

Abstract
High-altitude pulmonary edema (HAPE) is a potentially fatal condition, occurring at altitudes greater than 3,000 m and affecting rapidly ascending, non-acclimatized healthy individuals. However, the lack of biomarkers for this disease still constitutes a bottleneck in the clinical diagnosis. Here, ultra-high performance liquid chromatography coupled with Q-TOF mass spectrometry was applied to study plasma metabolite profiling from 57 HAPE and 57 control subjects. 14 differential plasma metabolites responsible for the discrimination between the two groups from discovery set (35 HAPE subjects and 35 healthy controls) were identified. Furthermore, 3 of the 14 metabolites (C8-ceramide, sphingosine and glutamine) were selected as candidate diagnostic biomarkers for HAPE using metabolic pathway impact analysis. The feasibility of using the combination of these three biomarkers for HAPE was evaluated, where the area under the receiver operating characteristic curve (AUC) was 0.981 and 0.942 in the discovery set and the validation set (22 HAPE subjects and 22 healthy controls), respectively. Taken together, these results suggested that this composite plasma metabolite signature may be used in HAPE diagnosis, especially after further investigation and verification with larger samples.
High altitude pulmonary edema (HAPE) is a life threatening clinical condition, mostly occurring in non-acclimatized healthy individuals who rapidly ascend to high altitude (above 3000 m)1. It is the major cause of death related to high altitude exposure. Currently, HAPE diagnosis mainly relies on patient interviews, physician’s examination, X-ray radiograph and computed tomography (CT) of the chest, and magnetic resonance imaging (MRI)2, as there are lack of objective laboratory-based tests. Current diagnostic methods are highly dependent on the clinician experience, which usually results in underdiagnosis, delayed diagnosis, and misdiagnosis due to the high heterogeneity of clinical symptoms. Therefore, the identification of metabolite biomarkers for HAPE would be of great clinical value in laboratory-based diagnosis of HAPE and understanding the pathophysiology of the disease.
Metabonomics is a top-down systems biology approach whereby metabolic responses to disease or treatment are analyzed and modeled3. Hence, metabonomics represents an excellent developing prospect for capturing diseases specific metabolic signatures as possible biomarkers4. Metabolite biomarkers have been successfully applied in the discrimination or diagnosis of various diseases such as cancer5, neurodegenerative diseases6, cardiovascular disease7, diabetes8, and so on. Previously, there was a report on identifying the molecular alterations associated with HAPE by 1H NMR- based metabonomics approach9, which not only provided valuable clues in dissecting the mechanisms of HAPE, but also exemplified the ability of metabonomics to identify diagnostic biomarkers for HAPE from clinical samples. Given that 1H NMR analytical technology cannot provide complete coverage of the human metabonome due to the diverse physicochemical properties of metabolites and the relatively low sensitivity of 1H NMR, the metabolite variations and the disturbances of metabolic pathways of HAPE are still far from complete. It is meaningful to apply complementary metabonomic platforms such as mass spectrometry to identify novel biomarkers of HAPE.
In this study, we therefore applied metabonomic method based on ultra-high performance liquid chromatography (UHPLC) coupled with Q-TOF mass spectrometry to profile metabolites in plasma samples from 57 HAPE subjects and 57 healthy controls. One of the purposes was to identify the differential plasma metabolites in HAPE patients relative to healthy controls. The other purpose was to optimize a simplified metabolite signature for HAPE diagnosis.
Materials and Methods
Participants
The study protocol was approved by the Human Ethics Committee of the Third Military Medical University, and written informed consent was obtained from all study volunteers prior to participation. All procedures involving the human subjects were carried out in accordance with the recommendations of the Helsinki Declaration.
A total of 57 HAPE subjects were enrolled as cases for the study, including 49 men and 8 women with a mean age of 34.79 ± 5.84 years, who developed the disease after traveling from the lowlands to Golmud district (altitude 2,780–4,500 m) in Qinghai, China. All subjects were recruited between March 2013 and December 2014 from the 22nd Hospital of the Chinese People’s Liberation Army, which is the largest hospital located in the city of Golmud in Qinghai. This hospital is the primary treatment center for individuals suffering from high altitude disease in this region, which is considered as a “checkpoint” to Tibet by many travelers. The HAPE patients enrolled in this study did not receive any medication therapy prior to sample collection. Fasting blood samples were collected from HAPE patients. The diagnosis of HAPE was based on standard criteria10 including cough, dyspnea, cyanosis at rest, absence of infection, the presence of pulmonary rales and cyanosis. In all cases, HAPE was confirmed by chest radiographic findings of infiltrates consistent with pulmonary edema. The control group consisted of 49 men and 8 women with a mean age of 34.66 ± 6.08 years. These individuals were selected according to a 1:1 case-matching scheme using the variables sex, age, blood pressure, BMI and method of ascent. All control subjects were non-natives of a high altitude environment who had not developed any symptoms or signs of HAPE or related illness after exposure to high altitude within 7 days. Blood samples from healthy volunteers were obtained under fasting conditions.
In total, the 57 HAPE patients and 57 healthy controls were enrolled into this study and then were divided into a discovery set and a validation set. The discovery set, composed of 35 HAPE subjects and 35 healthy controls, was used to identify plasma diagnostic markers for HAPE; the remaining subjects were used to establish the validation set to independently validate the diagnostic generalizability of these biomarkers. The detailed demographic and clinical data of the participants are presented in Table 1. Any patients with previous history of cardiopulmonary diseases and other metabolic diseases such as diabetes, hypertension, obesity and heart disease, as identified by self-reported medical history or full examination carried out after HAPE recovery, were excluded from study participation.
Table 1. Demographic and clinical details of recruited subjects.
| Discovery Set | Validation set | |||||
|---|---|---|---|---|---|---|
| Control | HAPE | p value | Control | HAPE | p value | |
| sample size | 35 | 35 | − | 22 | 22 | − |
| sex (M/F) | 31/4 | 31/4 | − | 18/4 | 18/4 | − |
| Age, year | 34.81 ± 6.03 | 35.00 ± 5.75 | 0.88 | 34.42 ± 6.29 | 34.45 ± 6.10 | 0.98 |
| Method of ascent | by train | by train | − | by train | by train | − |
| Systolic blood pressure, mm Hg | 119.57 ± 3.75 | 120.56 ± 3.51 | 0.25 | 120.76 ± 3.86 | 119.91 ± 3.60 | 0.45 |
| Diastolic blood pressure, mm Hg | 78.27 ± 4.06 | 78.15 ± 4.00 | 0.89 | 77.87± 4.22 | 78.22 ± 3.74 | 0.77 |
| Body mass index, kg/m2 | 22.45 ± 2.29 | 22.27 ± 2.12 | 0.73 | 22.39 ± 2.45 | 22.58 ± 2.19 | 0.78 |
| arterial oxygen saturation, % | 92.34 ± 3.89 | 77.48 ± 5.99 | <0.0001 | 92.39 ± 3.39 | 76.54 ± 5.67 | <0.0001 |
| Pulse rate, rate/min | 81.47 ± 5.82 | 94.95 ± 10.82 | <0.0001 | 81.47 ± 5.82 | 94.95 ± 10.82 | <0.0001 |
Chemicals and reagents
HPLC-grade Methanol and acetonitrile (ACN) were purchased from Merk (Darmstadt, Germany). Formic acid was obtained from Fluka (Buchs, Switzerland). Sphingosine and palmitoylcarnitine were purchased from Acros Organics (NewJersey, USA). Glutamine, methionine, hypoxanthine, inosine, valine and isoleucine were obtained from Shanghai Jingchun Reagent Co. Ultrapure water was prepared with a Milli-Q water purification system (Millipore, Bedford, MA, USA).
Sample preparation
Fasting venous blood (with EDTA as an anticoagulant) was obtained from all the above-mentioned individuals. The plasma was separated immediately by centrifugation (3000 × g, 10 min). The harvested plasma samples were stored at −80 °C, and transported to Shanghai for further experiments. Prior to the analysis, a volume of 400 μL of methanol was added to 100 μL of plasma. After vigorous shaking for 1 min and incubation on ice for 10 min, the mixture was centrifuged at 14,000 × g for 15 min at 4 °C to precipitate the protein. All the supernatant was removed (without removing any particles left at the bottom of the vial). The supernatant was evaporated to dryness with a gentle nitrogen stream. The dry residue was reconstituted in 100 μL of ACN/water (7:3, v/v), then centrifuged again at 14,000 × g for 10 min at 4 °C.
As part of the system conditioning and quality control (QC) process, a pooled QC sample was prepared by mixing equal volumes (100 μL) from each of the 114 samples as they were being aliquoted for analysis. It was processed as real samples and then was inserted through the analytical run at intervals of 8–13 real samples to be analyzed eleven times. The QC samples were sufficiently spread out through the whole run as to ensure its validity.
UHPLC-Q-TOFMS analysis
UHPLC analysis was performed on Agilent 1290 Infinity LC system (Agilent, Germany). Chromatographic separation was carried out at 40 °C on an ACQUITY UPLC BEH C18 column (2.1 mm × 100 mm, 1.7 μm, Waters, Milford, MA). The column oven was set at 40 °C. The mobile phase consisted of 0.1% formic acid (A) and ACN modified with 0.1% formic acid (B), using a gradient elution of 5%B at 0–2 min, 5%–95% B at 2–13 min, 95% B at 13–15 min. The total run time was 20 min including equilibration. The flow rate was 350 μL/min and the injection volume was 4 μL.
An Agilent 6530 Accurate-Mass Quadrupole Time-of-Flight (Q-TOF) mass spectrometer (Agilent, USA) was used in the study. The Q-TOF mass spectrometer was operated in electrospray ionization source (ESI) positive ion mode with a capillary voltage of 3.5 kV, drying gas flow of 11 L/min, and a gas temperature of 350 °C. The nebulizer pressure was set at 45 psig. The fragmentor voltage was set at 120 V and skimmer voltage was set at 60 V. All analyses were acquired using a mixture of 10 mM purine (m/z 121.0508) and 2 mM hexakis phosphazine (m/z 922.0097) as internal standards to ensure mass accuracy and reproducibility. Data were collected in centroid mode and the mass range was set at m/z 50–1000 using extended dynamic range. Potential biomarkers were analyzed by MS/MS in the Q-TOF. Nitrogen was used as the collision gas. MS/MS analysis was performed on the mass spectrometer set at different collision energy of 10−50 eV according to the stability of each metabolites. MS spectra were collected at 2 spectra/s, and MS/MS spectra were collected at 0.5 spectra/s, with a medium isolation window (~4 m/z). A negative ion scan was only employed when metabolite identification was carried out.
Data Handling
The raw data in instrument specific format (.d) were converted to common data format (.mzData) files using a conversion software program (file converter program available in Agilent MassHunter Qualitative software), in which the isotope interferences were eliminated. The program XCMS (version, 1.40.0) (http://masspec.scripps.edu/xcms/xcms.php) was used for nonlinear alignment of the data in the time domain and automatic integration and extraction of the peak intensities11. XCMS parameters were default settings (major default parameters: profmethod = bin; method = matchedFilter; step = 0.1) except for the following: full width at half maximum (FWHM) = 8, bandwidth (bw) = 10 and snthresh = 5, due to narrower peaks obtained by the use of the column packed with 1.7 μm particles. The variables presenting in at least 80% of either group were extracted12, and the variables with a retention time less than 0.5 min (near to the dead time) were excluded due to a high degree of ion suppression that they suffered13. Variables with less than 30% relative standard deviation (RSD) in QC samples14 were then retained for further multivariate data analysis because they were considered stable enough for prolonged UHPLC–Q-TOFMS analysis. For each chromatogram, the intensity of each ion was normalized to the total ion intensity, in order to partially compensate for the concentration bias of metabolites between samples and to obtain the relative intensity of metabolites. The resulting three-dimensional matrix, including retention time and m/z pairs (variable indices), sample names (observations), and normalized ion intensities (variables), was exported to multivariate data analysis.
The normalized data was introduced to SIMCA-P V11.0 (Umetrics, Sweden) for principal component analysis (PCA) and partial least squares discriminant analysis (PLS-DA) after mean-centering and pareto scaling, a technique that increased the importance of low abundance ions without significant amplification of noise. The quality of the models was evaluated with the relevant R2 and Q2 as well discussed elsewhere15. T-test was performed in succession to reveal the statistical differences for the variables between healthy and HPAE individuals.
Results
Plasma metabolic profiling by UHPLC-MS
The separation conditions of plasma on UHPLC-MS system were optimized in terms of peak shape and reproducibility. The representative chromatograms of plasma metabolomes in ESI positive mode are shown in Supplementary Fig. S1. The stability of the analytical method is very important to obtain valid metabonomic data. To validate the system performance during the analysis of real samples, a pooled QC sample was applied16, which was a representative “mean” sample including all the analytes during the analysis. The QC sample was processed as real samples and then was inserted amongst the real sample queue to be analyzed eleven times. PCA results of the QC sample demonstrated that the peak areas deviation was less than 2 SD, indicating that the data from the UHPLC-MS were statistically acceptable (supplemental Fig. S2). In addition, it was found that the variation and of the retention times are 0.02–0.06 min for metabolites of interest in QC samples, and the relative standard derivations (RSD) for peak areas of metabolites of interest are 4.2%–13.1% in QC samples (see Table 2 for data). All the results demonstrated the robustness of the method. This confirms that the significant differences observed between the two groups by multivariate statistical analysis were more likely to be a result of genuine subtle changes in metabolites, rather than products of artifacts arising from technical errors.
Table 2. Potential biomarkers and their metabolic pathways.
| No. | m/z | tR(min) | Formula | Metabolite | Ratioa | VIPb | Trendc | Related pathway | %RSDd |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 147.0761 | 0.81 | C5H10N2O3 | Glutaminee | 0.67 | 2.65 | ↓* | Alanine, aspartate and glutamate metabolism | 12.7 |
| 2 | 150.0581 | 0.97 | C5H11NO2S | Methioninee | 1.75 | 1.41 | ↑* | Methionine metabolism | 10.3 |
| 3 | 118.0860 | 1.10 | C5H11NO2 | Valinee | 0.62 | 6.07 | ↓* | Valine, leucine and isoleucine biosynthesis | 8.3 |
| 4 | 137.0456 | 1.14 | C5H4N4O | Hypoxanthinee | 2.02 | 2.97 | ↑* | Purine metabolism | 7.9 |
| 5 | 269.0875 | 1.28 | C10H12N4O5 | Inosinee | 2.14 | 1.39 | ↑* | Purine metabolism | 13.1 |
| 6 | 132.1008 | 1.31 | C6H13NO2 | Isoleucinee | 0.64 | 9.75 | ↓* | Valine, leucine and isoleucine biosynthesis | 4.2 |
| 7 | 300.2887 | 9.54 | C18H37NO2 | Sphingosinee | 1.81 | 1.41 | ↑* | Sphingolipid metabolism | 8.9 |
| 8 | 400.3420 | 10.46 | C23H45NO4 | Palmitoylcarnitinee | 1.60 | 2.39 | ↑* | Sphingolipid metabolism | 7.5 |
| 9 | 520.3379 | 10.62 | C26H50NO7P | LysoPC(18:2)f | 0.60 | 9.73 | ↓* | Phospholipid metabolism | 6.7 |
| 10 | 426.3574 | 10.66 | C26H51NO3 | C8−ceramidef | 1.74 | 4.46 | ↑* | Sphingolipid metabolism | 4.8 |
| 11 | 280.2631 | 10.83 | C18H33NO | Linoleamidef | 1.76 | 1.98 | ↑* | Fatty acid metabolism | 10.3 |
| 12 | 570.3546 | 11.00 | C30H52NO7P | LysoPC(22:5)f | 0.60 | 3.01 | ↓* | Phospholipid metabolism | 9.5 |
| 13 | 546.3549 | 11.14 | C28H52NO7P | LysoPC(20:3)f | 0.61 | 4.81 | ↓* | Phospholipid metabolism | 5.4 |
| 14 | 256.2638 | 13.03 | C16H33NO | Palmitic amidef | 1.59 | 1.50 | ↑* | Fatty acid metabolism | 10.8 |
aThe ratio of relative amounts of HAPE group to control group.
bVariable Importance in Projection.
cChange trend compared with control group. (↑): up-regulated. (↓): down-regulated.
dVariation of the biomarker concentrations in QC samples expressed as relative standard deviation (%RSD).
eMetabolites validated with standard sample.
fMetabolites putatively annotated. *p value < 0.01.
Multivariate statistical analysis of metabolites
The normalized data sets contained 1218 ions. To determine whether the metabolite fingerprints in plasma differed between the healthy and HAPE subjects, we first evaluated separation between healthy and HAPE subjects using unsupervised principal component analysis (PCA). The obvious separation was achieved between HAPE group and healthy group (R2 = 0.75) (Fig. 1A). To further search ion peaks that can discriminate between the two groups, the supervised PLS-DA model was established in that it was more focused on the actual class discriminating variation compared to the unsupervised PCA model. A clear separation between healthy group and HAPE group was observed in the PLS-DA score plot by the first two components (Fig. 1B) (R2 = 0.97, Q2 = 0.93). To validate the model, permutation tests with 99 iterations were further performed. These permutation tests compared the goodness of fit of the original model with the goodness of fit of randomly permuted models. As shown in Fig. 1C, the validation plot indicates that the original model is valid. The criteria for validity are as follows: all the permuted R2 (cum) and Q2 (cum) values to the left are lower than the original point to the right, and the blue regression line of the Q2 (cum) points has a negative intercept17,18.
Figure 1. Multivariate data analysis.

(A) PCA score map derived from UHPLC-Q-TOFMS spectra concerning healthy (■) and HAPE ( ) individuals. (B) PLS-DA score map derived from UHPLC-Q-TOFMS spectra concerning healthy (■) and HAPE (
) individuals. (B) PLS-DA score map derived from UHPLC-Q-TOFMS spectra concerning healthy (■) and HAPE ( ) individuals. (C) Validation plot obtained from 99 permutation tests. (D) S-plot of the PLS-DA model.
) individuals. (C) Validation plot obtained from 99 permutation tests. (D) S-plot of the PLS-DA model.
Identification of differential plasma metabolites in HAPE
Metabolites were carefully screened before being approved as potential biomarkers. First, significant original variables were extracted from the S-plot, which is a covariance-correlation-based procedure, and thus the risk of false positives in metabolite selection was reduced19. The S-plot (Fig. 1D), derived from the first component of the combined model, explains most of the variables in data set, in which the ions furthest away from the origin contribute significantly to the clustering of the two groups and may be regarded as potential biomarkers (in two shaded areas of Fig. 1D). Next, the variable importance for projection (VIP) reflecting the importance of variables has been applied to filter the important metabolites in the model. The most important 30 variables were first selected according to their VIP value. Furthermore, the fold change of the relative intensity from the differential metabolite between the two groups was set as 1.5. Unpaired Student’s t-tests were performed as the final testing procedure, and the critical p-value was set to 0.05 for significantly differential variables. Following the criterion above, 14 metabolite ions were selected as potential biomarkers related to HAPE.
The detailed method for the compound identification was described in the author previous work20. In brief, the corresponding quasi-molecular ion peak was found according to the accurate mass and retention time in the extracted ion chromatogram (EIC), and then the most probable molecular formula were calculated by Agilent MassHunter software. Fig. S3 A and S3 B show the EIC and MS spectrum of a typical ion whose m/z is 400.3420. Then, MS/MS analysis of m/z 400.3420 in plasma was performed using UHPLC-Q-TOFMS in the same chromatographic and mass spectrometric conditions (Fig. S3 C). With its fragmentation information and the freely accessible databases such as HMDB (http://www.hmdb.ca) and METLIN (http://metlin.scripps.edu), the major fragment ions m/z 341.2685, 144.1016, 85.0290 and 60.0813 represent the fragments of [C20H37O4]+, [C7H14NO2]+, [C4H5O2]+ and [C3H9N]+, respectively. Therefore, the m/z 400.3420 was identified as palmitoylcarnitine according to the elemental composition, retention time and fragmentation information. Finally, the MS/MS spectrum of the commercial standard palmitoylcarnitine was used to confirm the identified compound. Other biomarkers have been similarly identified and are listed in Table 2 and the structures and MS/MS spectra of the metabolites are presented in Supporting Information Fig. S4. Among these metabolites, the high level of C8-ceramide, palmitoylcarnitine, hypoxanthine, linoleamide, palmitic amide, methionine, sphingosine and inosine, and the low level of isoleucine, valine, glutamine, lysoPC(18:2), lysoPC(20:3) and lysoPC(22:5) were observed in HAPE subjects relative to healthy controls. (Fig. 2)
Figure 2. HAPE subjects possess increased/decreased metabolites.
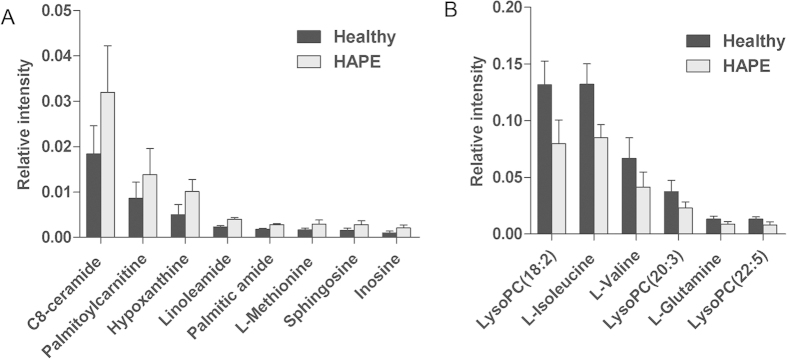
(A) The relative signal intensities of the increased metabolites in HAPE subjects. (B) The relative signal intensities of the decreased metabolites in HAPE subjects. Data are expressed as mean ± S.D.
Identification of a simplified HAPE metabolite signature
14 candidate plasma biomarkers of HAPE were identified in the above analysis. However, diagnosis based on quantification of so many metabolites would not be economical and convenient in clinical practice. It would be more practical in diagnosing HAPE to identify a simplified plasma metabolite signature. Therefore, the 14 differential metabolites were used as candidates for selection of a simplified HAPE metabolite signature. It is well-known that changes in more important positions of a network will trigger a more severe impact on the pathway than changes occurring in marginal or relatively isolated positions21. The metabolic pathway impact analysis with MetaboAnalyst 3.0 revealed that these differential metabolites are important for the organism response to HAPE and are responsible for multiple pathways22,23. Therefore, it was used to optimize a plasma metabolite signature for HAPE. The metabolic networks are directed graph as Fig. 3. It was revealed that the identified metabolites are responsible for sphingolipid metabolism, alanine, aspartate and glutamate metabolism, methionine metabolism, D-Glutamine and D-glutamate metabolism, valine, leucine and isoleucine biosynthesis, purine metabolism, fatty acid metabolism and phospholipid metabolism. The impact-value threshold calculated from pathway topology analysis was set to 0.10 24, and two unique pathways including sphingolipid metabolism and alanine, aspartate and glutamate metabolism was filtered out as potential targets pathway for HAPE. Potential metabolite signature was then identified from the two metabolic pathways. Among the 14 differential metabolites, C8-ceramide and sphingosine belong to sphingolipid metabolism, and glutamine belongs to alanine, aspartate and glutamate metabolism. Therefore, it was speculated that these three metabolites should yield the higher predictive power for future diagnostic applications. The relative concentrations of these three plasma metabolite biomarkers for HAPE are presented in Fig. 2.
Figure 3. The pathway impact of HAPE on plasma metabolites with MetaboAnalyst 3.0.
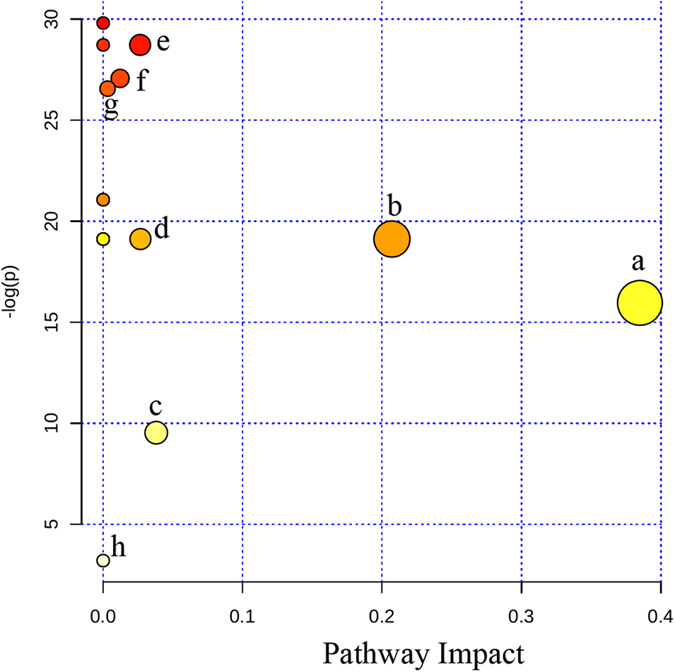
(a) sphingolipid metabolism; (b) alanine, aspartate and glutamate metabolism, (c) methionine metabolism; (d) D-Glutamine and D-glutamate metabolism; (e) valine, leucine and isoleucine biosynthesis; (f) purine metabolism; (g) phospholipid metabolism; (h) fatty acid metabolism.
To further validate the potential diagnostic effectiveness of the simplified metabolite signature, the ROC-curve was plotted using relative intensities of metabolites. The three representative metabolites including C8-ceramide, sphingosine and glutamine were selected as a panel of candidate markers. Logistic regression was used to combine the three variables into a multivariable. The prediction model is as follows: P = 1/[1 + exp(−(−2.39 + 204.16 × (C8-ceramide) + 3015.73 × (sphingosine) − 829.96 × (glutamine)))]. The results indicated that a panel of three metabolites generated an AUC of 0.981 with a sensitivity of 91.43% and a specificity of 94.29% and 0.942 with a sensitivity of 86.36% and a specificity of 81.82 for the discovery and validation sets, respectively (Fig. 4A). According to the highest prediction sensitivity (91.43%) and specificity (94.29%) of the ROC curves on the discovery set, an optimal cutoff value of 0.4988 was obtained. Based on this cutoff value, it was found that 65 out of 70 samples (92.8%) in the discovery set as well as 37 out of 44 samples (84.1%) in validation set could be accurately predicted (Fig. 4B). This finding indicated that this simplified plasma metabolite signature was a “good” classifier of HAPE patients and healthy controls.
Figure 4. ROC curves based on the binary logistic regression model by the combination of three plasma metabolites (C8-ceramide, sphingosine and glutamine), and their prediction plots based on the optimal cutoff value from ROC curves.

(A) The HAPE samples from the discovery set were applied to construct a binary logistic regression model based on the combination of plasma C8-ceramide, sphingosine and glutamine, and the ROC curves of the discovery set ((A), left) and validation set ((A), right) were obtained from the above established prediction model. (B) The optimal cutoff value with the highest sensitivity and specificity in the ROC curves of the training set was obtained (0.4988) and applied to evaluate the prediction capacity (92.8% for discovery set ((B), left) and 84.1% for validation set ((B), right)) of the current model, where 0 and 1 on the x axis represent healthy controls and HAPE patients, respectively, and blue circle represent samples.
Discussion
HAPE is a severely life-threatening acute mountain sickness that endangers the lives of people climbing or migrating to high altitudes. Currently, the lack of disease biomarkers constitutes a bottleneck in the clinical diagnosis of HAPE. Here, using UHPLC-Q-TOFMS-based plasma metabonomic approach, 14 potential biomarkers related to HAPE was identified. Compared to previous 1H NMR-based study, UHPLC-Q-TOFMS-based metabolomic approach provided larger coverage of HAPE-related metabonome including sphingolipid, phospholipids, fatty acid amides and several amino acids. Based on metabolic pathway impact analysis, a metabolite signature consisting of three plasma metabolite biomarkers C8-ceramide, sphingosine and glutamine was further identified as an effective diagnostic pattern, yielding an AUC of 0.981 in the discovery set and 0.942 in the validation set. Our results suggest that this metabolite signature may be helpful in the development of objective laboratory-based diagnostic tools for HAPE.
To understand the underlying molecular functions of these plasma metabolite biomarkers, metabolic pathway analysis was conducted. The 14 metabolites were found to be primarily involved in (a) sphingolipid metabolism (b) alanine, aspartate and glutamate metabolism, (c) methionine metabolism, (d) D-Glutamine and D-glutamate metabolism, (e) valine, leucine and isoleucine biosynthesis, (f) purine metabolism, (g) fatty acid metabolism, (g) phospholipid metabolism and (h) fatty acid metabolism. By relating the metabolic pathways, the metabolic network of HAPE-related potential biomarkers was constructed (Fig. 5). The disturbed metabolic pathways are discussed in detail below.
Figure 5. Schematic overview of the disturbed metabolic pathways associated with HAPE.

The metabolites are shown in color: red represents increased metabolites, green represents decreased metabolites and the open circles represent no detected or changed metabolites in our experiment. PRPP, phosphoribosyl pyrophosphate; GS, Glutamine synthetase
The significantly higher levels of C-8 ceramide and sphingosine were observed in HAPE subjects relative to healthy controls, suggesting the sphingolipid metabolism is upregulated in HAPE subjects. It was reported that ceramide-challenged pulmonary endothelial cells exhibit decreased barrier function, independent of apoptosis25,26, which may contribute to lung inflammation and pulmonary edema27. A recent study also reported that exogenous sphingosine-1-phosphate boosted acclimatization in rats exposed to acute hypobaric hypoxia28. In addition, the level of palmitoylcarnitine was significantly increased in HAPE subjects relative to healthy controls, which illustrated that HAPE facilitated the process of sphingolipid biosynthesis. These consistent set of findings suggested that reestablishing the sphingolipid homeostasis was an important drug target for improving physiological acclimatization of subjects venturing into high altitude.
The low level of glutamine, a key metabolite in the pathway of alanine, aspartate and glutamate metabolism and D-Glutamine and D-glutamate metabolism, was observed in HAPE subjects. Since the biosynthesis of glutamine depends on glutamine synthetase, we speculated that the decrease in activities of glutamine synthetase should be a reason for the decrease of glutamine. In agreement with this presumption, several studies have consistently reported that the activity of glutamine synthetase was decreased in animal model exposed to high altitude (4000 m)29.
A significantly higher level of methionine was observed in HAPE subjects relative to healthy controls, suggesting the methionine metabolism pathway is perturbed in HAPE subjects. Although the metabolic mechanism is not yet well defined, it is plausible that the metabolic disorder results from dysregulation of proteolysis, oxidative catabolism, and gluconeogenesis30. Further study on its underlying mechanisms is underway in our laboratory.
The levels of inosine and hypoxanthine were significantly increased in HAPE subjects relative to healthy controls. Inosine and hypoxanthine are the products of adenosine metabolic degradation. It was previously reported that adenosine was released by hypoxic canine lung tissue and the levels of inosine and hypoxanthine showed sustained significant increases31, which is consistent with our results. In addition, an increased purine metabolism flux induced by acute systematic hypoxia has also been observed in a recently reported LC-MS study32. These data suggests that perturbed purine metabolism is implicated in HAPE.
The low levels of valine and isoleucine, correlated with valine, leucine and isoleucine biosynthesis, were observed in HAPE subjects. Valine and isoleucine, two branched-chain amino acids, may be an important alternative energy substrate. It seems that the reduction in ATP production due to the inhibition of citrate cycle induced by the hypobaric hypoxia of high altitude could lead to the utilization of branched-chain amino acids as energy compensation. Branched-chain amino acids have been suggested as a useful supplementation in the treatment of lung disease33 and in trekking at high altitude34. These results together with our findings suggest that HAPE is associated with disturbances in branched-chain amino acids metabolism.
Fatty acid amides (FAMs) are a group of endogenous lipid signaling molecules found in the brain and blood of mammals. Linoleamide and palmitic amide were FAMs and a significantly higher levels of them were observed in HAPE subjects relative to healthy controls. Although the possible role of linoleamide and palmitic amide in HAPE could not be directly elucidated, the physiological functions of the other FAM such as endocannabinoid anandamide could give an indirect clue to them. It was previously reported that endocannabinoid anandamide could mediate hypoxic pulmonary vasoconstriction via fatty acid amide hydrolase (FAAH)-dependent metabolites and hypoxia could cause elevated anandamide in the lung35. Meanwhile, exaggerated hypoxic pulmonary vasoconstriction is one of the pathological features of HAPE1. In line with these previous reports, we speculated that linoleamide and palmitic amide may also be an important mediator of hypoxic pulmonary vasoconstriction and be involved in the generation of pulmonary hypertension. The mechanism of action of the two FAMs still carried out in our laboratory.
Lysophosphatidylcholines (LysoPCs) including LysoPC(18:2), LysoPC(20:3) and LysoPC(22:5) were obviously decreased in plasma from HAPE subjects relative to healthy controls, suggesting the phospholipid metabolism is implicated in stroke patients. LysoPC is an important signaling molecule with diverse biological functions and can mediate many cell signaling pathways in monocytes/macrophages and specific receptors36,37, so that it participates in inflammatory response. The regulation of phospholipid metabolites may have important implications in inflammation response following HAPE induced-by hypobaric hypoxia.
Conclusion
In conclusion, an UHPLC−Q-TOFMS based metabonomic approach has been developed to profile HAPE-related metabolic changing in plasma. Fourteen potential biomarkers have been identified as being primarily involved in sphingolipid metabolism, alanine, aspartate and glutamate metabolism, methionine metabolism, D-Glutamine and D-glutamate metabolism, valine, leucine and isoleucine biosynthesis, purine metabolism, fatty acid metabolism and phospholipid metabolism. Using metabolic pathway impact analysis and metabolite enrichment analysis, we identified a panel of plasma metabolite biomarkers relating to HAPE, of which the combination of plasma C8-ceramide, sphingosine and glutamine could discriminate HAPE patients from healthy controls with high accuracy. It suggested that this composite urinary metabolite signature may have diagnostic and/or prognostic values for HAPE, which deserve to be further investigated in larger populations with accurately characterized patients and to explore their corresponding mechanisms related to HAPE.
Additional Information
How to cite this article: Guo, L. et al. Three plasma metabolite signatures for diagnosing high altitude pulmonary edema. Sci. Rep. 5, 15126; doi: 10.1038/srep15126 (2015).
Supplementary Material
Acknowledgments
This work was supported by the Special Projects of Scientific Research in Health Service under the Ministry of Health (No. 2010002012 and CWS11J309) and the National Natural Science Foundation of China (No. 81402888).
Footnotes
Author Contributions L.G. and H.L. performed experiments. G.T. and L.T. analyzed data and wrote the main manuscript text. P.L. provided plasma samples of HAPE. L.H. and Q.R. designed and supervised experiments, and submitted the manuscript. All authors reviewed the manuscript.
References
- Bhagi S., Srivastava S. & Singh S. B. High-altitude pulmonary edema: review. J Occup Health. 56, 235–243 (2014). [DOI] [PubMed] [Google Scholar]
- Pennardt A. High-altitude pulmonary edema: diagnosis, prevention, and treatment. Curr Sports Med Rep. 12, 115–119 (2013). [DOI] [PubMed] [Google Scholar]
- Fiehn O. Metabolomics–the link between genotypes and phenotypes. Plant Mol Biol. 48, 155–171 (2002). [PubMed] [Google Scholar]
- Sreekumar A. et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 457, 910–914 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Armitage E. G. & Barbas C. Metabolomics in cancer biomarker discovery: Current trends and future perspectives. Journal of Pharmaceutical and Biomedical Analysis 87, 1–11 (2014). [DOI] [PubMed] [Google Scholar]
- Jove M., Portero-Otin M., Naudi A., Ferrer I. & Pamplona R. Metabolomics of Human Brain Aging and Age-Related Neurodegenerative Diseases. Journal of Neuropathology and Experimental Neurology 73, 640–657 (2014). [DOI] [PubMed] [Google Scholar]
- Rasmiena A. A., Ng T. W. & Meikle P. J. Metabolomics and ischaemic heart disease. Clinical Science 124, 289–306 (2013). [DOI] [PubMed] [Google Scholar]
- Zhang A. H., Qiu S., Xu H. Y., Sun H. & Wang X. J. Metabolomics in diabetes. Clinica Chimica Acta. 429, 106–110 (2014). [DOI] [PubMed] [Google Scholar]
- Luo Y., Zhu J. & Gao Y. Metabolomic analysis of the plasma of patients with high-altitude pulmonary edema (HAPE) using 1H NMR. Mol Biosyst. 8, 1783–1788 (2012). [DOI] [PubMed] [Google Scholar]
- Hultgren H. N. & Marticorena E. A. High altitude pulmonary edema. Epidemiologic observations in Peru. Chest. 74, 372–376 (1978). [DOI] [PubMed] [Google Scholar]
- Smith C. A., Want E. J., O’Maille G., Abagyan R. & Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 78, 779–787 (2006). [DOI] [PubMed] [Google Scholar]
- Bijlsma S. et al. Large-scale human metabolomics studies: a strategy for data (pre-) processing and validation. Anal Chem. 78, 567–574 (2006). [DOI] [PubMed] [Google Scholar]
- Chen J. et al. Metabonomics study of liver cancer based on ultra performance liquid chromatography coupled to mass spectrometry with HILIC and RPLC separations. Anal Chim Acta. 650, 3–9 (2009). [DOI] [PubMed] [Google Scholar]
- Gika H. G., Theodoridis G. A., Wingate J. E. & Wilson I. D. Within-day reproducibility of an HPLC-MS-based method for metabonomic analysis: application to human urine. J Proteome Res. 6, 3291–3303 (2007). [DOI] [PubMed] [Google Scholar]
- Yin P. et al. A metabonomic study of hepatitis B-induced liver cirrhosis and hepatocellular carcinoma by using RP-LC and HILIC coupled with mass spectrometry. Mol Biosyst. 5, 868–876 (2009). [DOI] [PubMed] [Google Scholar]
- Sangster T., Major H., Plumb R., Wilson A. J. & Wilson I. D. A pragmatic and readily implemented quality control strategy for HPLC-MS and GC-MS-based metabonomic analysis. Analyst. 131, 1075–1078 (2006). [DOI] [PubMed] [Google Scholar]
- Mahadevan S., Shah S. L., Marrie T. J. & Slupsky C. M. Analysis of metabolomic data using support vector machines. Anal Chem. 80, 7562–7570 (2008). [DOI] [PubMed] [Google Scholar]
- Pasikanti K. K. et al. Noninvasive urinary metabonomic diagnosis of human bladder cancer. J Proteome Res. 9, 2988–2995 (2010). [DOI] [PubMed] [Google Scholar]
- Wiklund S. et al. Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Anal Chem. 80, 115–122 (2008). [DOI] [PubMed] [Google Scholar]
- Tan G. et al. Metabonomic profiles delineate the effect of traditional Chinese medicine sini decoction on myocardial infarction in rats. PLoS One. 7, e34157 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W. et al. Combined metabonomic and quantitative real-time PCR analyses reveal systems metabolic changes in Jurkat T-cells treated with HIV-1 Tat protein. J Proteome Res. 11, 5109–5123 (2012). [DOI] [PubMed] [Google Scholar]
- Xia J., Mandal R., Sinelnikov I. V., Broadhurst D. & Wishart D. S. MetaboAnalyst 2.0--a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 40, W127–133 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J. & Wishart D. S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat Protoc. 6, 743–760 (2011). [DOI] [PubMed] [Google Scholar]
- Wang X., Yang B., Sun H. & Zhang A. Pattern recognition approaches and computational systems tools for ultra performance liquid chromatography-mass spectrometry-based comprehensive metabolomic profiling and pathways analysis of biological data sets. Anal Chem. 84, 428–439 (2012). [DOI] [PubMed] [Google Scholar]
- Goggel R. et al. PAF-mediated pulmonary edema: a new role for acid sphingomyelinase and ceramide. Nat Med. 10, 155–160 (2004). [DOI] [PubMed] [Google Scholar]
- Petrache I., Petrusca D. N., Bowler R. P. & Kamocki K. Involvement of ceramide in cell death responses in the pulmonary circulation. Proc Am Thorac Soc. 8, 492–496 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache I. et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 11, 491–498 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla S. et al. Exogenous sphingosine-1-phosphate boosts acclimatization in rats exposed to acute hypobaric hypoxia: assessment of haematological and metabolic effects. PLoS One. 9, e98025 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radak Z. et al. The effect of high altitude and caloric restriction on reactive carbonyl derivatives and activity of glutamine synthetase in rat brain. Life Sci. 62, 1317–1322 (1998). [DOI] [PubMed] [Google Scholar]
- Tissot van Patot M. C. et al. Human placental metabolic adaptation to chronic hypoxia, high altitude: hypoxic preconditioning. Am J Physiol Regul Integr Comp Physiol. 298, R166–172 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentzer R. M. Jr., Rubio R. & Berne R. M. Release of adenosine by hypoxic canine lung tissue and its possible role in pulmonary circulation. Am J Physiol. 229, 1625–1631 (1975). [DOI] [PubMed] [Google Scholar]
- Lou B. S., Wu P. S., Liu Y. & Wang J. S. Effects of acute systematic hypoxia on human urinary metabolites using LC-MS-based metabolomics. High Alt Med Biol. 15, 192–202 (2014). [DOI] [PubMed] [Google Scholar]
- Menier R., Talmud J., Laplaud D. & Bernard M. P., Branched-chain aminoacids and retraining of patients with chronic obstructive lung disease. The Journal of sports medicine and physical fitness 41, 500–504 (2001). [PubMed] [Google Scholar]
- Shimizu M. et al. Energy expenditure during 2-day trail walking in the mountains (2,857 m) and the effects of amino acid supplementation in older men and women. European Journal of Applied Physiology. 112, 1077–1086 (2012). [DOI] [PubMed] [Google Scholar]
- Wenzel D. et al. Endocannabinoid anandamide mediates hypoxic pulmonary vasoconstriction. Proc Natl Acad Sci USA 110, 18710–18715 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong C. Q. et al. Expression of the lysophospholipid receptor family and investigation of lysophospholipid-mediated responses in human macrophages. Biochim Biophys Acta. 1682, 112–119 (2004). [DOI] [PubMed] [Google Scholar]
- Kabarowski J. H. G2A and LPC: regulatory functions in immunity. Prostaglandins Other Lipid Mediat 89, 73–81 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.