324–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.