

Introduction
Many lines of research have established a relationship between the gut microbiome and patients with liver disease. For example, patients with cirrhosis have increased bacteremia, increased blood levels of lipopolysaccharide (LPS), and increased intestinal permeability. Patients with cirrhosis have bacterial overgrowth in the small intestine. Selective intestinal decontamination with antibiotics is beneficial for patients with decompensated cirrhosis.
In experimental models of chronic liver injury with fibrosis, several toll-like receptors (TLR) are required to make mice sensitive to liver fibrosis. The presumed ligand for the TLRs are bacterial products derived from the gut microbiome, and TLR knockout mice are resistant to liver inflammation and fibrosis.
We and others have characterized the association between pre-clinical models of liver disease in mice with the microbial diversity in their gut microbiome. In each model, including intra-gastric alcohol, bile duct ligation, chronic carbon tetrachloride (CCL4), administration and genetic obesity, there is a significant change in the gut microbiome from normal control mice. However, there is not a single clear bacterial strain or pattern that distinguish mice with liver injury from controlled mice [1].
So how can the gut microbiota affect liver disease? We can identify at least six (6) changes that would result in liver injury, inflammation, and/or fibrosis. These include: 1) changes in caloric yield of diet; 2) regulation of gut permeability to release bacterial products; 3) modulation of choline metabolism; 4) production of endogenous ethanol; 5) regulation of bile acid metabolism, and; 6) regulation in lipid metabolism.
Changes in caloric yield of diet
Many different type of experiments using gnotobiotic (germ-free) mice have determined that different gut microbiota can extract and yield different caloric contents to their host. One of the most dramatic studies[2] demonstrated that the obese phenotype of the human donor can be replicated by a fecal microbiota transplant into a gnotobiotic mouse. In each case, twins that are disparate for weight transmitted their lean or obese phenotype to germ free (gnotobiotic) mice through fecal transplant of their respective microbiota.
Regulation of gut permeability to release bacterial products
This has been an area of intense research in which different TLRs have been assessed for their roles of liver disease. For each TLR, the presumed ligand is a bacterial product from the gut microbiota. In particular, hepatic stellate cells (HSC) have TLRs 2, 4, and 9. Kupffer cells, the resident macrophage in the liver, have TLRs 2, 3, 4, and 9, and hepatocytes have TLRs 1-9. Since increased intestinal permeability will result in bacterial products in the portal blood, the cells of the liver will be first to be exposed to these ligands. An earlier observation for the importance of TLRs in chronic liver disease was the observation that TLR4 mutant mice have reduced liver fibrosis following chronic CCl4 or BDL[3]. The ligand for TLR4 is the LPS on gram negative bacteria. Furthermore, reducing the gut microflora by a non-absorbable antibiotic cocktail decreases plasma LPS levels in liver injury and results in decreased hepatic injury and fibrosis. This again is consistent with the LPS ligand interacting with the TLR4 receptor and resulting in inflammation and fibrosis in the liver. A series of experiments in these mice is consistent with the model that LPS from the gut microflora activates the TLR4 on the hepatic stellate cells (HSCs). This results in down regulation of the TGF beta pseudoreceptor BAMBI. By removing BAMBI, the HSCs are now responsive to TGF beta produced by Kupffer cells and/or recruited macrophage which then activates the HSCs to become myofibroblast and result in myofibroblast proliferation and extracellular matrix production, resulting in liver fibrosis (Figure 1).
Figure 1.
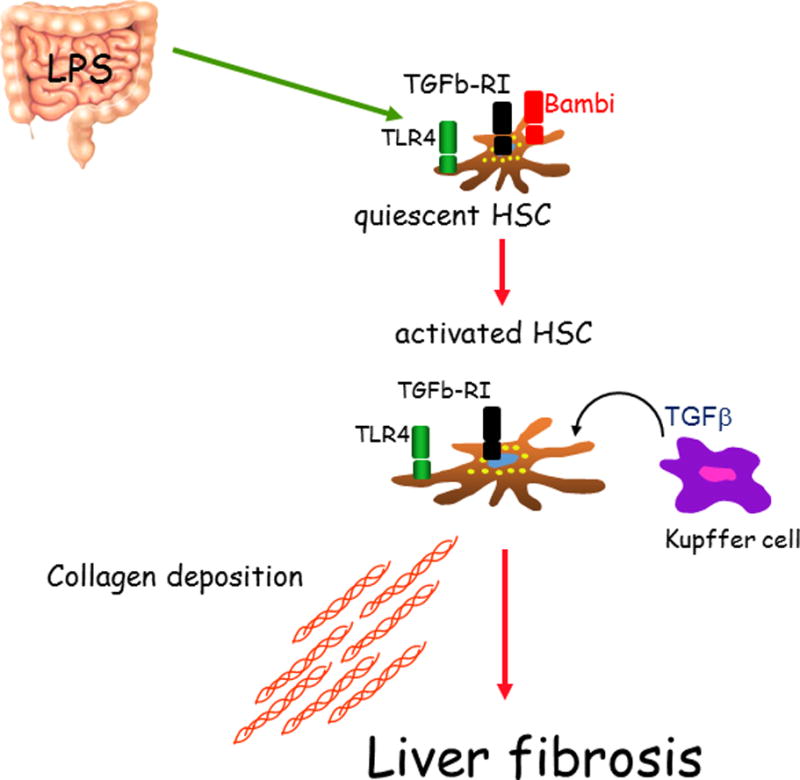
A proposed model by which in the binding of LPS to its receptor TLR4 leads to liver fibrosis.
A ligand for TLR9 is the CpG DNA from bacteria, and TLR9 is required for liver fibrosis but through a different mechanism than TLR4. Using a model of choline deficient/amino acid supplemental diet (CDAA) as a model for non-alcoholic steatohepatitis (NASH), we demonstrated that TLR9 knockout mice were resistant to hepatic steatosis, inflammation and fibrosis. A series of studies are consistent with the model that the TLR ligands (bacterial DNA) activate TLR9 on Kupffer cells/recruited macrophage. This results in the release of IL-1 beta, which in turn activate the IL-1 receptor on HSCs, resulting in their activation. The IL-1 receptor on hepatocytes is also activated, resulting in lipid accumulation, insulin resistance, and hepatocyte injury. The combined results of macrophage IL-1 beta on hepatocytes and HSCs produce hepatic injury, inflammation, and fibrosis[4].
Similarly, TLR2 is required for hepatic fibrosis following common bile duct ligation (BDL). Again, the mechanism is different from TLR4 and TLR9. TLR2 knockout mice have less bacterial translocation following BDL[5]. This is because TLR2 is required for increased intestinal permeability, presumably through activation by bacterial cell wall ligands. A series of experiments demonstrated that TLR2 on intestinal monocytes is activated by ligand resulting in production of TNF alpha. TNF alpha in turn activates the TNRF 1 on intestinal epithelial cells. This produces the previously demonstrated activation of Rho A, phosphorylation of MLC, and then increased intestinal permeability. Then, increased bacterial products from the gut microflora are available to the liver to act as ligands to activate injury, inflammation, and fibrosis.
Modulating choline metabolism
Several lines of evidence demonstrate the importance of the host gut microbiome in choline metabolism and subsequent liver injury. In particular, choline deficient diets in mice produce NASH, as discussed above. The choline metabolizing enzyme in the host PEMT converts choline into phosphatidylcholine. Phosphatidylcholine in turn is required for VLDL secretion in hepatocytes. Thus if choline is deficient, then phosphatidylcholine is decreased, and lipids accumulate in the hepatocytes. Interestingly, there are SNPs (single nucleotide polymorphisms in the human PEMT gene that result in an enzyme that produces less product, and these SNPs are associated with NAFLD.
Finally, choline is metabolized by certain gut bacteria to TMA, making it unavailable for the required synthesis across of phosphatidylcoholine. Therefore, the gut microbiota can produce a relative choline deficiency, resulting in a hepatic steatosis (NAFLD).
An interesting study[6], demonstrated that when volunteers were placed on a choline deficient diet, those people with the PEMT SNP with deceased activity had increased risk of developing a choline deficient fatty liver. In addition, the microbiota from the people with fatty liver was different from the microbiota from patients who did not develop fatty liver. This demonstrates an interaction between the gut microbiome and the host genome in the development of NAFLD.
Production of endogenous ethanol
Using principal component analysis, the gut microbiota of normal weight children, obese children, and children with NASH can be distinguished [7]. Measurement of blood ethanol levels in these children demonstrated increased ethanol in the children with NASH compared to the non-obese children and the obese children without NASH. The somewhat controversial hypothesis is that these gut microbiota in the children with NASH produce endogenous ethanol, and this ethanol can induce NASH.
Regulation of bile acids
The gut microbiota is responsible for deconjugating bile acids and converting primary bile acids into secondary bile acids. Cirrhotics have a lower abundance of bacteria with 7-alpha dehydroxylating activity, the enzyme which converts primary to secondary bile acids. Bile acids in turn have direct actions on bacteria (bacteriastatic), and also act as ligands for FXR and TGR5. Therefore, the metabolism of bile acids by gut microbiota can result in changes in signaling which can result in hepatic injury.
Regulation of liver metabolism
An early observation was that mice in a germ-free environment (gnotobiotic mice) gained less weight than conventional mice on the same diet[8]. Analysis of the mRNA and protein levels in these mice demonstrated that the conventional gut microbiota promotes fat storage by suppressing the enzyme FIAF in the intestine. FIAF in the intestinal epithelium suppresses LPL activity which decreases triglyceride storage in adipocytes. Therefore, when the gut microbiota suppresses FIAF, there is increased LPL activity and increased triglyceride storage resulting in weight gain.
Alcoholic liver disease
There is a long history of studying the role of the gut microflora in alcoholic liver disease. Therefore, the most extensively studied preclinical model of liver injury and fibrosis is the intragastric ethanol-feeding model of alcoholic liver disease in mice. Enteric alcohol administration increases intestinal bacterial overgrowth[9]. This intragastric ethanol feeding also changes the microbial diversity of the gut microbiome, resulting in fewer Firmicutes and more Bacteroidetes. In particular, the probiotic Lactobacillus is greatly decreased in the alcohol model. Alcohol causes a decrease in the intestinal antimicrobial peptides, in particular Reg 3g. However, treatment with a prebiotic FOS improves alcoholic steatohepatitis by inducing Reg 3g expression and reducing intestinal bacterial overgrowth.
Chronic ethanol administration reduces the bacterial biosynthesis of saturated long chain fatty acids (LCFAs). This was determined by a metagenomic analysis of the gut microbiome in mice treated with chronic alcohol administration or controls. Many of the enzymes in the biosynthetic pathway of the saturated long chain fatty acids are decreased in the alcohol fed mice. Metabolomic analysis of the intestinal contents of these mice demonstrated that, in fact, there was a decrease in the saturated long chain fatty acids including those long chain fatty acids that can only be produced by bacterial synthesis. Supplementation with saturated long chain fatty acids to the mice receiving the intragastric ethanol resulted in a decrease of alcoholic liver disease. This was manifested by a decrease in steatosis of the liver, decreased ALT, and decreased oxidative stress. Supplementation with long chain fatty acids also decreased the alcohol-induced intestinal permeability. Furthermore, supplementation of the diet with long chain fatty acids, reversed the alcohol induced dysbiosis, and in particular resulted in increased Lactobacillus.
In patients who are abusing alcohol, the bacterial genes that regulate fatty acid synthesis are decreased, just as in the preclinical model. Furthermore, in alcoholics, the concentration of Lactobacillus in the gut microbiome is dependent on the concentration of the saturated long chain fatty acids[10]. This has led to a model in which chronic ethanol administration results in the decreased synthesis of saturated long chain fatty acids by bacteria. The decrease of the saturated long chain fatty acids results in dysbiosis, including decreased lactobacilli, which use saturated long chain fatty acids as fuel. This dysbiosis with decreased anti-inflammatory bacteria results in intestinal inflammation and gut permeability dysfunction. This produces translocation of bacterial products from the intestinal lumen into the portal blood and liver which produces alcoholic liver disease.
Liver injury in gnotobiotic mice
In mice in a gnotobiotic environment, liver injury induced by either carbon tetrachloride or TAA results in increased liver fibrosis compared to mice with conventional gut microbiola[11]. Similarly, Myd88/Trif double knockout mice are more susceptible to CCl4 induced liver fibrosis. From these two experiments, we concluded that the conventional gut microbiota have a protective role in liver injury even though large quantities of bacterial products are powerful induces of liver injury.
In conclusion, experimental and clinical liver diseases are associated with bacterial overgrowth, dysbiosis, and increased intestinal permeability. Bacterial products from the gut activate TLR 2, 4, and 9 to promote inflammation, steatosis, and fibrosis. Bacterial enzymes modify bile acids that effect the liver via FXR and TGR5. Experimental alcoholic liver disease is characterized by inhibition of the host antimicrobial peptide Reg3g and dysbiosis with decreased synthesis of long chain fatty acids. Experimental alcoholic liver disease is improved by a diet supplemented with a prebiotic FOS, with saturated long chain fatty acids, or with a probiotic lactobacillus rhamnosus GG. Overall, conventional gut microbiota have a protective role in liver injury.
Acknowledgments
Supported by National Institutes of Health (R01 GM041804-26, P50 AA011999-16, P42 ES010337-13, U01 AA021856-02, R01 DK101737-01A1, U01 AA022614-01A1, R01 DK099205-01A1; Schnabl: R01 AA020703, U01 AA02185).
References
- 1.Fouts DE, Torralba M, Nelson KE, et al. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J Hepatol. 2012;56(6):1283–1292. doi: 10.1016/j.jhep.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ridaura VK, Faith JJ, Rey FE, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341(6150):1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13(11):1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 4.Miura K, Kodama Y, Inokuchi S, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139(1):323–334. e327. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartmann P, Haimerl M, Mazagova M, et al. Toll-like receptor 2-mediated intestinal injury and enteric tumor necrosis factor receptor I contribute to liver fibrosis in mice. Gastroenterology. 2012;143(5):1330–1340. e1331. doi: 10.1053/j.gastro.2012.07.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spencer MD, Hamp TJ, Reid RW, et al. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology. 2011;140(3):976–986. doi: 10.1053/j.gastro.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu L, Baker SS, Gill C, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57(2):601–609. doi: 10.1002/hep.26093. [DOI] [PubMed] [Google Scholar]
- 8.Backhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101(44):15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan AW, Fouts DE, Brandl J, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology. 2011;53(1):96–105. doi: 10.1002/hep.24018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen P, Torralba M, Tan J, et al. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology. 2015;148(1):203–214. e216. doi: 10.1053/j.gastro.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazagova M, Wang L, Anfora AT, et al. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J. 2015;29(3):1043–1055. doi: 10.1096/fj.14-259515. [DOI] [PMC free article] [PubMed] [Google Scholar]