

Abstract
Objective
Skeletal muscle weakness is a prominent clinical feature in patients with rheumatoid arthritis (RA), but the underlying mechanism(s) is unknown. Here we investigate the mechanisms behind arthritis-induced skeletal muscle weakness with special focus on the role of nitrosative stress on intracellular Ca2+ handling and specific force production.
Methods
Nitric oxide synthase (NOS) expression, degree of nitrosative stress and composition of the major intracellular Ca2+ release channel (ryanodine receptor 1, RyR1) complex were measured in muscle. Changes in cytosolic free Ca2+ concentration ([Ca2+]i) and force production were assessed in single-muscle fibres and isolated myofibrils using atomic force cantilevers.
Results
The total neuronal NOS (nNOS) levels were increased in muscles both from collagen-induced arthritis (CIA) mice and patients with RA. The nNOS associated with RyR1 was increased and accompanied by increased [Ca2+]i during contractions of muscles from CIA mice. A marker of peroxynitrite-derived nitrosative stress (3-nitrotyrosine, 3-NT) was increased on the RyR1 complex and on actin of muscles from CIA mice. Despite increased [Ca2+]i, individual CIA muscle fibres were weaker than in healthy controls, that is, force per cross-sectional area was decreased. Furthermore, force and kinetics were impaired in CIA myofibrils, hence actin and myosin showed decreased ability to interact, which could be a result of increased 3-NT content on actin.
Conclusions
Arthritis-induced muscle weakness is linked to nitrosative modifications of the RyR1 protein complex and actin, which are driven by increased nNOS associated with RyR1 and progressively increasing Ca2+ activation.
Keywords: Rheumatoid Arthritis, Arthritis, Cardiovascular Disease
Introduction
In addition to the primary symptoms arising from inflammatory processes in the joints, muscle weakness and impaired work capacity are commonly reported by patients with rheumatoid arthritis (RA). The degree of muscle impairment appears to depend on the severity of the disease, and reductions in muscle strength up to 70% have been reported in patients with severe RA.1 Although decreased muscle strength is traditionally thought to be caused by a loss of muscle mass, impaired intrinsic contractility is also present in patients with RA.2 In line with this, tetanic force per cross-sectional area was markedly reduced in whole muscles from mice with collagen-induced arthritis (CIA),3 a mouse model that displays many pathological characteristics of human RA.4
Skeletal muscle produces low levels of reactive oxygen and nitrogen species (ROS/RNS) that are necessary for normal metabolism, gene regulation and modulation of cellular signalling.5 However, high levels of ROS/RNS have been associated with damage of cellular components and contractile dysfunction.6–9 Many studies suggest that overproduction of nitric oxide (NO) contributes to the pathogenesis of chronic arthritis and it is known that NO synthase (NOS) inhibitors reduce joint inflammation in experimental models of arthritis.10 Mammalian skeletal muscle fibres can express three isoforms of NOS: the neuronal (nNOS), inducible (iNOS) and endothelial (eNOS) isoform. Synthesis of NO from eNOS and nNOS depends on increases in the free cytosolic Ca2+ concentration ([Ca2+]i).11
Ca2+ is required for muscle contraction, and tightly controlled Ca2+ homeostasis is crucial for normal muscle function. The major Ca2+ release channel in skeletal muscle fibres, the ryanodine receptor 1(RyR1) protein complex, is highly sensitive to ROS/RNS and its channel activity is dramatically altered by redox modifications.12 For instance, increases in RNS impair RyR1-mediated Ca2+ homeostasis and contribute to contractile dysfunction in muscle disorders.6–9 Moreover, there are several important interactions between Ca2+ and nNOS: the activity and transcription of nNOS are stimulated by Ca2+; nNOS can be co-localised with RyR and in disease states nNOS may move from the sarcolemma to the sarcoplasmic reticulum (SR).13
Here we investigate the mechanisms behind arthritis-induced skeletal muscle weakness with special focus on the role of nitrosative stress on intracellular Ca2+ handling and myofibrillar force production.
Materials and methods
Ethical approval
The Ethics Committee of the Karolinska Institutet and the Regional Research Ethics Committee of Southern Sweden approved the study. All animal experiments complied with the Swedish Animal Welfare Act, the Swedish Welfare ordinance and applicable regulations and recommendations from Swedish authorities. The study was approved by the Stockholm North Ethical Committee on Animal Experiments.
Patients with RA and healthy controls
The study was performed with seven patients with RA who fulfilled the 1987 American College of Rheumatology criteria14 and a control group of seven age-matched (age range 45–75 years) without inflammatory disease. Four of the patients with RA were recruited from a phase IV clinical trial (Clinicaltrials.gov, NCT01270087) of patients with active disease despite standard treatment with non-biological disease-modifying antirheumatic drugs. The other three patients had early RA (<24 months’ disease duration) and were on stable medication since at least 3 months. Muscle biopsies from patients with RA and healthy controls were obtained at rest from musculus femoris vastus lateralis or musculus tibialis anterior using a semiopen biopsy technique. Muscle tissue samples were flash frozen in liquid nitrogen and stored at −80°C.
Induction and evaluation of CIA
CIA was induced in female DBA/1 mice as described previously.3 Briefly, a preparation of 100 μg of type II collagen (CII) obtained from bovine nasal cartilage and 300 μg of Mycobacterium tuberculosis in 0.1 mL of emulsion was injected subcutaneously into the base of the tail. On day 28, the animals were boosted with a subcutaneous injection of 100 μg of CII in Freund's incomplete adjuvant. Control mice were injected with 0.1 mL saline on both occasions. Mice were housed in standard cages with a 12 h light/dark cycle. They had free access to food and water, which were placed on the floor of the cages. Mice were monitored daily to ensure that a sufficient health and nutritional status was maintained. The development of erythema and swelling of the toe, metatarsophalangeal and ankle joints were followed on a daily basis. Individual paws were scored for inflammation on a scale of 0–3 as follows: 0=normal, 1=one joint affected, 2=two joints affected and 3=whole paw affected. Mice were killed by rapid neck disarticulation and muscles were mainly excised from hind limbs of CIA mice with severe inflammation (score 3) and from saline-treated control mice; in some experiments, we also used muscles from CIA mice with mild inflammation (score 0–1).
Measurement of [Ca2+]i and force in mechanically dissected single fibres
Single intact flexor digitorium brevis (FDB) fibres (fast-twitch, type II) were obtained by dissection and thereafter mounted in a chamber between a force transducer and an adjustable holder. Fibres were superfused with Tyrode solution of the following composition (mM): 121 NaCl, 5 KCl, 0.5 MgCl2, 0.4 Na2HPO4, 1.8 CaCl2, 0.1 EDTA, 24 NaHCO3 and 5.5 glucose, and fetal calf serum (0.2%). The solutions was bubbled with 95% O2–5% CO2 (pH 7.4). The fibre length was adjusted to obtain maximum tetanic force. The diameter of the fibre at this length was used to calculate the cross-sectional area. Experiments were performed at room temperature (∼24°C). The fibre was stimulated with supramaximal electrical pulses (0.5 ms in duration) delivered via platinum electrodes placed along the long axis of the fibre. [Ca2+]i was measured with the fluorescent Ca2+ indicator indo-1. Indo-1 was microinjected into the isolated fibre, which was then allowed to rest for at least 20 min. The mean fluorescence of indo-1 at rest and during tetanic contractions was measured and converted to [Ca2+]i using an intracellularly established calibration curve.15
Immunoblotting
Muscles were homogenised in lysis buffer of the following composition (mM): 20 HEPES (pH 7.6), 150 NaCl, 5 EDTA, 1 Na3VO4 and 25 KF, and 5% glycerol (v/v), 0.5% Triton X-100 (v/v) and protease inhibitor cocktail (1 tablet/50 mL; Roche). The protein content was determined using the Bradford assay. Crude total muscle homogenate were separated by electrophoresis and transferred onto membranes. Membranes were incubated with primary antibody (anti-RyR1, 1:5000, Pierce and Abcam; anti-dihydropyridine receptor (DHPR α2-subunit) 1:1000, Abcam; anti-3-nitrotyrosine (3-NT), 1:1000, Abcam; anti-nNOS, -eNOS and -iNOS, 1:1000, Abcam; anti-actin, 1:1000, Abcam; anti-malondialdehyde (MDA), 1:1000, Abcam; anti-S-nitrosothiol (SNO), 1:500, Sigma; Oxyblot protein oxidation detection kit, 1:150, Millipore) and infrared-labelled secondary antibodies (IRDye 680 and IRDye 800, 1:5000, Licor). Immunoreactive bands were visualised using the Odyssey Infrared Imaging System. Band densities were analysed with Image J and normalised to RyR1 or actin expression.
Immunoprecipitation
The lysate (500 μg) was incubated at 4°C overnight with anti-RyR1 antibody in 500 μL of modified RIPA buffer consisting of (in mM) 10 Tris–HCl (pH 7.5), 150 NaCl, 5 NaF, 1 Na-Orthovanadate, 1% Triton X-100 and one tablet of protease inhibitor. The immune complexes were incubated with Dynabeads Protein G (Invitrogen) for 2 h at 4°C, after which the beads were washed three times with 100 mM Na-acetate solution (pH 5.0). Proteins were separated by electrophoresis and immunoblots were performed as described above and quantified relative to RyR1 expression.
Myofibrillar force
Small pieces of the muscle samples were homogenised following standard procedures,16 which resulted in a solution containing isolated myofibrils. The myofibrils were transferred into an experimental chamber placed on the top of an inverted microscope equipped with a system of atomic force cantilevers (AFC).17 A laser is shined upon and reflects from a precalibrated AFC, which acts as a force transducer. The cantilever deflection caused by myofibril activation is detected and recorded using a photo-quadrant detector. Since the stiffness of the AFC (K) is known and we measure the amount of cantilever displacement (Δd), the force (F) can be calculated as F=K • Δd.
Myofibrils were chosen for mechanical testing based on striation pattern and number of sarcomeres in series (between 10 and 30). Under high magnification, the contrast between the dark bands of myosin (A-bands) and the light bands of actin (I-bands) provided a dark-light intensity pattern, representing the striation pattern produced by the sarcomeres, which allowed measurements of sarcomere length during the experiments. Using micromanipulators, the myofibrils were attached between the AFC and a rigid glass needle. A computer-controlled, multichannel fluidic system connected to a double-barrelled pipette was used for activation of the myofibril in solutions with different Ca2+ concentrations (15°C). Length changes during the experiments were induced with a rigid micro-needle connected to a piezoelectric motor.
Once the myofibrils were fully activated and maximal force for a given Ca2+ concentration was obtained, they underwent shortening (amplitude 30% of sarcomere length; speed 10 µm/s) during which the force declined and then rapidly redeveloped to reach a new steady state.
The maximal force produced by the myofibrils was calculated after force development stabilisation. Forces were averaged for a period of 2 s reach steady state. All forces were normalised by the myofibril cross-sectional area. For each contraction, rates of force development (KAct) and redevelopment (KRedev) were analysed with a two-exponential equation (a*(1-exp(-K*t)-exp(-l*t))+b), and the fast phase of relaxation (KRelax) was analysed with a single-exponential equation (a*exp(-K*(t-c))+b). For both equations, F is force, t is time, K is the rate constant for force development, a is the amplitude of the exponential, and b and c are constants.
Statistics
Data are presented as mean±SE. Statistical difference between two groups was established with unpaired or paired Student t test. A p value less than 0.05 was regarded as statistically significant.
Results
Increased nNOS protein expression in muscles of CIA mice and patients with RA
The total nNOS protein expression was markedly increased in fast-twitch (FDB) muscles from CIA mice (figure 1A and C), which is in accordance with previous data from slow-twitch (soleus) muscles.3 The protein expression of eNOS did not differ between the CIA and control FDB muscles and iNOS expression was not detected in either CIA or control muscles (figure 1A and C), which is also in agreement with previous results from soleus muscles.3 CIA muscles were studied 16±1 day post the second injection (see ‘Methods’), hence we do not expect iNOS expression since it has been shown to increase directly after injection and thereafter return to baseline within ∼24 h.18 We also compared nNOS protein expression in vastus lateralis or tibialis anterior muscles (mixed muscle fibre types) from patients with RA and healthy controls and observed a higher expression in muscles from RA patients (figure 1B–C). Interestingly, nNOS appears to run lower on the gel in patients with RA compared with healthy controls, which could be due to post-translational modifications as reported previously.19 Together, data from both the animal model and human biopsies show that arthritis is accompanied with higher nNOS expression in muscles.
Figure 1.
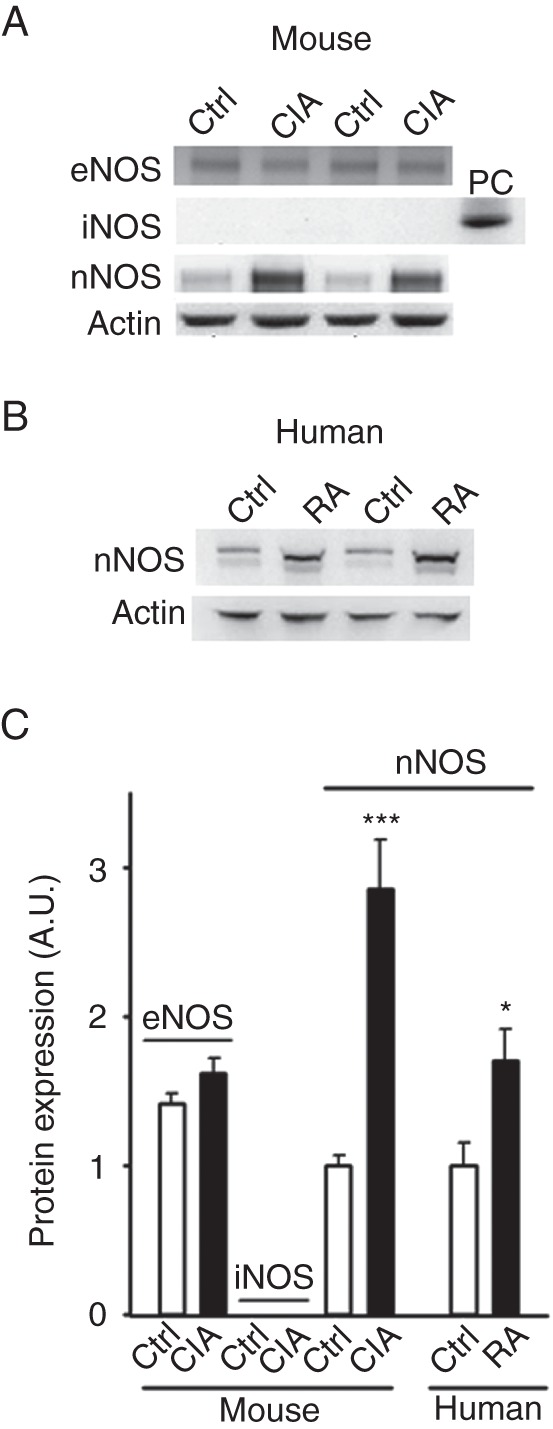
Increased protein expression of nNOS in skeletal muscles from collagen-induced arthritis (CIA) mice and patients with rheumatoid arthritis (RA). The levels of eNOS, iNOS, nNOS in mouse muscles (A) and nNOS in human muscles (B) were determined by western blotting. iNOS was not detected in control (Ctrl) or CIA, but was detected in LPS-stimulated cells (positive control, PC). (C) Mean levels (±SEM) of NOS isoforms were quantified and normalised to the actin content. 4–7 muscles per group. *p<0.05; ***p<0.001 versus controls.
Increased amount of nNOS and nitrosative stress on the RyR1 complex in muscle fibres from CIA mice
Immunoprecipitation of RyR1 was used to assess the link between nNOS and the RyR1 macromolecular complex in CIA muscles. There was a marked increase in the amount of nNOS bound to the RyR1 channel complex in all three CIA muscle types tested; fast-twitch extensor digitorum longus (EDL) and FDB and slow-twitch soleus (figure 2A–B). There was no difference in total RyR1 or DHPR protein expression between CIA and control muscles (see online supplementary figure S1). Further, NO produced in the presence of superoxide (O2•−) will rapidly react to form the powerful RNS peroxynitrite (ONOO−), which can react with proteins and have deleterious effects.20 Intriguingly, nNOS can produce both NO and O2•− and may in this way directly increase ONOO− formation.21 3-NT can be used as a footprint of peroxynitrite production in vivo.20 3-morpholinosydnonimine-N-ethylcarbamide (SIN-1) is a peroxynitrite donor,22 and control experiments were performed where FDB muscles were incubated with increasing concentrations of SIN-1 and this resulted in increasing 3-NT signal (figure 2C–D). Importantly, figure 2E–F shows that proteins of the RyR1 macromolecular complex from CIA muscle, especially in proteins at ∼150 kDa, have a marked increase in the 3-NT content. This means that proteins of the macromolecular complex that regulate RyR1 activity, rather than the large RyR1 monomer (∼500 kDa) itself, appear to be targets of RNS under these conditions.23 One tentative target, the transverse tubular voltage sensor DHPR α2-subunit (∼150 kDa) was addressed by immunoprecipitation. These experiments showed similar 3-NT content on this DHPR subunit, as well as on proteins bound to it, in CIA and control muscles (see online supplementary figure S2). Thus, the exact nature of the 3-NT-modified proteins associated with RyR1 remains to be established.
Figure 2.

nNOS associated with the RyR1 complex is accompanied by increased nitration. (A) Representative western blots of nNOS co-immunoprecipitated with RyR1 in lysates of slow-twitch soleus muscles and fast-twitch EDL and FDB muscles of control and collagen-induced arthritis (CIA) mice. (B) Mean levels of nNOS co-immunoprecipitated with RyR1. Representative western blot (C) and mean data (D; n=3) of 3-NT protein binding with increasing concentrations of SIN-1 in FBD control fibres. Representative 3-NT western blot of RyR1 immunoprecipitates from FDB muscles of control and CIA mice (E) and mean data (proteins larger than ∼ 64 kDa were quantified) of 3-NT normalised to RyR (F; n=4). Data are presented as mean±SEM; *p<0.05, **p<0.01 versus controls.
Nitrosative stress on the RyR1 complex is associated with increased tetanic [Ca2+]i
To assess whether changes in the RyR1 channel complex affected SR Ca2+ handling, we compared basal and tetanic [Ca2+]i and force production in CIA and control muscle fibres. We observed no difference in basal [Ca2+]i between CIA and control fibres (87±13 vs 79±0.6 nM, n=6, p=0.65). On the other hand, tetanic [Ca2+]i was significantly higher over the whole range of stimulation frequencies (15–120 Hz) in fibres from CIA mice (representative original records at 120 Hz in figure 3A and mean data in figure 3B). Intriguingly, tetanic [Ca2+]i was almost twice as high in CIA fibres than in control fibres at the higher stimulation frequencies (50–120 Hz).
Figure 3.

Increased tetanic Ca2+ accompanied by muscle weakness in muscle fibres from collagen-induced arthritis (CIA) mice. (A) Typical examples (120 Hz stimulation frequency, 350 ms train duration) of [Ca2+]i (left) and specific force (right) in control (—) and CIA (---) fibres. (B) [Ca2+]i is significantly increased over a wide range of stimulation frequencies in fast-twitch FDB muscle fibres from CIA mice (•) compared with controls (○, Ctrl) (n=9–10). (C) Representative Commassie stained gels from fast-twitch muscles of CIA and control mice. (D) Mean expression of myosin and actin protein expression as measured from Commassie stained gels and normalised to total protein content (n=6). (E) Force per cross-sectional area was decreased in CIA muscle fibres (n=9–10). (F) Force–[Ca2+]i curves, generated by plotting the mean force against the mean [Ca2+]i at different frequencies. Data are mean±SEM; *p<0.05; **p<0.01; ***p<0.001 versus controls.
Intrinsic muscle weakness in muscle fibres from CIA mice
No atrophy or loss in actin or myosin content was observed in muscles from CIA mice (figure 3C–D). In fact, the isolated CIA single fibres in which [Ca2+]i and force were measured had a slightly larger cross-sectional area than the control fibres (647±24 vs 486±38 µm2, n=9–10, p<0.01). However, specific force (ie, force per cross-sectional area) was lower in CIA than in control fibres (figure 3A and E). To further analyse the decreased specific force in CIA fibres, we plotted mean forces obtained in the two groups at different frequencies against the corresponding [Ca2+]i values (figure 3F). Force–[Ca2+]i curves were then constructed by fitting the mean data points to the following equation:
where P is the measured force, Pmax the predicted maximum force at saturating [Ca2+]i, Ca50 the [Ca2+]i at 50% Pmax and N describes the steepness of the force–[Ca2+]i relationship. This analysis showed a markedly lower Pmax (266 vs 388 kN/m2) and higher Ca50 (0.75 vs 0.54 µM) in CIA than in control fibres, whereas the steepness of the relationships was similar (N 2.46 vs 2.94). Thus, the decreased force in CIA fibres was due to a combination of two myofibrillar impairments: decreased ability to generate force and reduced Ca2+ sensitivity. The mechanism(s) underlying these impairments would be post-translational modifications because we detected no differences in the total content of the two major myofibrillar proteins, actin and myosin (figure 3C–D).
To further study mechanisms behind the increased tetanic [Ca2+]i and decreased force in CIA fibres, some fibres were exposed to caffeine (5 mM), a potent RyR1 agonist.12 The rationale behind these experiments is that tetanic stimulation in the presence of caffeine will release virtually all Ca2+ stored in the SR and tetanic [Ca2+]i will reach a level high enough to maximally activate the contractile machinery.24 Figure 4A shows representative original records of [Ca2+]i, and force from 120 Hz tetani produced in the absence and presence of caffeine and mean data are shown in figure 4B–C. The results show a markedly larger effect of caffeine on [Ca2+]i in control than in CIA fibres, and in the presence of caffeine, tetanic [Ca2+]i was no longer higher in the CIA fibres (5.08±0.35 vs 4.96±0.30 µM, p=0.79) (figure 4B). This indicates that the increased tetanic [Ca2+]i in CIA fibres (see figure 3B) is not caused by increased SR Ca2+ storage, but rather facilitated RyR1 Ca2+ release. The increased tetanic [Ca2+]i with caffeine had no notable effect on force production in either CIA or control fibres (figure 4C), which can be explained by tetanic force at 120 Hz being close to maximal already before caffeine application (see figure 3E).
Figure 4.
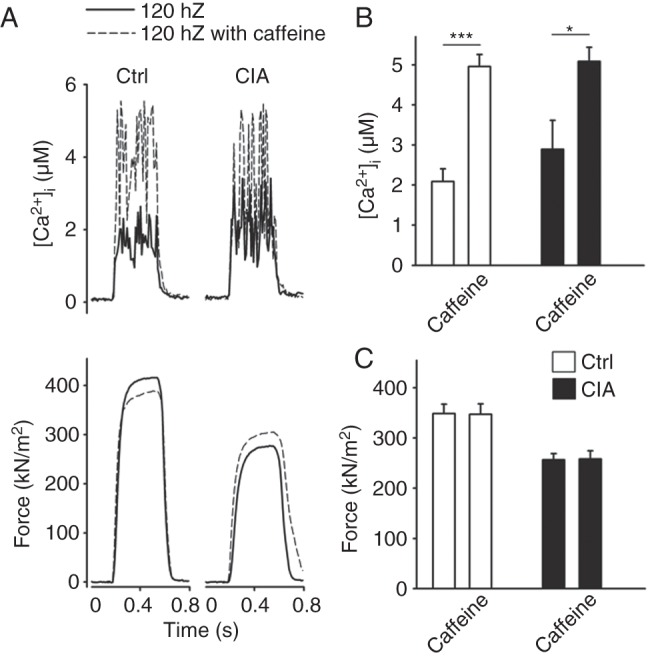
Sarcoplasmic reticulum Ca2+ storage is similar in collagen-induced arthritis (CIA) and control fibres. (A) Typical example (120 Hz stimulation frequency, 350 ms train duration) of [Ca2+]i (upper panel) and specific force (lower panel) in the presence (---) or absence (—) of caffeine (5 mM, 2 min exposure). Mean values (±SEM, n=6) of [Ca2+]i (B) and specific force (C) in the presence or absence of caffeine in CIA and control FDB fibres. *p<0.05, **p<0.001 versus controls.
Nitrosative stress of contractile proteins behind muscle weakness
Muscles from mice with RA are significantly affected by weakness, which is caused by impairments in the contractile machinery (see figure 3–4). Figure 5A and C shows that actin, but not myosin, in CIA muscles is attacked by ONOO− as shown by the increased nitration (3-NTActin). Malondialdehyde (MDA) is a common marker of oxidative stress and the same holds for oxyblot that measures the amount of carbonylation.25 No difference in MDA content (figure 5B, D) or carbonylation on actin and myosin was observed between the CIA and control group (oxyblotActin 1.01±0.26 vs 1.02±0.23; oxyblotMyosin 0.94±0.13 vs 1.02±0.23 A.U., n=5–6).
Figure 5.

Nitration of actin and decreased actin–myosin interaction underlie weakness in CIA muscles. Western blots assessing 3-NT (A) and MDA (B) accumulation on actin and myosin in fast-twitch muscles from CIA and control mice. Hydrogen peroxide (200 µM, 20 min) was used as positive control (PC). Mean data of 3-NT (C) and MDA (D) accumulation on actin and myosin normalised to total protein (n=6). (E, F) Typical records of active force from atomic force cantilever measurements in myofibrils in pCa2+ of 5.5 and 4.5, respectively. (G) Mean values of the active isometric forces produced by myofibrils at a sarcomere length of 2.5 µm and pCa2+ 4.5 (n=12). (H) Mean values of the rates of force development during initial activation (KAct), after shortening (KRedev), and the fast rate of relaxation following deactivation (KRelax) (n=6). (I) Myofibril force-pCa2+ relation, generated by plotting the mean force against the pCa2+ at a sarcomere length 2.5 µm. There was a rightward shift in the curve from CIA myofibrils (pCa2+50(Ctrl): 5.65 vs pCa2+50(CIA): 5.5; p<0.05), whereas the steepness was not significantly affected (NCtrl: 4.87 vs NCIA: 4.56). Data are mean±SEM; *p<0.05; **p<0.01; ***p<0.001 versus controls.
Increased 3-NT content on both the RyR1 complex and on actin in muscles from CIA mice suggests that the redox balance is specifically shifted towards increased ONOO− production in these muscles (figure 2D–E, 5A, C). Since 3-NT binding is a covalent post-translational protein modification, it should not be affected by acute exposure to either oxidising or reducing agents. Thus, if the force decrease in CIA muscle depends on actin nitration, this force depression should be little affected by exposure to the oxidant tert-butyl hydroperoxide (t-BuOOH) or the reducing agent dithiothreitol (DTT), which both have relatively large effects on force production under control conditions.15 26–28 Accordingly, neither 10 min exposure to t-BuOOH (10 µM) nor DTT (1 mM) had any significant effect (p>0.5) on force generation in CIA muscle fibres at any stimulation frequency (eg, force at 120 Hz before and after t-BuOOH was 197±30 vs 191±4 kN/m2 (n=3), and before and after DTT 260±20 vs 242±19 kN/m2 (n=3)).
Some experiments were performed on fast-twitch muscles from CIA mice displaying only mild inflammation (score 0–1). The protein expression of nNOS, as well as eNOS, in these muscles was similar to controls. Moreover, force production was similar in these CIA muscles and control muscles (see online supplementary figure S3).
Finally, the actin–myosin interactions were studied in more detail by bypassing the normal activation (ie, DHPR-RyR1 coupling and Ca2+ release) and directly activate myofibrils and measure force production with atomic force cantilevers. Figure 5E–G shows that the active force in myofibrils from CIA mice was markedly lower than in myofibrils of healthy controls at both pCa (-log[Ca2+]) 4.5 and 5.5. Moreover, the force–pCa2+ relationship was shifted to the right in CIA myofibrils (figure 5H), that is, their myofibrillar Ca2+ sensitivity was decreased, whereas the steepness of the relationship was not affected. After being fully activated, myofibrils were rapidly shortened. The rates of force development during the initial activation (KAct) and after the shortening (KRedev) were lower in CIA myofibrils (figure 5I). To assess whether slowed cross-bridge kinetics occurred also in intact muscle fibres from CIA mice, we measured the rates of activation and relaxation in tetanic contractions with maximal activation, that is, in the presence of caffeine (see figure 4). Average force records then showed slightly lower rates of activation (∼15%) and relaxation (∼30%) in CIA than in control fibres (see online supplementary figure S4).
Discussion
We here demonstrate that increased nNOS protein levels in skeletal muscle are present in patients with RA as well as in the CIA mouse model of RA. We also show markedly increased formation of nNOS-RyR1 complexes in skeletal muscles from CIA mice. These changes were accompanied by a substantial increase in tetanic [Ca2+]i. However, specific force was reduced by approximately one-third in individual fast-twitch muscle fibres of CIA mice. We show that this muscle weakness can be explained by decreased myofibrillar force production, which could be a consequence of increased levels of nitrosative stress on actin.
We have previously shown a force decrease of similar magnitude in slow-twitch CIA soleus (type I) muscles, although in those muscles myosin and troponin I were affected by reversible nitrosylation.3 Here in fast-twitch (type II) muscles, proteins are also nitrosylated, but we observed no differences in SNO content between CIA and healthy controls (see online supplementary figure S5). Instead, irreversible 3-NT protein modifications induced by ONOO−-derived radicals20 on the RyR1 complex and actin were observed. Altered footprints and modifications of ROS/RNS between muscle types are expected considering their different characteristics and metabolic features, for example, slow-twitch fibres have high oxidative capacity and more superoxide dismutase (SOD, preferentially mitochondrial SOD) than fast-twitch fibres with high glycolytic capacity and a less effective ROS/RNS defence system.5 29 Limitations in human muscle biopsy material prevented us from comparing the RyR1 channel complex and actin in patients with RA and healthy controls. However, Salanova and colleagues recently demonstrated co-localisation of RyR1 with nNOS in skeletal muscle from healthy human subjects,30 and it was previously shown that nNOS can shuttle between the sarcolemma and the SR.13 Moreover, despite using a rather heterogeneous group of patients with RA, we observed a marked increase in nNOS, which suggests that this is a central (mal)adaptation in patients with arthritis.
Our results indicate a central causative role of increased ONOO− formation and protein nitration in the weakness observed in CIA muscles. Increased ONOO− production occurs as a consequence of temporally and spatially coordinated increases in NO and O2•−. We here show an increased nNOS expression in muscles both from CIA mice and patients with RA, which would increase NO production. The source of O2•− is more uncertain and was not directly assessed in the present study. Intriguingly, nNOS can produce both NO and O2•− and may in this way directly increase ONOO− formation.21 Although other sources of O2•− formation cannot be excluded, nNOS-derived O2•− generation is an attractive candidate for increased ONOO− production because NO and O2•− will then be produced at the same site, which minimises the necessity of diffusion of these reactive molecules. Moreover, muscles of CIA mice with mild inflammation showed no increase in nNOS protein expression and their force production was similar to controls. This shows that increased nNOS is not an obligate consequence of inflammation and hence strengthens the causative link between nNOS and muscle weakness in arthritis. However, the ultimate proof of the proposed link is missing and would require usage of other experimental models, for example, force measurements in nNOS-deficient muscles from subjects displaying severe arthritis.
We show similar impairments in contractile function in isolated muscle fibres and isolated myofibrils from CIA mice: ∼30% decrease in force at saturating [Ca2+]; decreased myofibrillar Ca2+ sensitivity without any change in the steepness of force–[Ca2+] relationship and slowed force development. These contractile impairments were caused by stable modification(s) since they were observed even after isolation of myofibrils and single-muscle fibres, and the contractile impairments in the latter preparation were not affected by subsequent exposures to an oxidising (t-BuOOH) or a reducing (DTT) agent. Intriguingly, stable deficiencies in the same contractile parameters were observed in skinned rat skeletal muscle fibres after exposure to the ONOO−-donor SIN-1.26 We propose that the impaired contractility in CIA muscles involves ONOO−-mediated 3-NT accumulation on actin (see figure 5A), which is a covalent modification and hence stable. This proposal is supported by results from in vitro motility assay experiments, which indicate a causative relationship between ONOO−-induced actin nitration and defective myofibrillar function.31 However, other modifications are likely to contribute to the impaired myofibrillar function in CIA muscles and these remain to be established.
In conclusion, we show that the contractile dysfunction in CIA skeletal muscle is associated with increased production of RNS and impaired actin–myosin force production. Figure 6 outlines a tentative vicious circle in which increases in nNOS and [Ca2+]i lead to a progressively larger actin nitration, which plays a key role in the development of muscle weakness. Pharmacological intervention targeting RyR1 to stabilise SR Ca2+ release may be a promising strategy to protect against arthritis-induced muscle weakness, for example, treatment with AICAR (5-aminoimidazole-4-carboxamide ribonucleotide) or S107 that have been shown to stabilise RyR1 activity.7–9
Figure 6.
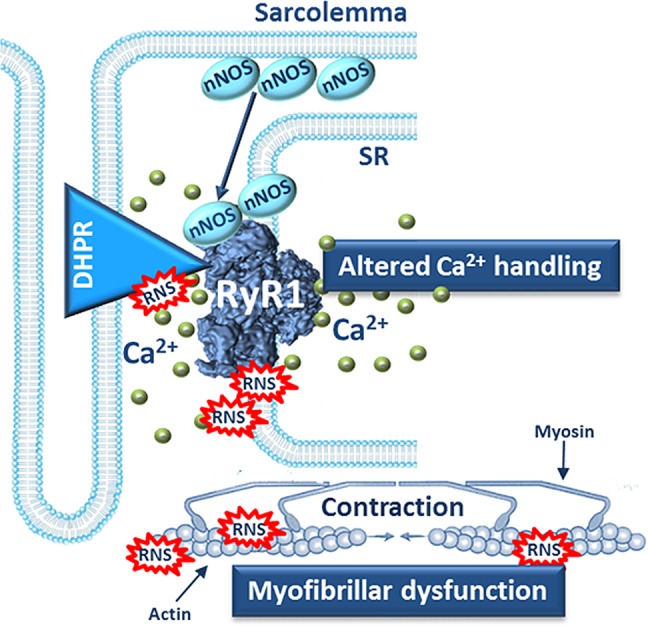
Schematic model of intracellular mechanisms underlying muscle weakness in arthritis. nNOS is globally increased in arthritic muscle and more nNOS is bound to the RyR1 protein complex. This leads to RNS-induced modifications of the RyR1 protein complex and the SR Ca2+ release during contractions increases, which further activates the Ca2+-sensitive nNOS and RNS production is augmented. The increased amounts of RNS attack myofibrillar proteins, such as actin, and cause contractile dysfunction and muscle weakness.
Supplementary Material
Acknowledgments
The authors would like to thank Eva Lindroos for assistance with the muscle biopsies. Dr Birgitta Nordmark, Rheumatology Unit, Karolinska University Hospital, and Dr Ulf Bergström, Department of Rheumatology, Skåne University Hospital, Malmö, for recruitment of patients.
Footnotes
Funding: This work was supported by The Swedish Research Council, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) grant #R01 AR062083 (HW), Canadian Institute for Health Research (CIHR), Karolinska Institutet Foundation, Swedish Foundation for Strategic Research, Magnus Bergvall Foundation, Swedish Heart and Lung Foundation, The French Muscular Dystrophy Association (AFM), Åke Wiberg Foundation, Swedish Rheumatism Association, Combine Sweden, Regional Agreement on Medical Training and Clinical Research (ALF) between Stockholm County Council and Karolinska Institutet.
Competing interests: None.
Ethics approval: Stockholm North Ethical Committee on Animal Experiments and Regional Research Ethics Committee of Southern Sweden.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Nordesjö LO, Nordgren B, Wigren A, et al. Isometric strength and endurance in patients with severe rheumatoid arthritis or osteoarthrosis in the knee joints. A comparative study in healthy men and women. Scand J Rheumatol 1983;12:152–6. [DOI] [PubMed] [Google Scholar]
- 2.Helliwell PS, Jackson S. Relationship between weakness and muscle wasting in rheumatoid arthritis. Ann Rheum Dis 1994;53:726–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamada T, Place N, Kosterina N,et al. Impaired myofibrillar function in the soleus muscle of mice with collagen-induced arthritis. Arthritis Rheum 2009;60:3280–89. [DOI] [PubMed] [Google Scholar]
- 4.Williams RO. Collagen-induced arthritis in mice. Methods Mol Med 2007;136:191–99. [DOI] [PubMed] [Google Scholar]
- 5.Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 2008;88:1243–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellinger AM, Reiken S, Carlson C,et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med 2009;15:325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Durham WJ, Aracena-Parks P, Long C,et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell 2008;133:53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersson DC, Betzenhauser MJ, Reiken S,et al. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab 2011;14:196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanner JT, Georgiou DK, Dagnino-Acosta A,et al. AICAR prevents heat-induced sudden death in RyR1 mutant mice independent of AMPK activation. Nat Med 2012;18:244–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCartney-Francis NL, Song X, Mizel DE, et al. Selective inhibition of inducible nitric oxide synthase exacerbates erosive joint disease. J Immunol 2001;166:2734–40. [DOI] [PubMed] [Google Scholar]
- 11.Förstermann U, Closs EI, Pollock JS,et al. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 1994;23:1121–31. [DOI] [PubMed] [Google Scholar]
- 12.Lanner JT, Georgiou DK, Joshi AD, et al. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol 2010;2:a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sears CE, Ashley EA, Casadei B. Nitric oxide control of cardiac function: is neuronal nitric oxide synthase a key component? Philos . Trans R Soc Lond B Biol Sci 2004;359:1021–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnett FC, Edworthy SM, Bloch DA,et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 15.Andrade FH, Reid MB, Allen DG, et al. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J Physiol 1998;509:565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rassier DE. Pre-power stroke cross bridges contribute to force during stretch of skeletal muscle myofibrils. Proc Biol Sci 2008;275:2577–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Labuda A, Grutter PH. Exploiting cantilever curvature for noise reduction in atomic force microscopy. Rev Sci Instrum 2011;82:013704. [DOI] [PubMed] [Google Scholar]
- 18.El-Dwairi Q, Comtois A, Guo Y, et al. Endotoxin-induced skeletal muscle contractile dysfunction: contribution of nitric oxide synthases. Am J Physiol 1998;274:C770–9. [DOI] [PubMed] [Google Scholar]
- 19.Brindicci C, Kharitonov SA, Ito M,et al. Nitric oxide synthase isoenzyme expression and activity in peripheral lung tissue of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010;181:21–30. [DOI] [PubMed] [Google Scholar]
- 20.Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 2007;6:662–80. [DOI] [PubMed] [Google Scholar]
- 21.Stuehr D, Pou S, Rosen GM. Oxygen reduction by nitric-oxide synthases. J Biol Chem 2001;276:14533–6. [DOI] [PubMed] [Google Scholar]
- 22.Mattart L, Calay D, Simon D,et al. The peroxynitrite donor 3-morpholinosydnonimine activates Nrf2 and the UPR leading to a cytoprotective response in endothelial cells. Cell Signal 2012;24:199–213. [DOI] [PubMed] [Google Scholar]
- 23.Lanner JT. Ryanodine receptor physiology and its role in disease. Adv Exp Med Biol 2012;740:217–34. [DOI] [PubMed] [Google Scholar]
- 24.Allen DG, Westerblad H. The effects of caffeine on intracellular calcium, force and the rate of relaxation of mouse skeletal muscle. J Physiol 1995;487:331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo S, Wehr NB. Protein carbonylation: avoiding pitfalls in the 2,4-dinitrophenylhydrazine assay. Redox Rep 2009;14:159–66. [DOI] [PubMed] [Google Scholar]
- 26.Dutka TL, Mollica JP, Lamb GD. Differential effects of peroxynitrite on contractile protein properties in fast- and slow-twitch skeletal muscle fibers of rat. J Appl Physiol 2011;110:705–16. [DOI] [PubMed] [Google Scholar]
- 27.Dutka TL, Verburg E, Larkins N,et al. ROS-mediated decline in maximum Ca2+-activated force in rat skeletal muscle fibers following in vitro and in vivo stimulation. PLoS ONE 2012;7:e35226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrade FH, Reid MB, Westerblad H. Contractile response of skeletal muscle to low peroxide concentrations: myofibrillar calcium sensitivity as a likely target for redox-modulation. FASEB J 2001;15:309–11. [DOI] [PubMed] [Google Scholar]
- 29.Higuchi M, Cartier LJ, Chen M, et al. Superoxide dismutase and catalase in skeletal muscle: adaptive response to exercise. J Gerontol 1985;40:281–6. [DOI] [PubMed] [Google Scholar]
- 30.Salanova M, Schiffl G, Rittweger J, et al. Ryanodine receptor type-1 (RyR1) expression and protein S-nitrosylation pattern in human soleus myofibres following bed rest and exercise countermeasure. Histochem Cell Biol 2008;130:105–18. [DOI] [PubMed] [Google Scholar]
- 31.Snook JH, Li J, Helmke BP, et al. Peroxynitrite inhibits myofibrillar protein function in an in vitro assay of motility. Free Radic Biol Med 2008;44:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.