

Abstract
Rationale
Non-coding gene variants at the SORT1 locus are strongly associated with LDL-C levels as well as with coronary artery disease (CAD). SORT1 encodes a protein called sortilin, and hepatic sortilin modulates LDL metabolism by targeting apoB-containing lipoproteins to the lysosome. Sortilin is also expressed in macrophages, but its role in macrophage uptake of LDL and in atherosclerosis independent of plasma LDL-C levels is unknown.
Objective
To determine the effect of macrophage sortilin expression on LDL uptake, foam cell formation, and atherosclerosis.
Methods and Results
We crossed Sort1−/− mice onto a ‘humanized’ Apobec1−/−; hAPOB Tg background and determined that Sort1 deficiency on this background had no effect on plasma LDL-C levels but dramatically reduced atherosclerosis in the aorta and aortic root. In order to test whether this effect was a result of macrophage sortilin deficiency, we transplanted Sort1−/−;LDLR−/− or Sort1+/+;LDLR−/− bone marrow into Ldlr−/− mice and observed a similar reduction in atherosclerosis in mice lacking hematopoetic sortilin without an effect on plasma LDL-C levels. In an effort to determine the mechanism by which hematopoetic sortilin deficiency reduced atherosclerosis, we found no effect of sortilin deficiency on macrophage recruitment or LPS-induced cytokine release in vivo. In contrast, sortilin deficient macrophages had significantly reduced uptake of native LDL ex vivo and reduced foam cell formation in vivo, whereas sortilin overexpression in macrophages resulted in increased LDL uptake and foam cell formation.
Conclusions
Macrophage sortilin deficiency protects against atherosclerosis by reducing macrophage uptake of LDL. Sortilin-mediated uptake of native LDL into macrophages may be an important mechanism of foam cell formation and contributor to atherosclerosis development.
Keywords: Atherosclerosis, low density lipoprotein cholesterol, macrophage, foam cell, receptor-mediated endocytosis
INTRODUCTION
Atherosclerotic cardiovascular disease is the leading cause of morbidity and mortality in the world. A central hallmark of atherosclerosis is the cholesterol-loaded macrophage or ‘foam cell.’ Despite decades of research, the molecular mechanisms by which arterial macrophages take up cholesterol-rich lipoproteins, such as low density lipoproteins (LDL), leading to the development of foam cells and atherosclerotic lesions remain to be fully elucidated. Kruth and colleagues have shown that macrophages internalize native LDL through a process of macropinocytosis, although LDL uptake cannot be fully accounted for by this process1. Gene deletion of known receptors of modified LDL, such as scavenger receptor A (SRA) and CD36, do not reduce foam cell formation or the development of atherosclerosis in mice2. Thus, pathways that mediate macrophage uptake of LDL leading to foam cell formation and atherosclerosis remain of substantial interest.
Unbiased genome-wide association studies (GWAS) of coronary artery disease (CAD) have the potential to identify new pathways involved in atherosclerosis. In one of the first GWAS for CAD, non-coding genetic variants at chromosome 1p13 were reported to be significantly associated with myocardial infarction and CAD3, a finding that has been widely replicated4, 5. The same variants have also shown to be significantly associated with plasma levels of LDL cholesterol6–8. The SORT1 gene, encoding the protein sortilin, appears to be the causal gene at the locus regulating LDL cholesterol levels9–11. Sortilin is a type I transmembrane trafficking receptor initially characterized by its ability to serve as a receptor for proneurotrophins9–11 and for its role as a sorting receptor for lysosomal hydrolases12, 13. Hepatic sortilin expression modulates VLDL production rates7, 9,10; in addition, hepatic sortilin binds LDL and promotes its cellular uptake and lysosomal degradation10. Sortilin is also expressed in macrophages, but little is known about its function in this cell type or its relationship to atherosclerosis.14, 15 We hypothesized that macrophage sortilin mediates macrophage LDL uptake. Through a combination of in vivo mouse studies and ex vivo macrophage studies utilizing Sort1−/− macrophages, we show here that macrophage sortilin promotes macrophage LDL uptake, foam cell formation, and atherosclerosis independent of plasma LDL-C levels.
METHODS
Detailed descriptions of all Methods can be found in the Online Supplement. Following is a summary of the key experimental approaches.
For studies of the effect of total body sortilin deficiency on atherosclerosis, we used the Apobec1−/−; hAPOB Tg mouse, in which the human apoB transgene is overexpressed in the liver and, in contrast to the wild-type mouse, is not edited, thus producing only the apoB-100 protein.21–23 These mice were crossed with total body Sort1−/− mice and experiments compared Sort1−/−;Apobec1−/−; hAPOB Tg mice to Sort1+/+;Apobec1−/−; hAPOB Tg littermates. Mice were fed a western-type diet for 18 weeks and assessed for atherosclerosis in the aortic roots and the entire aorta by en face quantitation. A detailed description of the atherosclerosis methods can be found in the Online Supplement.
For studies of hematopoietic sortilin deficiency on atherosclerosis, we transplanted donor Sort1−/−;Ldlr −/− and Sort1+/+;Ldlr −/− bone marrow into irradiated recipient Ldlr−/− mice. Six weeks post bone marrow transplantation the mice were placed on a western diet for 18 weeks and then assessed for atherosclerosis.
For studies of macrophage LDL uptake, both thioglycollate-elicited peritoneal macrophages and bone-marrow derived macrophages were used. The macrophages were incubated with125I-LDL for five hours and uptake and degradation were assessed.
Statistical analyses were done using 2-tailed paired student’s t test and 1 way ANOVA with a Bonferroni correction (for LPS experiment).
RESULTS
Sortilin deficiency in hematopoetic cells protects against atherosclerosis
Total body Sort1 deficiency on an Ldlr−/− background is associated with reduced plasma cholesterol levels, confounding attempts to address its role in atherosclerosis independent of LDL-C levels. We crossed Sort1−/− mice onto the background of an atherosclerosis-prone Apobec1−/−; hAPOB Tg mouse model, which has a human-like lipoprotein profile, and fed the mice a western type diet for 18 weeks. On this genetic background, total and LDL cholesterol levels were not different in Sort1−/− mice compared with Sort1+/+ mice (Figure 1a,b). After 18 weeks on diet, Sort1−/− mice had a 68% reduction in en face aorta lesion area (P <0.0001 Figure 1c,d) and an 87% reduction in aortic root lesion area (P <0.0001 Figure 1e,f) compared with Sort1+/+ mice, demonstrating a major effect of sortilin deficiency in reducing atherosclerosis despite no effect on plasma cholesterol in this model.
Figure 1. Whole body Sort1 deficiency reduces atherosclerosis independent of plasma lipid level.
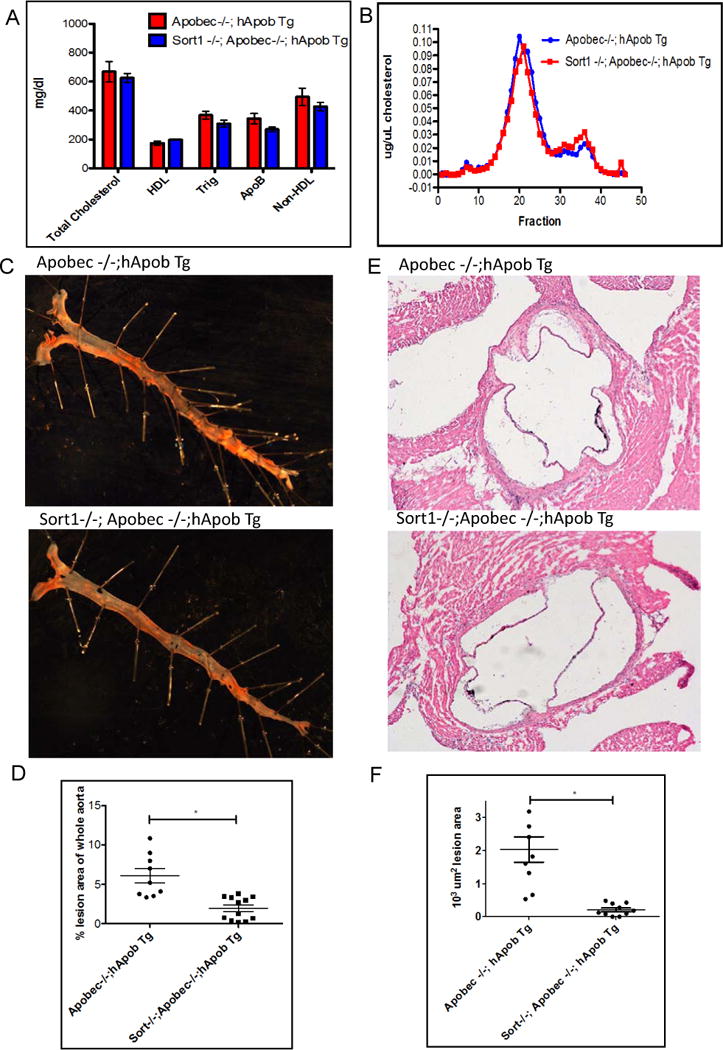
A. Eight week old Apobec −/−; hApob Tg and Sort −/−;Apobec −/−; hApobTg mice (n=10 per group) placed on a western diet for 18 weeks and retroorbital bleeds were performed after a 4 hr fast. Plasma was isolated by centrifugation and samples were run individually on a Cobas Mira Autoanalyzer (Roche Diagnostic Systems). B. Samples were pooled and run on FPLC. C. Whole aortas were dissected and tissues harvested. Aortas were stained with Oil Red O. D. Quantification of lesions on aorta E. Aortic roots were sectioned & stained with hematoxylin eosin (H&E) F. Quantification of atherosclerotic lesion area at aortic root P value <0.01
Macrophages express sortilin and we hypothesized that macrophage sortilin deficiency might account specifically for the reduced atherosclerosis. In order to test this hypothesis, irradiated Ldlr−/− mice were transplanted with bone marrow from Sort1−/−;Ldlr−/− mice or Sort1+/+;Ldlr−/− mice and 6 weeks after transplantation were started on a western type diet and fed for 18 weeks. Bone marrow engraftment was 74% (Online Figure Ia). Body, liver and spleen weights, plasma cholesterol, peripheral blood counts, and hepatic Sort1 expression were similar between groups (Online Figure I). Mice transplanted with Sort1−/−;Ldlr−/− bone marrow had a 69% reduction in en face aortic lesion area (P<0.00001) and a 34% reduction in aortic root lesion area (P < 0.01) compared to mice transplanted with Sort1+/+;Ldlr−/− bone marrow (Figure 2a–d), suggesting that hematopoietic, and potentially macrophage sortilin influences the development of atherosclerotic disease.
Figure 2. Sort1 hematopoietic deficiency reduces atherosclerosis.
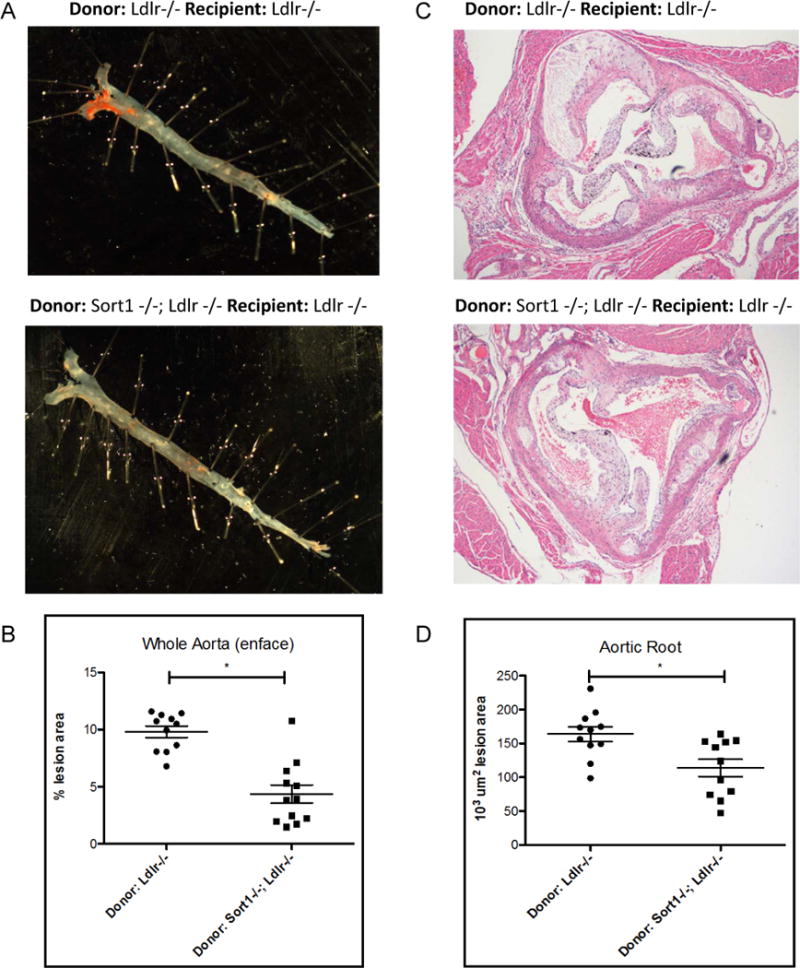
A. After 18 weeks on western diet recipient Ldlr−/− mice carrying donor Ldlr−/− or Sort1−/−;Ldlr−/− (n=11 per group) bone marrow transplanted mice aortas were dissected and stained with Oil Red O B. Quantification of lesions on aorta P<0.00001 C. Representative aortic roots stained with H&E D. Quantification of aortic roots P value <0.01
Sortilin deficiency has no effect on thioglycollate-elicited monocyte recruitment or LPS-induced inflammatory response in vivo
Monocyte recruitment is a key determinant of the macrophage content of atherosclerotic lesions. To determine if Sort1 deficiency affects macrophage recruitment, Sort1−/− and Sort1+/+ mice were injected i.p. with thioglycollate to elicit an inflammatory response. Three days after injection, peritoneal macrophages were harvested and counted. There was no difference in macrophage counts between Sort1+/+ and Sort −/− mice (Online Figure IIa). Monocyte recruitment and atherosclerosis development is strongly influenced by inflammation and cytokine production. To determine if Sort1−/− mice have reduced cytokine levels, cytokine multiplexing assays were performed on Sort1−/− and Sort1+/+ mice injected with lipopolysaccharide (LPS). Cytokine levels post LPS injection were found to be similar between Sort1−/− and Sort1 +/+ (Online Figure IIb,c).
Macrophage sortilin deficiency reduces LDL uptake and foam cell formation
To determine if macrophage sortilin deficiency reduces foam cell formation, primary bone marrow macrophages were isolated from Sort1−/−; Ldlr−/− and control Sort1+/+; Ldlr−/− mice, cells were differentiated with M-CSF for 7 days, incubated with 1 mg/mL LDL for 5 hours, and Oil Red O staining was performed. Sort1−/−;Ldlr−/− macrophages had a clear and consistent reduction in Oil Red O staining (Figure 3a). Sort1 deficient macrophages had a significant 28% reduction in total cellular cholesterol, a 25% reduction in free cholesterol, and a 32% reduction in cholesteryl ester (P<0.05; Figure 3b–d). In vivo foam cell formation assays were performed by isolating thioglycollate-elicited peritoneal macrophages from Sort1+/+;Apobec−/−;hApob Tg and Sort1−/−;Apobec−/−;hApob Tg mice fed a western type diet for 18 weeks. Consistent with the in vitro loading experiments, macrophages isolated from Sort1−/− mice had reduced Oil Red O staining and a significant 33% reduction in cellular cholesterol content compared to macrophages isolated from Sort1+/+ mice (P<0.05; Figure 3e–f). These studies indicated that sortilin-deficient macrophages have reduced capacity to form foam cells when exposed to high levels of LDL.
Figure 3. Sort1 deficiency reduces macrophage cellular cholesterol levels and Oil Red O staining in vivo and in vitro.
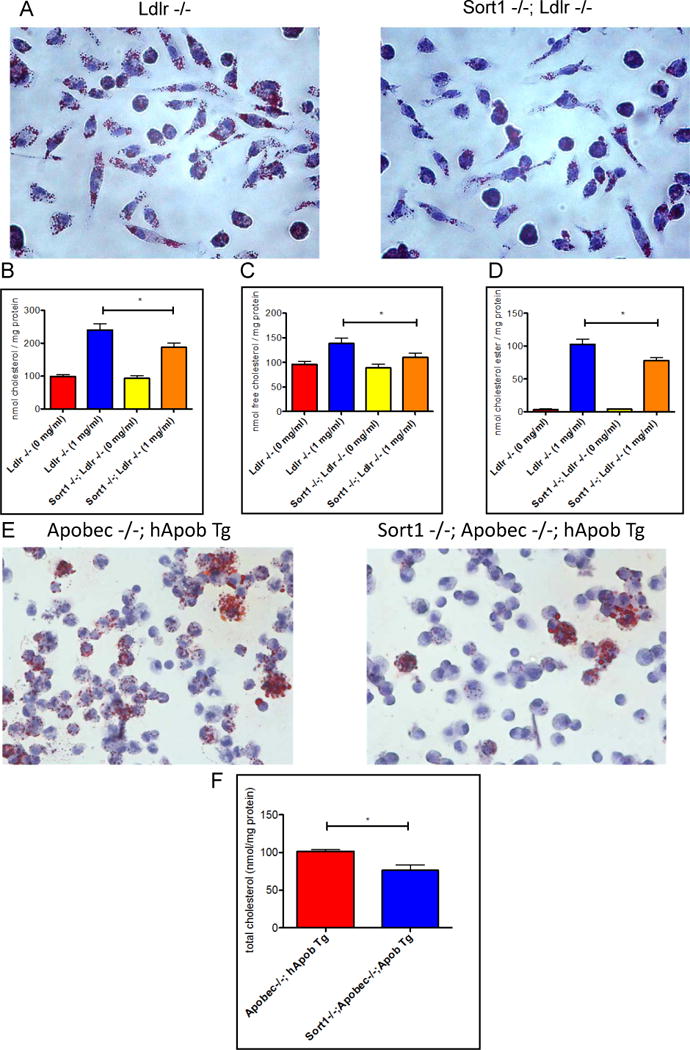
A. Bone marrow-derived macrophages were stained with Oil Red O (n=3) B. Cellular total cholesterol C. Free cholesterol D. Cholesterol ester measured by gas chromatography (GC/MS). E. Peritoneal macrophages from mice fed a western diet for 18 weeks were isolated and stained with Oil Red O F. Cellular total cholesterol of peritoneal macrophages measured by GC/MS. All experiments were replicated.
As sortilin can act as a receptor for LDL in hepatocytes, we hypothesized that sortilin promotes the internalization of LDL by macrophages. To test the response of sortilin expression to increasing cholesterol concentration in macrophages, thioglycollate-elicited peritoneal macrophages were isolated from wild-type mice and incubated for 24 hours in lipoprotein-deficient serum, lipoprotein-deficient serum supplemented with 25-hydroxycholesterol to reduce intracellular cholesterol content, or with lipoprotein deficient serum supplemented with high concentrations of LDL. In contrast to the LDL receptor, whose expression was reduced by co-incubation with LDL, Sort1 mRNA abundance increased over 400-fold with LDL incubation (P <0.05; Figure 4a) and sortilin protein also increased significantly with LDL incubation (Figure 4b).
Figure 4. LDL loading increases sortilin in macrophages, where it mediates uptake of LDL.
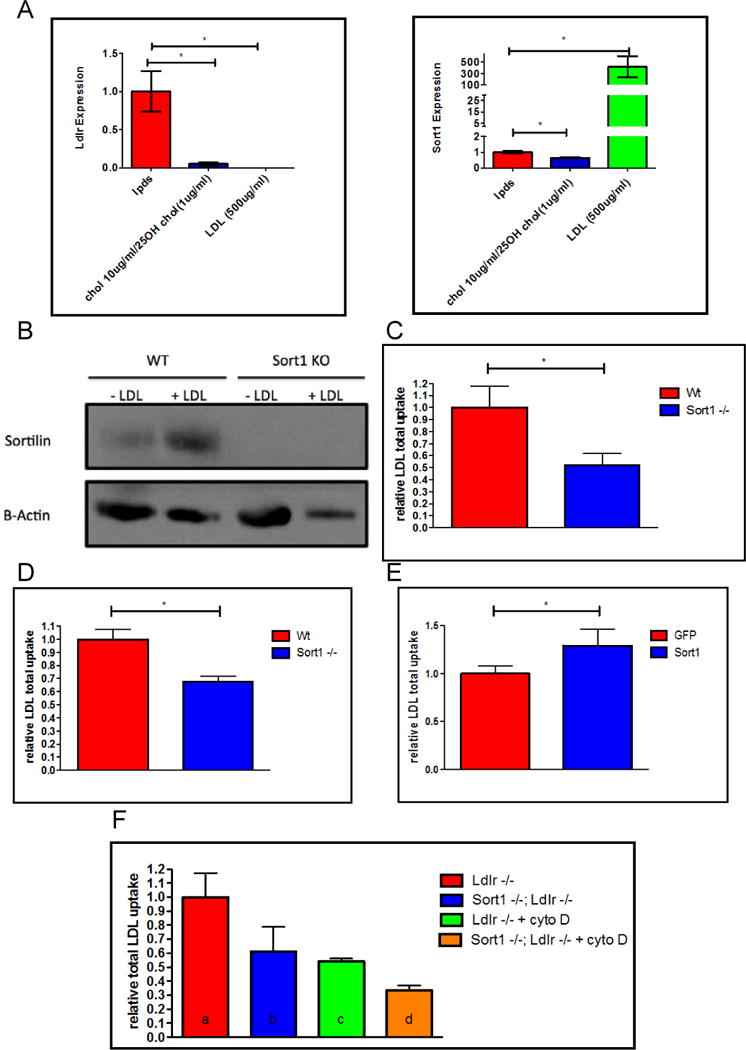
A. Thio-elicited peritoneal macrophages were incubated in conditions listed above. RNA expression and B. Westerns C. I-125 LDL uptake study from thioglycollate-elicited macrophages from Sort1 +/+ and Sort1 −/− mice (n=6) D. I-125 LDL uptake study from bone marrow derived macrophages from Sort1+/+ and Sort1−/− mice (n=6)”. and F. M-CSF differentiated macrophages from Ldlr −/− and Sort1−/−; Ldlr −/− mice. 250ug/ml LDL was incubated and 4ug/ml cytochalasin D (as indicated) for 5 hours (n=6).
To test the hypothesis that sortilin is able to promote the uptake of LDL into macrophages, 125I-LDL uptake studies were performed in thioglycollate-elicited and bone marrow derived macrophages from Sort1+/+ and Sort1−/− mice. Sort1 deficiency was associated with a 48% and 33% percent reduction in LDL uptake, respectively (P <0.05 for both; Figure 4c–d). We next tested if this effect on LDL uptake was independent of the LDL receptor. Bone marrow derived macrophages were isolated from Sort1+/+;Ldlr−/− and Sort1−/−;Ldlr−/− mice and 125I-LDL uptake studies were performed. Sort1 deficiency was associated with a 39% percent reduction in LDL uptake in the absence of the LDLR (P<0.05; Figure 4f).
To further confirm that sortilin deficiency confers atheroprotection by eliminating a receptor dependent pathway for LDL uptake and not by modulating macropinocytosis, LDL uptake studies were performed in bone marrow derived macrophages in the presence of cytochalasin D, a potent inhibitor of actin polymerization and macropinocytosis. Under these conditions, while LDL uptake is reduced, substantial residual LDL uptake still takes place.1 Sort1 deficiency was associated with a 38% reduction in LDL uptake in the presence of cytochalasin D (P< 0.05; Figure 4f). These studies indicate that macrophage sortilin deficiency reduces macrophage uptake of LDL and formation of foam cells and this effect is independent of the LDL receptor and of macropinocytosis.
Finally, to determine whether increased macrophage sortilin results in increased LDL uptake, J774 cells were transduced with lentivirus encoding GFP or Sort1 and LDL uptake studies were performed. Sort1 overexpression in macrophages led to a 29% increase in LDL uptake (P< 0.05; Figure 4e).
DISCUSSION
Genetic variation at the 1p13 SORT1 locus is strongly associated both with CAD, as well as with plasma LDL-C levels. We have previously shown that sortilin is a cell surface receptor for LDL on hepatocytes and its elevated expression in liver reduces LDL-C at least in part by facilitating LDL clearance from blood. Sortilin is expressed in macrophages, which actively take up LDL, leading us to investigate the role of macrophage sortilin in LDL uptake, foam cell formation, and atherosclerosis. After a series of studies of atherosclerosis in mice and LDL uptake in macrophages, we conclude that macrophage sortilin promotes LDL uptake, foam cell formation, and atherosclerosis and that deficiency is protective against atherosclerosis at least in part by reducing LDL uptake.
Macrophage uptake of modified LDL can be mediated by scavenger receptors such as SRA and CD36. However, deletion of SRA or CD36 does not reduce macrophage uptake of native LDL1, nor does it ameliorate atherosclerosis in hypercholesterolemic mice.17 Even CD36−/−; SRA −/− mice still contain abundant lipid laden macrophages in vessel wall and develop atherosclerosis.18 Kruth has shown that macrophages can take up native LDL through fluid-phase macropinocytosis, but there remains substantial LDL uptake even when this pathway is inhibited.1 Our data establish macrophage sortilin as the first receptor-mediated pathway of uptake of native LDL leading to foam cell formation and promoting atherosclerosis development. We also found that increasing concentrations of extracellular LDL causes an upregulation of macrophage Sort1 mRNA and protein. Because a function of macrophages is to phagocytose LDL, it is very possible that increased exposure of macrophages to LDL triggers the transcriptional upregulation of sortilin, which then mediates increased LDL uptake. The mechanisms of this upregulation of macrophage Sort1 by LDL require further exploration.
While this manuscript was under preparation, Mortensen et al. reported that sortilin deficiency reduced atherosclerosis in the ApoE −/− mouse model.19 While the fundamental observation is consistent with our data, these authors suggested a different mechanism, namely that decreased proinflammatory cytokines may have been responsible for the reduced atherosclerosis. We performed our cytokine assays prior to initiation of atherosclerotic disease, while Mortensen et al measured the cytokine profile after disease was present. In addition, these authors did not see a reduction in LDL uptake by sortilin deficient macrophages, although they used an ATTO dye conjugated to the LDL that may have influenced the interaction with sortilin. We also used a different mouse model, the Apobec1−/−; hAPOB Tg mouse, in which human apoB-100 containing LDL is the dominant lipoprotein,in a human-like lipoprotein profile, while Mortensen et al used the Apoe−/− mouse, which is characterized primarily by mouse apoB-48 containing remnant lipoproteins. Overall, the top-line results of the two studies, which used very different mouse atherosclerosis models, are highly comparable, whereas the mechanisms responsible for the reduced atherosclerosis may be complex and multifactorial.
In summary, our findings indicate that SORT1 deficiency in macrophages reduces LDL uptake and macrophage cholesterol loading independent of the LDL receptor or macropinocytosis, and protects against the development of atherosclerosis. The macrophage sortilin pathway is a novel pathway of macrophage cholesterol loading that quantitatively contributes to atherosclerosis.
Supplementary Material
Novelty and Significance.
What Is Known?
Genetic variants at the SORT1 locus are associated with plasma low density lipoprotein cholesterol (LDL-C) levels as well as myocardial infarction and coronary artery disease (CAD).
The gene SORT1 encodes a protein called sortilin, which in hepatocytes can mediate the uptake and degradation of LDL.
What New Information Does This Article Contribute?
Deletion of sortilin in macrophages reduces foam cell formation and atherosclerosis without influencing plasma LDL-C levels.
Macrophage sortilin is a new receptor-mediated pathway promoting uptake of native LDL by macrophages promoting foam cell formation and atherosclerosis.
The mechanisms linking genetic variation at the SORT1 locus with CAD have not been fully elucidated. Using atherosclerosis-prone mice with a human-like lipoprotein profile, we found that deletion of sortilin in the whole body or specifically in macrophages substantially reduced atherosclerosis without affecting plasma cholesterol. We found that macrophage sortilin mediates the uptake of native (unmodified) LDL leading to foam cell formation, a novel receptor-mediated process for macrophage uptake of LDL. This pathway could potentially be targeted as an approach to reducing risk of atherosclerosis.
Acknowledgments
We would like to thank Aisha Wilson, Edwige Edoard, Mao-Sen Sun, Amrith Rodrigues and Antonino Picataggi for animal/cell help; Kiran Musunuru and Qiurong Ding for human SORT1 and GFP plasmids. Deborah Cromley for help with lipid measurements; Margaret Nickel and Kevin Trindade for LDL isolation; Human Immunology Core for running Bioplex Pro Assay; Karen Vo for help with Hemavet; Edward Fisher, Peter Davies, Ellen Pure, Richard Assoian, Sissel Lund-Katz, Michael Phillips, Yanqing Gong, Robert Bauer, and Sony Tuteja-Stevens for scientific discussions.
SOURCES OF FUNDING: This work was supported by R01HL109489 from the NHLBI (DJR) and T32GM008076-30 from the NIGMS (KP).
Nonstandard Abbreviations and Acronyms
- LDL
low-density lipoprotein cholesterol
- LDLR
low-density lipoprotein receptor
- Tg
transgenic
- ApoB
Apolipoprotein B
- CAD
coronary artery disease
- GFP
green fluorescent protein
- GWAS
genome wide association studies
Footnotes
DISCLOSURES
None.
References
- 1.Barthwal MK, Anzinger JJ, Xu Q, Bohnacker T, Wymann MP, Kruth HS. Fluid-phase pinocytosis of native low density lipoprotein promotes murine m-csf differentiated macrophage foam cell formation. PloS one. 2013;8:e58054. doi: 10.1371/journal.pone.0058054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinberg D. The ldl modification hypothesis of atherogenesis: An update. Journal of lipid research. 2009;50(Suppl):S376–381. doi: 10.1194/jlr.R800087-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. The New England journal of medicine. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kathiresan S, Voight BF, Purcell S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nature genetics. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willer CJ, Sanna S, Jackson AU, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nature genetics. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kathiresan S, Melander O, Guiducci C, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nature genetics. 2008;40:189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musunuru K, Strong A, Frank-Kamenetsky M, et al. From noncoding variant to phenotype via sort1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kjolby M, Andersen OM, Breiderhoff T, Fjorback AW, Pedersen KM, Madsen P, Jansen P, Heeren J, Willnow TE, Nykjaer A. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell metabolism. 2010;12:213–223. doi: 10.1016/j.cmet.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Strong A, Ding Q, Edmondson AC, Millar JS, Sachs KV, Li X, Kumaravel A, Wang MY, Ai D, Guo L, Alexander ET, Nguyen D, Lund-Katz S, Phillips MC, Morales CR, Tall AR, Kathiresan S, Fisher EA, Musunuru K, Rader DJ. Hepatic sortilin regulates both apolipoprotein b secretion and ldl catabolism. The Journal of clinical investigation. 2012;122:2807–2816. doi: 10.1172/JCI63563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strong A, Patel K, Rader DJ. Sortilin and lipoprotein metabolism: Making sense out of complexity. Current opinion in lipidology. 2014;25:350–357. doi: 10.1097/MOL.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Canuel M, Bhattacharyya N, Balbis A, Yuan L, Morales CR. Sortilin and prosaposin localize to detergent-resistant membrane microdomains. Experimental cell research. 2009;315:240–247. doi: 10.1016/j.yexcr.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 13.Canuel M, Korkidakis A, Konnyu K, Morales CR. Sortilin mediates the lysosomal targeting of cathepsins d and h. Biochemical and biophysical research communications. 2008;373:292–297. doi: 10.1016/j.bbrc.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 14.Wahe A, Kasmapour B, Schmaderer C, Liebl D, Sandhoff K, Nykjaer A, Griffiths G, Gutierrez MG. Golgi-to-phagosome transport of acid sphingomyelinase and prosaposin is mediated by sortilin. Journal of cell science. 2010;123:2502–2511. doi: 10.1242/jcs.067686. [DOI] [PubMed] [Google Scholar]
- 15.Wong I, Liao H, Bai X, Zaknic A, Zhong J, Guan Y, Li HY, Wang YJ, Zhou XF. Probdnf inhibits infiltration of ed1+ macrophages after spinal cord injury. Brain, behavior, and immunity. 2010;24:585–597. doi: 10.1016/j.bbi.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Kruth HS. Receptor-independent fluid-phase pinocytosis mechanisms for induction of foam cell formation with native low-density lipoprotein particles. Current opinion in lipidology. 2011;22:386–393. doi: 10.1097/MOL.0b013e32834adadb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, Silver JM, McKee M, Freeman MW. Loss of receptor-mediated lipid uptake via scavenger receptor a or cd36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. The Journal of clinical investigation. 2005;115:2192–2201. doi: 10.1172/JCI24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manning-Tobin JJ, Moore KJ, Seimon TA, Bell SA, Sharuk M, Alvarez-Leite JI, de Winther MP, Tabas I, Freeman MW. Loss of sr-a and cd36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:19–26. doi: 10.1161/ATVBAHA.108.176644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mortensen MB, Kjolby M, Gunnersen S, Larsen JV, Palmfeldt J, Falk E, Nykjaer A, Bentzon JF. Targeting sortilin in immune cells reduces proinflammatory cytokines and atherosclerosis. The Journal of clinical investigation. 2014;124:5317–5322. doi: 10.1172/JCI76002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tangirala RK, Tsukamoto K, Chun SH, Usher D, Pure E, Rader DJ. Regression of atherosclerosis induced by liver-directed gene transfer of apolipoprotein a-i in mice. Circulation. 1999;100:1816–1822. doi: 10.1161/01.cir.100.17.1816. [DOI] [PubMed] [Google Scholar]
- 21.Kassim SH, Li H, Vandenberghe LH, Hinderer C, Bell P, Marchadier D, Wilson A, Cromley D, Redon V, Yu H, Wilson JM, Rader DJ. Gene therapy in a humanized mouse model of familial hypercholesterolemia leads to marked regression of atherosclerosis. PLoS One. 2010;10:e13424. doi: 10.1371/journal.pone.0013424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kassim SH, Li H, Bell P, Somanathan S, Lagor W, Jacobs F, Billheimer J, Wilson JM, Rader DJ. Adeno-associated virus serotype 8 gene therapy leads to significant lowering of plasma cholesterol levels in humanized mouse models of homozygous and heterozygous familial hypercholesterolemia. Hum Gene Ther. 2013;24:19–26. doi: 10.1089/hum.2012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Somanathan S, Jacobs F, Wang Q, Hanlon AL, Wilson JM, Rader DJ. AAV vectors expressing LDLR gain-of-function variants demonstrate increased efficacy in mouse models of familial hypercholesterolemia. Circ Res. 2014;115:591–599. doi: 10.1161/CIRCRESAHA.115.304008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.