

Abstract
Background
Current management of acute inhalational carbon monoxide (CO) toxicity includes hyperbaric or normobaric O2 therapy. However, efficacy has not been established. The purpose of this study was to establish therapeutic proof of concept for a novel injectable antidote consisting of the combination of hydroxocobalamin and ascorbic acid into a reduced form (B12r) as demonstrated by clinically-significant increase (> 500 ppm) in CO2 production, reduced carboxyhemoglobin (COHgb) half-life (COHgb t1/2), and increased cerebral O2 delivery and attenuation of CO-induced microglial damage in a preclinical rodent model of CO toxicity.
Methods
B12R-mediated conversion of CO to CO2 and COHgb t1/2 in human blood were measured by gas analysis and Raman resonance spectroscopy. Rats were exposed to either air or CO, then injected with saline or B12r. Cognitive assessment was tested in a Morris water maze. Brain oxygenation was measured with Licox. Brain histology was assessed by fluorescent antibody markers and cell counts.
Results
B12r resulted in significant CO2 production (1170 ppm), compared to controls. COHgb t1/2 was reduced from 33 min (NS) to 17.5 (p < 0.001). In rat models, severe CO-induced brain hypoxia (PbtO2 18 mmHg) was followed by significant reduction in τ25 to 12 min for B12r rats vs 40 min for NS-treated rats (p < 0.0001). There was major attenuation of CO-induced microglial damage, although cognitive performance differences were minimal.
Conclusion
Our preclinical data suggest that the novel synergism of hydroxocobalamin with ascorbic acid has the potential to extract CO through conversion to CO2, independently of high-flow or high-pressure O2. This resulted in a clinically-significant off-gassing of CO2 at levels 5 to 8 times greater than controls, a clinically-significant reduction in COHgb half-life, and evidence of increased brain oxygenation and amelioration of myoglial damage in rat models. Reduced hydroxocobalamin has major potential as an injectable antidote for CO toxicity
Keywords: antidote, hydroxocobalamin, ascorbic acid, carbon monoxide toxicity
Carbon monoxide (CO) exposure is the leading cause of unintentional poisoning death and long-term morbidity in the US. In 2012 alone, there were over 13,000 cases reported to US poison centers, with 143 serious outcomes and 54 deaths1. However serious delayed sequelae such as persistent and recurrent neurological deficits may occur in up to 30% of patients following apparent recovery from acute symptoms; these delayed neurological complications (DNS) may not become evident until weeks or months after exposure2. Reduction in O2 delivery to the tissues is thought to result from binding of CO to hemoglobin (Hgb) to form carboxyhemoglobin (COHgb). This results in conformational changes in Hgb that reduces its ability to offload O2 to the tissues and subsequently causes decreased O2 utilization, mitochondrial dysfunction, and hypoxic injury. As a result, current acute-care management involves either normobaric (NBO) or hyperbaric (HBO) oxygen therapy; however, efficacy for prevention of DNS is uncertain and has not been evaluated systematically3. Further limitations include unpredictable availability of equipment and delays between point of exposure, recognition of signs and symptoms, and initiation of therapy4. No antidotes for CO toxicity currently exist.
To be clinically useful, proposed antidotes for CO toxicity must be readily available, field-deployable, targeted, rapidly effective, and safe. We propose a solution of hydroxocobalamin (OHCbl), an FDA-approved antidote for cyanide poisoning, and ascorbic acid (vitamin C), a safe and powerful reducing agent. Other chemically-reduced forms of OHCbl (B12r) have been demonstrated to convert CO to CO2 in simple solutions in vitro5. We hypothesized B12r could also facilitate conversion of CO to CO2 in blood, resulting in the rapid reduction of the total body CO load via respiratory off-gassing of CO2. The speed of this reaction should avert or reduce CO-induced DNS by early prevention of inflammatory changes associated with elevated intracellular CO levels. Antidote effectiveness can therefore be defined as both the demonstration of either irreversible binding or conversion of CO, and clinically-significant reduction of delayed neurological and cognitive deficits. We performed a two-part test of this hypothesis to establish therapeutic proof of concept: (1) in vitro CO removal from blood as demonstrated by a clinically-significant increase (> 500 ppm) in CO2 production and reduction of carboxyhemoglobin (COHgb) half-life (t1/2), and (2) in vivo demonstration in a preclinical rodent model of cognitive function, increased cerebral oxygen delivery, and attenuation of CO-induced microglial damage.
Methods
Reduced OHCbl (B12r) was produced by combining 300 mg analytical grade hydroxocobalamin (OHCbl) and 300 mg ascorbic acid (AA) (Sigma-Aldrich, St. Louis, MO) in 5 mL deoxygenated 0.9% NaCl solution (NSdeox) in 100% N2 environment to prevent auto-oxidation6.
In vitro experiments: B12r–mediated CO reduction in blood
IRB-exempt waste human venous blood was obtained from VCU Apheresis clinic; 600 mL was collected into standard blood collection bags and anti-coagulated with 70 mL of CPD-A1. Blood was used within 24-48 hours following collection. Blood was circulated through a closed-loop hollow-fiber membrane oxygenator (Pediatric Quadrox-iD®, Maquet, Hirrlingen, Germany) and roller pump (Stöckert/Shiley®, Soma Technology Inc, Bloomfield CT) at 250 mL/min, and maintained at 37°C with a countercurrent water-flow heat exchanger (DC 10, Thermo Haake, Fisher Scientific). The system was equilibrated with medical air (20-22% v/v O2; <400 ppm CO2; 78-80% v/v N2) then ‘poisoned’ with 6000 ppm CO in research grade air (0.5838% v/v CO, balance air) for 20 minutes; all air flow rates were 178 mL/min. The system was then injected with 5 mL of either B12R or one of three negative controls: NSdeox, AA (350 mg in 5 mL NSdeox), or B12A (350 mg OHCbl in 5 mL NSdeox). CO2 concentration (volume %) was sampled at 10 Hz (BIOPAC Inc., Galeta CA) over 30 minutes from the time of B12r injection or when carboxyhemoglobin (COHgb) concentration reached 50%; in-flow gas was then switched back to to medical grade air. Gas-out concentration of CO2 was continuously measured at 10 Hz for 30 min post-infusion. Signals were amplified (CO2100C interface; BIOPAC Systems, Goleta, CA), and analog-digital conversions were performed online (AcqKnowledge™ v. 4 software; BIOPAC Systems, Goleta, CA). Median CO2 concentration for each solution was calculated from the normalized area under the curve (AUC) of the CO2-time response curves6.
Confirmation that CO2 was derived from B12R-mediated conversion of CO (and not an unidentified exogenous source) was obtained by radiocarbon tracing in separate trials. Twenty-mL of 13C labeled CO (Cambridge Isotope Laboratories, Tewksbury, MA) was injected into the system 30 min prior to infusion with 5 mL B12R. Gas samples were taken at baseline and 20 min post-infusion; the difference in the 13CO2/12CO2 ratio between baseline and post-B12r infusion was quantified by infrared spectral analysis (POCone®, Otsuka Electronics Co., Japan)6.
Carboxyhemoglobin (COHgb) concentrations were determined by Resonance Raman (RR) spectroscopy. CO-poisoned blood with a COHgb concentration of 50% was treated with either high-flow atmospheric pressure O2 alone, or with a combination of high-flow O2 and B12r solution. Blood samples were obtained at baseline and every 10 min for 120 min. RR spectra of COHgb were obtained for 20 µL subsamples sealed into melting-point capillary tubes; excitation lines were obtained from a 406.7 nm krypton-ion laser excitation source (Coherent Saber) and attenuated output power of 0.07-0.08 mW, collected using a 600-mm single-grating monochromator, and imaged using a back-illuminated CCD camera (Python CCD, Princeton Instruments, Trenton, NJ). Scans were completed in 3-5 min7. COHgb half-life t1/2 was calculated as (ln 2)/λ, where λ is the rate constant for the decay function Pt=P0 · exp(-λ·t); Pt is peak height at time t, and P0 is initial peak height. Calculations were performed in PROC NLIN (SAS 9.4).
In Vivo Studies
Ethics statement and animals
This study was approved in advance by the Institutional Animal Care and Use Committee (IACUC) of Virginia Commonwealth University (IACUC Protocol # AD10000569), and conforms to the Public Health Service Policy on Humane Care and Use of Laboratory Animals (2002). All rats were obtained from Harlan Laboratories (Indianapolis, IN) at 5-8 weeks of age. Before experimentation, animals were housed in pairs in ventilated cages and maintained at 25°C and 12L:12D, with ad lib access to food (commercial rat chow) and water. Animals were weighed daily for a minimum of 5 days prior to surgical procedures.
Brain oxygen tension
The experimental design was a 2 × 2 factorial on the factors exposure (medical air SHAM or CO) and intervention (NS or B12r). Thirty male adult Sprague-Dawley rats (mass 315-370 g) were randomly allocated to one of four groups (SHAM-NS, SHAM-B12r, CO-NS, CO-B12r) with EXCEL random numbers algorithm; 10 animals were allocated to each CO group, and 5 to each sham group.
All surgical procedures were aseptic. Animals were anesthetized with isoflurane (4% for induction, 2% for maintenance, balance medical air). Core temperature was monitored with a rectal probe and maintained at 36-38°C for the duration of each trial with a thermostatically-controlled feedback heating blanket (Harvard Apparatus, Holliston, MA). The head was stabilized in a stereotaxic frame, the skull exposed along the midline, and two 2–mm burr holes were drilled 1-mm posterior to bregma and 3-mm lateral to midline. The duramater was gently incised to allow placement of calibrated Licox® brain oxygenation (CC.1.R.) and temperature (CB.8) probes (Integra Neuroscience, Plainsboro, NJ); probes were inserted 2.3 mm into the parenchyma. Animals were allowed to stabilize for 30 min. Animals were then exposed for 30 minutes to either 2500 ppm CO or medical air, followed by a single dose of intervention solution, either B12r at 100 mg/kg or the weight-based equivalent volume of NS (2 mL/kg, IP); total volumes were 0.6-0.85 mL. Brain oxygen tension PbtO2 was recorded every 5 min from initial exposure to 60 min post-infusion. Animals were then euthanized with sodium pentobarbital (Euthasol®, 40 mg/kg IV, Virbac Animal Health, Fort Worth, TX).
Post-infusion change of PbtO2 over time t for both CO-exposure groups was described by the parametric nonlinear mixed-effects model PbtO2 = a ·[1 − b· exp(-k · t)] + Z(t), where a is the maximum value of PbtO2, b is a scaling parameter, k is the rate constant, and Z(t) describes the random effects component for each animal. Differences between control and antidote were evaluated by contrasts on each parameter estimate9. Primary outcome was time to achieve PbtO2 of 25 mmHg (τ25); this is the approximate hypoxic brain tissue threshold established for humans10, and is approximately 75% of baseline PbtO2 levels (33-35 mmHg) for rats. Calculations were performed in PROC NLMIXED (SAS 9.4).
Spatial learning
Forty-two male young adult Long-Evan rats (average initial weight 219 g; average terminal weight 300 g) were randomly assigned (RANDOM.ORG) to one of three treatment groups: medical air only (SHAM, n = 12), CO exposure with NS infusion (CO-NS, n = 19) or CO-exposure with B12r antidote infusion (CO-B12r, n = 21). CO-exposed animals received 2500 ppm CO (0.25% CO, 27% O2, balance N2) for 60 min, followed by 6000 ppm CO (0.6% CO, 27% O2, balance N2) for 10 min or until loss of righting response; animals were then removed from the exposure chamber and immediately administered B12r antidote or NS (2 mL/kg IP, 0.6-0.8 mL). Animals were allowed to recover in temperature-controlled recovery cages until they regained normal response to stimuli, and then returned to their primary housing cage.
Twenty-four hours following experimental exposures described above, animals began four-stage Morris Water Maze (MWM) testing. Deficits in MWM performance are associated with damage to specific regions of the brain involved with spatial navigation and learning, such as the hippocampus11,12. Tests were conducted in a standard water maze pool (diameter 183 cm; depth 63.5 cm) with a submerged platform 2.5 cm below the water surface; non-toxic white paint was added as a water opacifier. The four test stages occurred on post-injury days 1, 3, 6, and 8. Each stage consisted of blocks of four swimming trials of 60 sec each, starting from one of four randomly-chosen compass positions, with a ten-minute inter-trial rest interval. Platform location was constant between trials, but moved to a new location for each stage. Testing was performed by a technician blinded to group assignment, and not involved with injury protocol or analyses. Animal movements were tracked and quantified with a ceiling-mounted video camera and computer-assisted tracking software (Med Associates Inc., St Albans, VT). Spatial learning was quantified by path efficiency (PE, %) estimated as the straight-line distance from start to platform divided by observed total swim path length7. Differences between treatments for median PE were assessed by nonlinear mixed-model analysis (PROC NLMIXED, SAS 9.413).
Immunohistochemistry
Nine male Long-Evans rats (300-350 g), three from each of the above groups were killed on post-injury-day 10 with Euthasol (150 mg/kg IP), and fixed with 4% formalin via transcardial perfusion. Neuronal tissue was preserved in 4% formalin for 24 hours, then sectioned into 40 µm sections from bregma +3 to -7 on a Leica VT1000® vibratome. Sections were simultaneously blocked and incubated with primary antibodies at 4°C for 24 h utilizing a double-staining protocol for ionized calcium-binding adapter molecule-1 (Iba-1) and glial fibrillary acidic protein (GFAP). Tissue sections were then rinsed and incubated at room temperature for 60 min with secondary antibodies (DyLight-488 and DyLight-549) specific to the IBA-1 and GFAP antibodies. Control sections using only single antibodies and blocking solutions were prepared to examine non-specific binding. After curing for 24 hours sections were examined with a Zeiss LSM 710 laser scanning confocal microscope. Cell counts were obtained using an automated cell counting routine in NIH ImageJ (www.imagej.nih.gov). Microglial activation state was determined by morphometric analysis based on cell body size, process length, and thickness14. Astrocyte status was determined by manual morphometric analysis.
Results
In vitro studies
Median CO2 concentration for B12r averaged 1170 ppm, compared to < 200 ppm for controls. This represents a five- to eight-fold increase in the gas-out concentration of CO2 (Fig. 1). We detected a 16.7% increase in the 13CO2/12CO2 ratio over baseline with B12r infusion whereas infusing a standard sample with 13CO caused no interference in the analysis of 13CO2. COHgb half-life t1/2 was 33 (95% CI 27, 42) min under O2 alone, but was reduced to 18 (95% CI 15, 21) min with B12r infusion, a difference of 15 min (p < 0.001).
Fig. 1.

Comparative CO2 production (ppm/min) over 30 min induced by B12r antidote (hydroxocobalamin B12 +ascorbic acid) added to whole human blood containing 50% COHgb. The shaded area is the difference between CO2 produced from ascorbic acid alone, and that produced by B12r. Little or no CO2 was produced by infusion of normal saline (NS) or oxidized hydroxocobalamin.
In vivo studies
PbtO2 of both sham-exposure groups of rats averaged 31 (95% CI 28, 35) mmHg throughout the procedure, with no change upon administration of antidote or NS control. Substantial brain hypoxia was induced with CO-exposure; PbtO2 averaged 18 mmHg (95% CI 17, 19 mmHg) at 30 min. After administration of either NS or B12r there was a nonlinear asymptotic increase to baseline PbtO2 by the end of the monitoring period (Fig. 2); maximum PbtO2 (33 mmHg) did not differ between CO-exposure groups (p = 0.61) or from sham groups. However τ25 differed significantly between CO-exposed groups (p <0.0001); τ25 averaged 40 (95% CI 36, 45) min for CO-NS controls, compared to only 12 (95% CI 10, 13) min for CO-B12r animals.
Fig. 2.

Brain oxygen tension (PbtO2, mmHg) measured in 30 Sprague-Dawley rats exposed to medical air (AIR) or CO, and injected with either saline (NS) or antidote (B12r). Solid lines are fitted equations; dotted lines show estimated threshold τ25 for each CO-exposure treatment.
Median path efficiencies obtained from Morris water maze testing are shown in Fig. 3. There were no statistical differences between treatments at any time point (p > 0.2) although weak differences in learning trajectories were suggested by examination of medians. Rats exposed to medical air only showed the expected daily increase in path efficiency; efficiencies increased by an average of 8-10% per day over eight days of testing. In contrast, both CO-exposed groups showed a plateau in performance with either no change (CO-B12r) or a modest decline (CO-NS 7%).
Fig. 3.

Median path efficiencies (straight line distance/observed swim path length) for rats exposed to medical air (SHAM) or CO and injected with either NS (CO-NS) or antidote (COB12) and tested in a Morris Water maze over 8 days.
Preliminary immunochemistry data suggest loss in overall cell count (Fig. 4.A), increased demyelination (Fig. 4.B) and reduction of microglial activity (Fig. 4.C) associated with CO- exposure; however B12r treatment appeared to partially reverse CO-induced damage. Myelinated axons were abundant in cortical layers 2-5 in control animals compared to CO-exposed animals. However whereas rats exposed to CO showed obvious deficit; CO- B12r rats showed partial preservation of myelinated axons (Fig. 4.B). CO-exposed rats also exhibited microglia with morphology consistent with activation; cells had enlarged cell bodies and thicker, less-branched processes, compared to microglia of either control or CO-B12r treated rats (fig. 4.C).
Fig. 4.
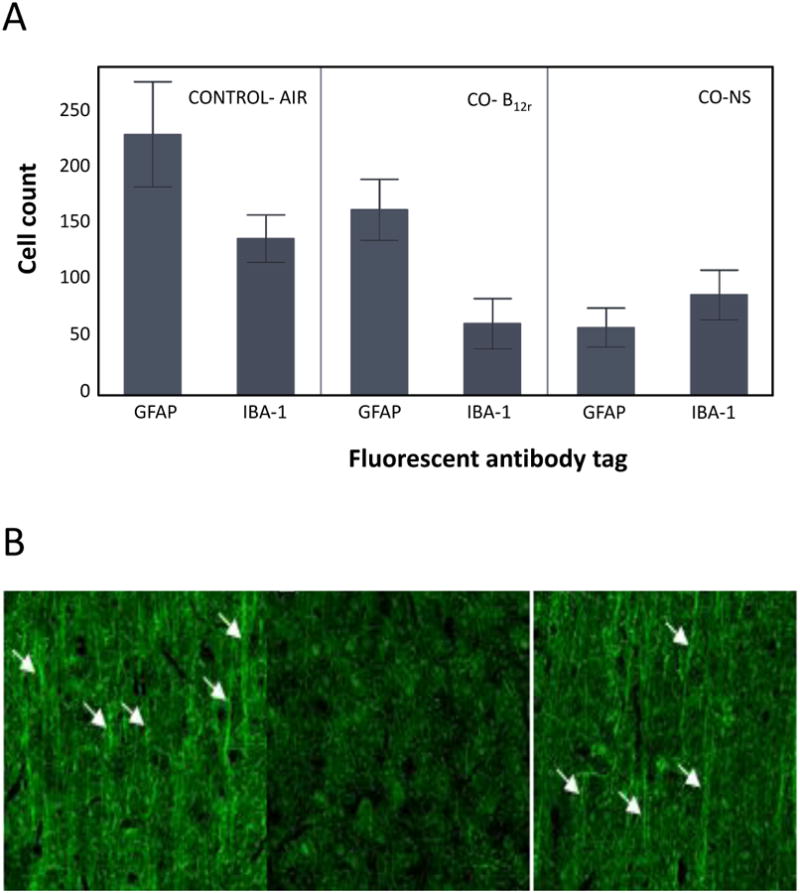
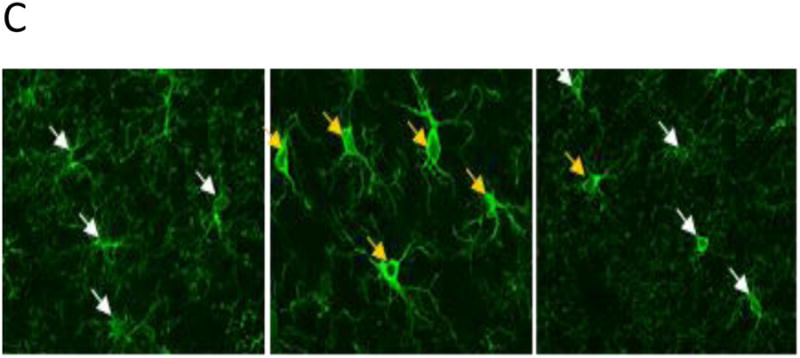
Immunochemistry of brain tissue of rats exposed to medical air (CONTROL) or CO with either antidote (B12r) or NS control solution. A. Mean (SD) microglial and astrocyte cell counts of GFAP and Iba-1 tagged cells; B. Relative myelination: Myelinated axons are indicated by white arrows. Note the paucity of myelin in the CO poisoned rats (center panel); C. Microglial activation. Yellow arrows highlight activated microglia based on increased Iba-1 staining density, thickened processes, and loss of arborization.
Conclusions
Victims of inhalational CO toxicity show a reduction of COHgb half-life from 5 hours on room air to 30-60 min with NBO and only 5 min with HBO. However neither of these interventions has been conclusively demonstrated to reduce the incidence of DNS, and may be difficult to deploy in a timely way. Our preclinical data suggest that the reduced form of hydroxocobalamin, with ascorbic acid as the reducing agent, results in a clinically-significant off-gassing of CO2 at levels 5 to 8 times greater than controls, a clinically-significant reduction in COHgb half-life, and evidence of increased brain oxygenation, and amelioration of myoglial damage in rat models. These data suggest a novel synergism of these two compounds when combined at high dose, with the potential to extract CO through conversion to CO2, independently of high-flow or high-pressure O2. Furthermore, these findings have significant clinical implications, as both hydroxocobalamin and ascorbic acid are safe and approved for use in humans, even at high doses15,16. Reduced hydroxocobalamin has major potential as an injectable antidote for CO toxicity.
Acknowledgments
Financial Support: Microscopy was performed at the VCU department of Anatomy and Neurobiology Microscopy Facility, supported in part, with funding from NIH-NINDS Center Core grant (5P30NS047463). We thank the VCU Apheresis Clinic staff, the perfusion team (especially H. McCarthy and B. Chui), K. Luu, G. DeCosta, L. Thompson, and especially J. Terner (Chemistry) for their generous assistance. This work was supported by internal funds from the departments of Surgery and Anesthesiology (VCU Health System) and by generous donations of supplies and equipment from the departments of Surgery, Gastroenterology, and Perfusion (VCU Health System). L. G. S. is an active-duty member of the US Air Force on civilian institute status from the AF Institute of Technology. J. D. R. is a reserve member of the US Navy on HPSP scholarship.
Footnotes
Authorship: J.D.R. had the original idea, and conceived and designed experiments. J. D. R, L. G. S, C. S. J., and A. H. N. performed experiments and collected data. P.S.R. assisted with experimental design and performed analyses. J.D.R. and P.S.R. drafted the manuscript. B.D.S. provided critical oversight at all stages, space and funding. All authors contributed to critical review of the manuscript for important intellectual content.
Disclosure: The authors declare no conflicts of interest.
References
- 1.Mowry JB, Spyker DA, Cantilena LR, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers' National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol Phila PA. 2013;51:949–1229. doi: 10.3109/15563650.2013.863906. [DOI] [PubMed] [Google Scholar]
- 2.Bhatia R, Chacko F, Lal V, Mittal BR. Reversible delayed neuropsychiatric syndrome following acute carbon monoxide exposure. Indian J Occup Environ Med. 2007;11:80–82. doi: 10.4103/0019-5278.34534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckley NA, Juurlink DN, Isbister G, Bennett MH, Lavonas EJ. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database of Systematic Reviews. 2011;(4) doi: 10.1002/14651858.CD002041.pub3. Art. No.: CD002041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buckley NA, Juurlink DN. Carbon monoxide treatment guidelines must acknowledge the limitations of the existing evidence. Am J Respir Crit Care Med. 2013;187:390. doi: 10.1164/rccm.201212-2262LE. [DOI] [PubMed] [Google Scholar]
- 5.Schrauzer GN, Lee LP. The reduction of vitamin B12a by carbon monoxide. Arch Biochem Biophy. 1970;138:16–25. doi: 10.1016/0003-9861(70)90278-x. [DOI] [PubMed] [Google Scholar]
- 6.Roderique JT. Unpublished M S thesis (Physiology), VCU Theses and Dissertations. Virginia Commonwealth University; 2013. Studies on the reaction of high-dose hydroxocobalamin and ascorbic acid with carbon monoxide: implications for treatment of carbon monoxide poisoning. [Google Scholar]
- 7.Somera LG. Unpublished M S thesis (Biology), VCU Theses and Dissertations, Paper 3306. Virginia Commonwealth University; 2014. Hydroxocobalamin treatment for carbon monoxide exposures: characterizing hemoglobin changes and testing for neurological sequelae. [Google Scholar]
- 8.Newcomb AH. Unpublished M S thesis (Physiology), VCU Theses and Dissertations, Paper 3560. Virginia Commonwealth University; 2014. Evaluation of the physiological effects of reduced hydroxocobalamin on acute carbon monoxide toxicity. [Google Scholar]
- 9.Brown H, Prescott R. Applied mixed models in medicine. Chichester UK: John Wiley & Sons, Ltd.; 2006. [Google Scholar]
- 10.Nortje J, Gupta AK. The role of tissue oxygen monitoring in patients with acute brain injury. Br J Anaes. 2006;97:95–106. doi: 10.1093/bja/ael137. [DOI] [PubMed] [Google Scholar]
- 11.D'Hooge R, De Deyn PP. Applications of the Morris water maze in the study of learning and memory. Brain Research Reviews. 2001;36:60–90. doi: 10.1016/s0165-0173(01)00067-4. [DOI] [PubMed] [Google Scholar]
- 12.Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang A, Liu N, Kuznetsova OM. Modeling the treatment effect on a median of a percent change from baseline in a lognormal variable using SAS PROC NLMIXED. Paper SP01. Pharma SUG 2009: Pharmaceutical Industry SAS® Users Group Conference; Portland OR. May 31-June 3, 2009. [Google Scholar]
- 14.Hutson CB, Lazo CR, Mortazavi F, Giza CG, Hovda D, Chesselet MF. Traumatic brain injury in adult rats causes progressive nigrostriatal dopaminergic cell loss and enhanced vulnerability to the pesticide paraquat. J Neurotrauma. 2011;28:1783–1801. doi: 10.1089/neu.2010.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mikirova N, Casciari J, Rogers A, Taylor P. Effect of high-dose intravenous vitamin C on inflammation in cancer patients. J Transl Med. 2011;10:189. doi: 10.1186/1479-5876-10-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uhl W, Nolting A, Golor G, Rost KL, Kovar A. Safety of hydroxocobalamin in healthy volunteers in a randomized, placebo-controlled study. Clin Toxicol Phila PA. 2006;44(Suppl 1):17–28. doi: 10.1080/15563650600811755. [DOI] [PubMed] [Google Scholar]