

Abstract
Genetically engineered mouse models (GEMMs) of cancer have affected virtually all areas of cancer research. However, the accelerated discovery of new cancer genes emerging from large-scale cancer genomics and new chemical entities pouring from the drug discovery pipeline have strained the capacity of traditional germline mouse models to provide crucial insights. This Review introduces new approaches to modelling cancer, with emphasis on a growing collection of non-germline GEMMs (nGEMMs). These offer flexibility, speed and uniformity at reduced costs, thus paving the way for much needed throughput and practical preclinical therapeutic testing models.
Genetically engineered mouse models (GEMMs) have been the mainstay of basic cancer biology research for the past two decades. Germline models allow for testing and understanding the mechanisms of oncogenic transformation, as well as for probing the kinetics and therapeutic responses of autochthonous tumours in an intact microenvironment1. Although most inherited cancer syndromes have been recapitulated with germline GEMMs, mimicking spontaneous tumorigenesis has become possible only in the past decade with the development of sophisticated multi-allelic mouse strains that have been guided by an improved understanding of the genetics of human counterparts.
Both the traditional and more advanced germline GEMMs have greatly contributed to understanding the different phases of tumorigenesis: initiation, maintenance, progression and regression (including minimal residual disease)2,3. Additional improvements include the development of germline inducible models in which a genetic element is activated only in a small proportion of cells in a specified tissue, usually by exogenous chemicals or viruses. Such knock-in systems enable the modelling of the stochastic nature of tumorigenesis4,5. However, even the most advanced germline GEMMs are not easily adapted to high-throughput translational research and drug testing owing to long timelines, difficulties in creating large cohorts in a short time frame, cost and space. In this Review, we discuss the advent and advantages of non-germline GEMMs (nGEMMs) for translational cancer research, with an emphasis on cancer gene validation and preclinical therapeutics.
Useful mouse models for translational research need to replicate the genetics and genomics, the context and the heterogeneity of human tumours. With respect to human relevance and predictability, a model should be driven by signature genetic events, with tumours occurring de novo in an immune-competent microenvironmental context, harbouring spontaneously acquired syntenic genomic alterations and variations representative of the heterogeneity, as well as exhibiting relevant clinical behaviour such as disease pathology and sites of metastasis. Furthermore, such models should develop the relevant tumours with high penetrance, reproducibility and synchronous kinetics, and should also be amenable to imaging modalities such as positron emission tomo graphy (PET), magnetic resonance imaging (MRI) and/or computerized tomography (CT), thus offering ease of cohorting for monitoring tumour growth and regression in live animals in multi-arm preclinical trial studies. Finally, particularly with respect to drug response testing, such models should ideally recapitulate the genomic instability characteristics of human cancers.
Traditional germline GEMMs and their limitations
Despite their success in modelling human cancers for biological studies, challenges remain for the widespread and economic use of germline GEMMs for preclinical therapeutic research and development. First, GEMMs have a long lead-time to generation and modification, and require the targeting of embryonic stem cells (ESCs), the generation of chimeras, germline transmission, complex intercrosses and colony expansion, as well as characterization of the phenotypes – a process that typically takes several years. Additionally, not all and in some cases only a minority of the mice generated from breeding will inherit the desired genotypes, owing to the multi-allelic intercrosses required to model the complex genetics of human cancers. This problem can be further exacerbated in cases in which homozygosity is lethal. Second, the vast majority of models have incomplete penetrance, coupled with a non-synchronous and often prolonged latency to tumour emergence, creating logistical and financial barriers for their use in preclinical therapeutic studies. Last, many GEMMs exhibit heterogeneity in their tumour phenotypes, including tissue type and location, which may or may not be desirable, and which increases the colony size that is required to generate statistically meaningful cohorts.
Even in situations in which all of these desirable features are achieved in a GEMM, limitations remain. For example, the adenomatosis polyposis coli (Apc)Min mouse accurately models a mutation that initiates 80% of human colon cancer, and the mice exhibit a highly replicable tumour multiplicity with a short (2-month) latency6. However, as the mutation is present in all tissues from birth, the ApcMin mouse only models tumorigenesis in patients with familial adenomatous polyposis (FAP), which represents less than 5% of all the patients with colorectal cancer. Although a wide variety of drug and supplement studies have been successfully carried out using the ApcMin mouse model7, their direct applicability to sporadic human colon cancer is unknown.
Despite these shortcomings, several germline GEMMs have proved useful for improving the understanding of tumour initiation and progression; these include the breakpoint cluster region (BCR)-ABL1 mouse model2, which enabled early translational drug studies for chronic myelogenous leukaemia. New concepts and understandings of therapeutic consequences have been elucidated in GEMMs. For example, experiments in a mouse model of pancreatic islet cell tumours have led to a new dosing schedule of standard chemotherapy combined with anti-angiogenic therapy8. In addition, the mouse model for mutS homologue 6 (MSH6) deficiency revealed an additional aspect of hereditary non-polyposis colorectal cancer (HNPCC) — namely, replication error-negative, lymphoma-correlated gastrointestinal tumours — that had previously not been captured in the traditional Amsterdam criteria and which is now being used in the clinic9. In summary, germline GEMMs of cancer have provided a useful starting point for disease modelling, but improvements are needed to facilitate their use as preclinical models for translational science.
Mosaic or conditional models: enhanced GEMMs
Mosaic models or conditional GEMMs are an extension of germline models in which a latent allele is pheno typically wild type until stimulated in a tissue- and time-specific manner with exogenous chemicals or viruses. These models include the Cre-lox and Flp-Frt systems (FIG. 1). Cre-lox models harbour genetic elements flanked by loxP sites that are modified by Cre recombinase. Traditionally, these alleles become constitutively activated or inactivated in compound transgenic animals by Cre recombinase under the control of a tissue-specific promoter. To bypass this constitutive feature, delivery of Cre can be temporally controlled in two different ways to result in the activation or inactivation of the LoxP alleles. In the first system, Cre can be delivered with a virus directly to an organ — as infectivity of the virus will not be 100%, a genetic mosaic will be created in the target tissue. For example, using adenovirus as the vector, Cre mosaic cancer models for lung and ovary have been established using a Kras lox-STOP-lox allele10–13 (FIG. 2). These mosaic systems have the advantage of reduced tumour burden, which typically leads to a prolonged lifespan that allows for advanced tumour progression compared with traditional GEMMs — a feature that can be useful for preclinical studies. Indeed, the KRAS-driven lung model has been effectively used to demonstrate the efficacy of the pan-PI3K inhibitor BEZ235 (REF. 14). However, the disadvantages of such viral delivery approaches include the unpredictability of delivery and efficiency, as well as the limited accessibility of certain target tissues for infection.
Figure 1. Conditional genetically engineered mouse models (GEMMs).
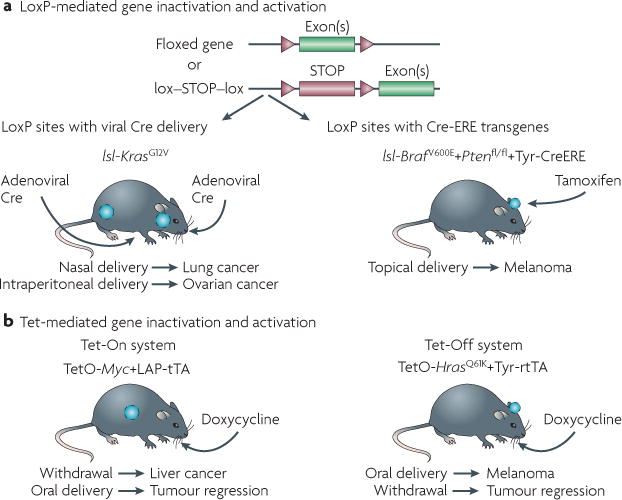
a | Activating Cre in a tissue-specific manner, by either viral delivery or Cre-oestrogen response element (ERE)-mediated tamoxifen administration. Examples are shown for lung cancer7 and ovarian cancer12, and melanoma11. b | Tetracycline (Tet)-mediated transgene inactivation or activation by doxycycline administration or withdrawal using the Tet-On or Tet-Off systems. Examples are shown for liver cancer22 and melanoma5. fl, flox; lsl, lox-STOP-lox; rtTA, reverse transactivator; tTA, transactivator.
Figure 2. Chimeric model generation.
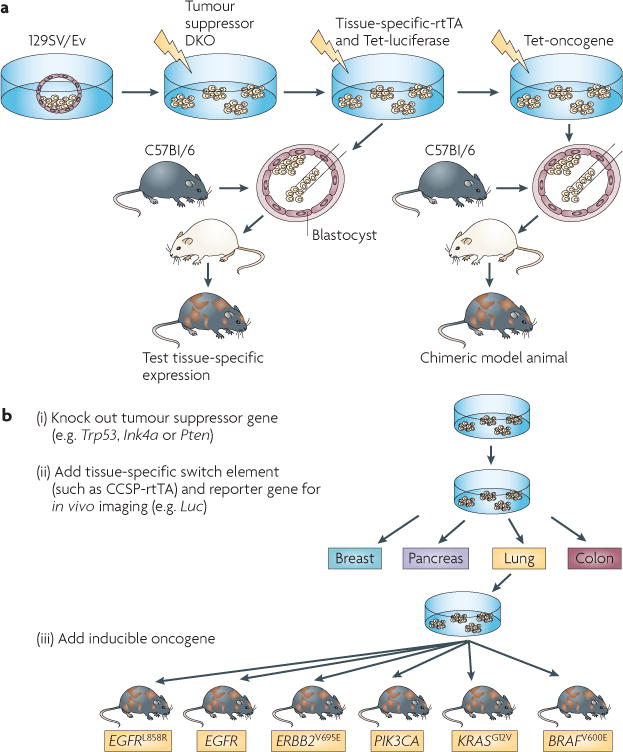
a | Genetically modified embryonic stem cells are transduced ex vivo with conditional oncogenic transgenes, injected into blastocysts and implanted into pseudopregnant mice to generate chimeras. b | By activating the respective transgenes in a tissue-specific manner, multiple tumour types can be induced in these chimeras in the context of normal stromal tissue derived from the wild-type blastocysts. Image is modified, with permission, from Nature Biotechnology REF. 31 © (2010) Macmillan Publishers Ltd. All rights reserved. DKO, double knockout; EGFR, epidermal growth factor receptor; Luc, luciferase; rtTA, reverse transactivator; Tet, tetracycline.
A second system makes use of a fusion of oestrogen response elements (EREs) to Cre15: the injection or application of tamoxifen analogues to the mouse activates Cre in both a tissue-specific and a time-specific manner. For example, a tyrosinase-driven CreERT2 allele excises lox-STOP-lox elements in a melanocyte-dependent manner when tamoxifen is topically administered16; this has been used to develop mouse models of melanoma17,18. These models illustrate the usefulness of the CreERT2 allele, as widespread tamoxifen application results in massive tumorigenesis across the whole mouse skin, whereas focal application results in well-defined and predetermined sites of tumour formation. This not only increases the lifespan of the mice for preclinical therapeutic analyses, but also provides biological reproducibility. Finally, the analogous Flp-Frt system allows for the possibility of two different recombinases (Cre and Flp) to be used in the same mouse for targeting different elements by excision in a temporally independent manner. Although this has not yet been extended to the manipulation of multiple genes in the same mouse, this dual system has been used in vivo to remove unwanted engineered elements, such as neomycin, from integrated targeting vectors19.
Another layer of control is provided by the flexible Tet-On and Tet-Off systems20. Each system is composed of two parts: the Tet operon promoter (TetO) that regulates the expression of the gene of interest, and either the transactivator (tTA) or the reverse transactivator (rtTA) transgenes, which are capable of binding to and regulating TetO. In the Tet-Off system, TetO is constitutively bound by tTA, which stimulates the expression of the gene of interest: when a tetracycline analogue (most commonly doxycycline) is introduced, it binds the tTA and prevents its interaction with TetO, thus shutting off expression of the gene. In the Tet-On system, rtTA is unable to bind TetO by itself, leaving the gene switched off. In the presence of doxycycline, rtTA binds to TetO and the gene is expressed. Therefore, the temporal control of gene expression is achieved by choosing when to feed the mouse doxycycline, and tissue specificity is governed by the promoter driving tTA or rtTA expression. The regulated induction of oncogene expression through doxycycline administration to mice that have undergone thymic maturation also mimics the somatic acquisition of genetic alterations that could otherwise be recognized by the immune system as foreign antigens. Unlike the Cre-lox system (which is irreversible after recombination) the Tet systems are reversible by withdrawal of doxycycline. This has also enabled the demonstration of a requirement for tumour maintenance in which, on doxycycline withdrawal, the transcriptional extinction of an oncogene such as HRAS or MYC leads to the almost complete regression of melanoma5 and lymphoma21, and liver22 or breast cancer23 (FIG. 2).
A variation on the mosaic models is the replication-competent avian leukosis virus long terminal repeat with splice acceptor (RCAS) avian retrovirus receptor system. Transgenic animals expressing the chicken receptor in a tissue-specific manner using tissue-specific promoters are infected with the RCAS virus that carries the oncogene of interest, sometimes inducible with the Tet system. Using the RCAS system, a series of brain tumour models, including models incorporating sonic hedgehog (SHH)24 or platelet-derived growth factor (PDGF)25, have been produced and used effectively in preclinical studies. For example, a vascular endothelial growth factor receptor (VEGFR) inhibitor25 or perifosine, a dual MAPK and AKT inhibitor, have been used in the PDGF model26. Similarly, lung cancer models that are driven by epidermal growth factor receptor (EGFR) mutations have been generated27, and erlotinib resistance in tumours carrying the EGFR-T790M mutation has been confirmed28. To overcome the limitations of the RCAS viral system, lentiviruses have recently been used for the somatic delivery of genes or switch elements29.
Overall, conditional GEMMs provide improved flexibility, statistical power and accuracy compared with the standard germline model design. However, despite their potential usefulness, a major hurdle for both traditional and conditional GEMMs has been the tremendous breeding efforts that are necessary to establish large cohorts for drug treatments that have enough power for statistical significance. Only a few drug development companies have established the means of producing such large cohorts of GEMMs. Nevertheless, when preclinical trials were carried out in lung cancer GEMMs, the results proved to be comparable to findings from human clinical trials that had parallel designs30, thus validating the use of GEMMs for therapeutic studies.
Non-germline GEMMs
The recent development of nGEMMs of cancer in various systems, including the lung31, breast32, liver and haematopoietic organs33–35, has revealed new insights into tumorigenesis. These models have been generated through the establishment of chimeric mice that develop spontaneous tumours in a tissue-specific manner or by the transplantation of cells (TABLE 1). Chimeric models require the implantation of genetically engineered ESCs into pre-implantation embryos, leading to chimeras that carry a mixture of predisposed cells that are derived from the ESCs and from wild-type host cells. In these settings, tumours develop in the context of normal tissue, recapitulating human tumorigenesis. Transplantation models are generated by implanting normal, stem or tumour cells into the respective adult tissue. The transplanted cells can be derived either from genetically engineered donor mice harbouring cancer-predisposing mutations or from mouse or human cells that have first undergone ex vivo engineering. Several of these systems have emerged with features that render them particularly useful for translational studies, as discussed below.
Table 1.
Comparison of the key features of GEMMs and nGEMMs
| Characteristics | Traditional GEMM | Conditional GEMM | Chimeric models | Mouse in mouse | Classic xenograft | Human in mouse |
|---|---|---|---|---|---|---|
| Penetrance* | Model-dependent | Model-dependent | Usually high | Usually high | Usually high | Low |
| Synchronicity‡ | Model-dependent, usually poor | Model-dependent, can be high | Model-dependent, can be high | High | High owing to cohorting | Poor |
| Replicability§ | Model-dependent | Model-dependent, can be high | High | High | Often depends on passaging conditions | Model-dependent |
| Immunocompetent host‖ | Yes | Yes | Yes | Either | No | No |
| Rapid cohort generation¶ | Model-dependent, usually no | Model-dependent, usually no | Yes | Yes | Yes | Dependent on human tissue donations |
| Relevant genetic aberrations | Can be engineered | Can be engineered | Can be engineered | Can be engineered | Confounded by culturing artefacts | Can be engineered |
| Relevant microenvironment# | Yes | Yes | Yes | Yes or partial | Possible by addition of stroma | Possible by addition of stroma |
| Familial or spontaneous model** | Familial | Spontaneous | Spontaneous | Spontaneous | Neither | Spontaneous |
| Genome instability‡‡ | Generally no, except when engineered specifically with mutations that cause genomic instability | Generally no, except when engineered specifically with mutations that cause genomic instability | Likely | Generally no, except when engineered specifically with mutations that cause genomic instability | Yes, but can be confounded with culturing artefacts | Yes |
GEMM, genetically engineered mouse model; nGEMM, non-germline GEMM.
Penetrance is the number of mice developing a relevant tumour.
Synchronicity is low variance in tumour latency.
Replicability is similarity in tumour phenotypes within a cohort.
Immunocompetent host is whether the host has an intact immune system.
Rapid cohort generation is the generation of tumour-bearing mice within a short time period.
Relevant microenvironment is the similarity of the surrounding stromal tissue to that in a human tumour.
Familial or spontaneous model is the type of human cancer being modelled.
Genome instability is the presence or absence of genomic instability in the tumours.
Chimeric models
The use of chimeric models is a new approach for improving clinical relevance, in which developing cancer cells are seeded in the context of normal surrounding tissue. This feature overcomes the genetic field effect of expressing an oncogene in the whole tissue of an adult animal, a limitation that is inherent to the GEMMs. The development of chimeric models begins with engineering tetracycline-inducible cancer-relevant alleles and imaging markers exclusively into mouse ESCs. For example, Zhou et al.31 described a model in which both alleles of the Cdkn2a locus (which encodes the tumour suppressors INK4A and ARF) were deleted and a tissue-specific rtTA, tetracycline-dependent oncogenes and a luciferase imaging marker were engineered into the ESCs. Injecting these genetically engineered ESCs into wild-type host blastocysts generated cancer-prone chimeric mice in which specific tumour types developed in the context of normal stroma31 – a situation that mirrors cancer development in humans. The importance of stromal influence on tumorigenesis is increasingly recognized36,37, making this an even more attractive feature of a chimeric model. Further tissue and temporal specificity can be achieved by the localized or timed application of induction agents.
As the transgenic alleles are engineered ex vivo, the generation of chimeric models does not require large breeding cohorts. They can therefore be produced rapidly and at a reduced cost compared with standard GEMMs. Even after the establishment of a GEMM, multi-allelic GEMMs often require management of heterozygous and homozygous breeding, leading to laborious genotyping efforts, the necessary generation of unwanted genotypes, considerable time investments and a substantial amount of space requirement, all of which add to the already challenging costs that are associated with mouse studies. It is therefore of no surprise that only a few studies have used GEMM breeding schemes involving four or more modified alleles38,39. By contrast, chimeric models considerably reduce the number of animals generated, as all mutations and allele modifications can be carried out ex vivo in ESCs, which can be rapidly expanded as well as readily frozen for storage in a cost-efficient manner. Producing the actual chimeras requires only generating or purchasing the host blastocysts, thawing the ESCs and injecting them into the blastocysts and housing the resulting chimeras. Therefore, large study cohorts can be generated in a time-, labour- and cost-effective manner.
In principle, two major challenges could limit the reliability of chimeric models: the functional validation of individual genetic elements and the maintenance of pluripotency of the ESC clones. Recent studies suggest that such challenges can be overcome if the ESCs are modified in a stepwise manner and functionally tested for the introduced genetic element at each step before selection for their ability to contribute to a host embryo. For example, chimeric models of lung cancer induced by ERBB2-V665E (also known as HER2-V665E), EGFR-L858R or KRAS-G12V gave rise to invasive adenocarcinomas31. Importantly, each initiating genetic lesion results in key differences in pathway activation. Overall, this approach satisfies the need for an immunocompetent host and for rapid cohort generation, in which the rate-limiting factor is the generation of ESCs and blastocysts. Importantly, every mouse will be primed for cancer induction, removing the dependence on Mendelian segregation for generating the desired genotypes. Conversely, the usefulness of the chimeric models depends on the capability of ESCs to populate the target organ; although it has not yet been observed, it is possible that there are cell lineages that do not lend themselves for ESC repopulation.
As for drug trials, particularly in epithelial cancers, chimeric nGEMMs can mimic the tumour–stroma interaction better than xenograft models, which should lead to improved understanding of drug–tumour interactions. For example, an allelic series of lung cancer chimeric models containing ERBB2V659E, KRASG12V or EGFRL858R oncogenes demonstrated that the resulting adenocarcinomas in normal lung tissue exhibited features of advanced malignancies. An experimental therapeutic trial carried out on the EGFR-L858R and KRAS-G12V chimeric models with an EGFR inhibitor (AV-4 1 2) accurately reflected the clinical observations31: chimeric mice harbouring tumours driven by EGFR-L858R exhibited a dramatic response to AV-412 treatment, with nearly complete tumour elimination and with lung volume returning close to normal values. KRAS-G12V-dependent chimeric lung tumours did not show any appreciable response to the treatment, and clinical symptoms did not improve. Interestingly, some EGFR-L858R tumours did not show a complete response to AV-412 treatment, probably reflecting the early emergence of drug resistance, which is consistent with clinical observations. A breast chimeric model and its application in translational research has also been described40.
Mouse in mouse transplantation models
The chimeric models described above are produced by the manipulation of ESCs followed by injection into blastocysts. A related approach involves the transplantation of genetically altered tissue-restricted stem and progenitor cells into primed syngeneic recipient mice, in which the stem or progenitor cells home to the appropriate tissue and the tumours arise surrounded by otherwise normal tissue. Such systems have been widely used in the haematopoietic system, in which haematopoietic stem and progenitor cells can be readily isolated from bone marrow or fetal livers and intravenously transplanted into lethally irradiated recipient mice41. As occurs following high-dose chemotherapy in human patients before bone marrow transplantation, irradiation creates a niche for the modified haematopoietic stem and progenitor cells to engraft42,43. Importantly, if placed into short-term culture before transplantation, the stem and progenitor cells can be transduced with retroviral vectors expressing oncogenes that recapitulate genetic changes that occur in human leukaemia and lymphoma44. Moreover, short hairpin RNAs (shRNAs) encoded from viral vectors can be introduced into precursor cells to mimic tumour suppressor gene loss41, greatly facilitating the modelling of tumour suppressor genes that would other wise require laborious knockout gene targeting strategies in GEMMs. Because it is possible to mix and match different retroviral vectors with different target stem and progenitor cell populations, leukaemias and lymphomas harbouring many genotypic combinations can be rapidly produced without extensive allelic intercrosses.
These transplantation systems can be rapidly established and are, despite their simplicity, good models of the human counterparts (as elaborated below). Owing to their flexibility, it has been possible to study many genes, or gene combinations, and to carry out structure–function studies on various oncogenes45–47. For example, a haematopoietic transduction–transplantation system was used to show that tumour-derived MYC mutants from human Burkitt’s lymphoma are more oncogenic than wild-type MYC because they evade the activation of the p53 tumour suppressor pathway48. Furthermore, similar systems have been used to rapidly produce mice for studies using conventional (see for example REFS 49,50) or targeted therapeutics51–53. Notably, the resulting leukaemias and lymphomas can be readily transplanted into syngeneic recipients, in which they home to the appropriate organs and produce secondary malignancies that are identical to the primary hosts. Indeed, the importance of syngeneic transplantation in preclinical therapeutics has previously been highlighted by the efficacy of studies using donor GEMM primary tumours, such as those that are derived from multiple different models of breast cancer54–56. Therefore, nGEMM systems can be readily propagated in vivo and transferred between metastatic sites, even in immunocompetent hosts.
Although the haematopoietic system is perhaps the most tractable tissue that lends itself to the transplantation approach, conceptually similar systems can be adapted to produce cancer types for which progenitor cells can be isolated, transduced and transplanted. To date, such models have been created for various cancer types, including that of breast57, brain58,59, ovary60 and liver34. By combining transplantation models with other genetic tools – for example, conditional gene expression or RNA interference (RNAi) — it is possible to rapidly study the role of candidate genes in tumour maintenance. Furthermore, by implanting cells that are engineered with somatic signature mutations into strains of different genetic backgrounds it is possible to explore the role of germline variability on biological phenotypes and/or the influence of tumour microenvironment. In one example, such studies have demonstrated that p53 loss is required for tumour maintenance, and that p53 reactivation leads to tumour regression by a combination of cell autonomous arrest programmes together with a new form of immune surveillance leading to the clearance of the former tumour cells, indicating the importance of a competent stroma61. It also becomes possible to carry out in vivo forward genetic screens (discussed below). However, one potential drawback of this system is that in some models, tumour phenotypes are not always replicated from mouse to mouse (TABLE 1). For example, liver progenitor cells engineered to express HRAS-V12 have a consistent latency and full penetrance, whereas those expressing MYC have a broad latency and ~40% penetrance34. This may be partly due to MYC not being sufficient to induce tumorigenesis alone and therefore it might rely on stochastic genetic changes to bring about full transformation. However, the effect of this potential shortcoming, if present, can be mitigated with a study design that is careful with the timing of treatment events.
Another application of transplantable models is the identification of cells-of-origin for particular malignancies by transducing and transplanting various progenitor cells for a particular phenotype. For example, Cdkn2a-null mouse neural stem cells or astrocytes transduced ex vivo to express constitutively active EGFR (the EGFRvIII mutant) have been shown to drive the development of high-grade gliomas when transplanted orthotopically into immunodeficient host mice58, showing that the neural stem cells would be the cell-of-origin for high-grade gliomas. More recently, neural stem cells isolated from the subventricular zone were further shown to be the cell-of-origin for glioblastoma62. Conceptually, many mouse tissues may be amenable to a similar modelling approach. Using a mouse origin for these progenitor cells provides the advantage of using an immunocompetent syngeneic host to ensure that tumour development is subjected to the same types of immune-mediated selective pressure as those seen in standard GEMMs, and as in human patients.
Transplantation nGEMMS offer major advantages in both genetic and drug screens. In germline GEMMs, large-scale genetic screens are carried out using retroviral or transposon mutagenesis and insertion sites are largely random. These screens are useful in constitutive germline models when used with mouse moloney leukaemia virus (MuLV; a common tool for retroviral insertion screens63) or in combination with mosaic models (for example, in colon cancer64). However, owing to their simplicity and ease-of-use, transplantation-based mosaic nGEMMs particularly lend themselves to the rapid validation of cancer genes. If one presumes that most of the genes that modulate cancer in humans will do so in mice, it becomes possible to use genomic information from human tumours to focus candidate-testing studies in mouse mosaic models for identifying, validating and characterizing functionally relevant cancer genes.
As one example illustrating the above approach, pools of shRNAs targeting the mouse orthologues of genes recurrently deleted in human hepatocellular carcinomas (HCCs) were tested for their ability to promote tumorigenesis in a mosaic mouse model of HCC65. In contrast to randomly selected shRNA pools, many deletion-specific pools accelerated hepatocarcinogenesis in the mice. Through further analysis, several new tumour suppressor genes were identified and validated, many of which had not been previously linked to cancer. These included the gene encoding the nuclear export protein exportin 4 (Xpo4) — which is also lost in other tumour types, including breast carcinoma65. In another example, a series of shRNA pools targeting a focused set of cancer-associated genes (a curated list of 1,000 cancer genes informed by multiple studies66) were introduced into haematopoietic progenitor cells derived from Eμ-Myc transgenic mice and screened for their ability to promote lymphomagenesis following engraftment into syngeneic recipients67,68.
Among the new tumour suppressors that were identified, Rad17, the product of which is involved in the oncogene-induced replicative stress response, proved to be a haploinsufficient tumour suppressor the complete inactivation of which is otherwise deleterious to embryogenesis67. Therefore, in this instance, shRNA screens could identify a tumour suppressor the heterozygous inactivation of which may promote tumorigenesis. Interestingly, although heterozygous deletions encompassing RAD17 occur in human cancers, it would have been difficult to pinpoint RAD17 as a relevant tumour suppressor through genomic approaches alone, as it does not particularly stand out using traditional computational methods. Surprisingly, some of the other tumour suppressors identified in this screen may have pro-oncogenic activities in other contexts, such as MEK1, a well-known mediator of the oncogenic MAPK pathway; and a surprising number of them encode secreted proteins — functional aspects that were not suspected a priori. This emphasizes the importance of tissue specificity, including tumour microenvironments, in determining the pro-cancer or anti-cancer functions of any particular gene.
These results establish the feasibility of in vivo genetic screens and illustrate how combining cancer genomics, functional genetic screens and mosaic mouse models can facilitate the functional annotation of the cancer genome. Current large-scale cancer genomics efforts are identifying many candidate genes; although extremely powerful by itself, genomic evidence is not sufficient to prove cancer relevance. Biological activities of these candidates must be functionally validated in various models, which is time intensive and expensive. Through functional genetic screens, it is possible to rapidly filter large numbers of candidate oncogenes and tumour suppressor genes for relevant biological activities to prioritize downstream follow-up studies. The ease of manipulation of mosaic models enables the modelling of the appropriate context for such genetic screens, an important parameter in a screen system given the context specificity of gene function.
This relatively high-throughput approach could be expanded to other mouse models or could include shRNA or open reading frame (ORF) libraries targeting genes that are affected by larger deletions, promoter methylation or point mutations. Furthermore, the concepts described above can be expanded in the future by incorporating more complex genetic models, viral vectors and/or selection schemes. For example, doxycycline-inducible vectors can dissect the role of genes in tumour maintenance versus initiation, for example, a cDNA library that can be turned off once the tumour is already established. Another possibility is carrying out a synthetic lethal screen with a known anti-oncogenic compound, in which the combination of the drug and the suppression of molecular targets can enhance the rational design of combination therapy. This broad range of possibilities highlights the flexibility of nGEMM systems for both basic and translational research (BOX 1).
Box 1. Genetic screens, and gene and drug discovery in models.
Types of screens
- Genetic modifier screen
- Straightforward screen of cDNA or short hairpin RNA, small interfering RNA or microRNA libraries for genes that enhance or suppress tumour initiation, growth and/or progression.
- Drug screen
- Similar to genetic modifier screens, but using a library of usually unknown, but rationally designed, chemical compounds.
- Second site suppressor screen
- Screen that seeks to find genes that, after withdrawal of the initiating oncogenic signal (such as Ras or MYC), can prevent regression of the tumour.
- Synthetic lethal screen
- Screen that looks for genetic elements or chemical compounds that singly have no effect on tumour viability, but together induce potent lethality.
- Molecular target screen
- Instead of looking for broad tumour phenotypes, the readout of this screen is the modulation of particular molecular targets.
Importantly, studies examining the relationship between cancer genotype and sensitivity to conventional chemotherapy suggest that nGEMMs can accurately model therapeutic response. For example, mice transplanted with lymphoma cells from Eμ-Myc mice, an nGEMM of human Burkitt’s lymphoma, are highly responsive to the chemotherapeutic drug cyclophosphamide, which parallels the situation in the primary transgenic mice and mirrors the situation in human patients33. Furthermore, disruption of p53 by RNAi produces an attenuated response to therapy and early relapse in these mice, recapitulating the association between p53 mutations and treatment failure found in various haematopoietic malignancies (see REF. 69). More recent work has dissected the roles of the DNA damage response factors in response to conventional chemotherapy in the mouse in a high-throughput manner that was not possible using traditional GEMMs70. Insights from such approaches may eventually enable the stratification of patients for the appropriate therapy in the clinic70.
In nGEMMs of acute myelogenous leukaemia (AML), mice with leukaemia expressing the runt-related transcription factor 1 (RUNX1; also known as AML1)–RUNX1T1 (also known as ETO) fusion protein, which mimics t(8;21) that occurs in human patients, show robust responses and even cures to frontline chemotherapy. Mice with leukaemia that express mixed-lineage leukaemia (MLL) fusions, which mimic translocations involving 11q23, are refractory to standard therapy, much like their human counterparts50.
Expanding beyond conventional chemotherapy, nGEMM mice harbouring myeloproliferative diseases that are triggered by the deletion of the tumour suppressor neurofibromin 1 (Nf1) are insensitive to MEK inhibitors (which target a downstream component of the Ras–MAPK pathway that becomes overactivated by NF1 loss), but full-blown AMLs with the same initiating lesion plus a randomly integrated retrovirus are surprisingly sensitive53. Nevertheless, these AMLs eventually acquire drug resistance that can be traced back to the integrated retroviruses, which have generated subpopulations of AML cells that are sensitive or resistant; MEK inhibition selects for the resistant subpopulations and reveals the role of genes such as RAS guanyl releasing protein 1 (Rasgrp1) in mediating drug resistance. Overall, mouse in mouse transplantation models offer a high degree of ex vivo engineering flexibility, the rapid generation of cohorts and the possibility of a fully competent immune environment. However, the direct applicability of mouse tumours to the clinic must be complemented with studies using human tissues.
Human in mouse (HIM) transplantation models
The analysis of GEMMs of cancer and the corresponding human cancers has established that many of the bona fide transforming or tumour suppressor genes and key transformation pathways are conserved between mouse and human cells71,72. At the same time, it is well known that cross-species differences exist on the cellular level, such as telomere dynamics73, or on the macro-environmental level, such as diet and carcinogen exposure. For example, inactivation of p53 (the most commonly inactivated tumour suppressor in humans) in the mouse has failed to recapitulate the epithelial cancer phenotypes: these mice do not develop the same range of tumours as that observed in patients with Li–Fraumeni syndrome. The reason for this difference became clear when it was appreciated that the mouse has long telomeres and promiscuous expression of telomerase74; therefore, mouse epithelial cells do not experience telomere-based crisis (when damaged telomeres require repair to prevent cell death), which is a potent cooperative driver of epithelial carcinogenesis in the setting of p53 deficiency75. When telomerase deficiency is introduced by inactivating telomerase reverse transcriptase (Tert), p53 inactivation led to the development of a range of epithelial tumours similar to those that occur in human patients with Li–Fraumeni syndrome73. Interestingly, even the knockin mutants of gain-of-function p53 alleles from some patients with Li-Fraumeni syndrome conspicuously failed to generate breast cancers, suggesting subtle species differences76. Historically, the solution to this problem has been the use of human tumour-derived cell lines. When transplanted into immune compromised mice, some of these lines have the ability to form tumours. Such xenografts have been a staple of cancer research for decades — and although they have provided a platform for many important discoveries, they have unfortunately failed to predict drug responses1. This failure might be due to in vitro propagation, an environment with different growth factors from those in the in vivo environment, as well as the subcutaneous grafting of the cells into non-physiological space.
To circumvent the species differences, recent advances have included the use of human primary, non-cancerous tissue transplanted into mice (as described below) (FIG. 3). In general, these approaches rely on the isolation of differentiated or tissue progenitor cells from donor tissue for ex vivo genetic engineering, followed by transplantation into the appropriate mouse tissue of an adult immune compromised mouse. This approach avoids artefacts being introduced during prolonged in vitro culturing and allows the experiment to start with euploid, non-transformed cells. One of the first models to use primary, non-cancerous human tissue was that of melanoma and skin models (REFS 77,78 and reviewed in REF. 79). Primary cells from human skin were isolated, cultured and transfected with retroviruses. The cells were allowed to form skin using three-dimensional culture in vitro and then grafted onto immune compromised mice. Transduction of keratinocytes with SHH resulted in basal cell carcinomas77, whereas transduction with HRAS-V12 in combination with cyclin-dependent kinase 4 (CDK4) or nuclear factor-κB inhibitor-α (IKBα) resulted in squamous cell carcinomas80. Transduction of melanocytes with an oncogenic mutant of NRAS in combination with the overexpression of TERT, dominant-negative p53 and an activating CDK4 mutant resulted in melanomas78. Another model incorporates human newborn foreskin grafts onto recombination activating gene 1 (Rag1)-deficient mice, inducing melanoma formation by a combination of treatment with DMBA and ultraviolet B (UVB) radiation81. Therefore, the modelling of skin tumours using human donor tissue was able to recapitulate all three major forms of skin cancer.
Figure 3. Human in mouse transplantation models.
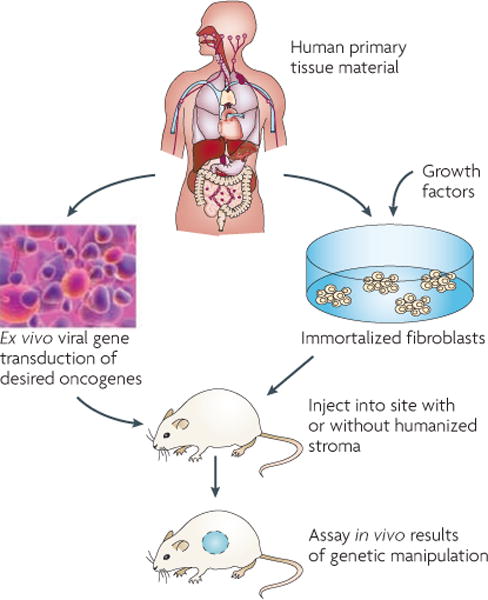
Pluripotent human stem cells are grown ex vivo on feeder cells and transduced with the oncogenic elements of interest. Without long culture times, the cells are injected into orthotopic sites on the mice with or without humanized stromal cells. Here, the tumour types of interest will develop from human cells within the context of the relevant mouse or human normal stroma.
Others have reported a leukaemia model using cord blood as donor tissue82. Umbilical cord cells were enriched for stem cells, transduced with a retrovirus containing the MLL-MLLT1 (also known as ENL) fusion gene and transplanted into irradiated mice, shortly after which acute lymphoblastic leukaemia formed within 140 days and featured hallmarks of the human disease. Transducing the cells with a retrovirus encoding the MLL-MLLT3 (also known as AF9) fusion gene resulted in various AMLs, as occurs in patients. This model allowed the authors to identify and characterize MLL leukaemia-initiating cells (LICs), which retained both myeloid and lymphoid lineage potential and remained responsive to microenvironmental cues. The properties of these cells provide a biological basis for several clinical hallmarks of MLL leukaemias82.
More recently, Kuperwasser and colleagues32 reported a breast cancer model in which primary human mammary epithelial cells were isolated from donor tissue as organoids, which are cells that retain some of the original three-dimensional context, that is, they are not a single cell suspension. These organoids were transplanted into the cleared mammary fat pad of immune compromised mice and formed structures that are indistinguishable from human mammary gland structures83. When the organoids are transfected with lentivirus harbouring KRASG12V or ERBB2V665E in combination with simian virus 40 (SV40) large T antigen in vitro without culture and transplanted into the mouse cleared fat pad, human mammary gland structures formed and developed tumours from these glandular structures, just as occurs in humans32. Tumorigenesis goes through stages identified in patients, such as ductal carcinoma in situ (DCIS). Interestingly, in these tumours, overexpression of TERT is not necessary, although this has not been observed following in vitro manipulation of human mammary epithelial cells. The developing tumours show hallmarks of basal-like breast cancer, a tumour with few treatment options. Therefore, this model allows the study of the development and therapeutic sensitivities of this important tumour type for the first time.
Although these human in mouse (HIM) model systems are clearly powerful in many ways, disadvantages remain, including the absence of a memory-based immune system and the reliance on virally introduced transgenes rather than endogenous genetic aberrations, as well as the potential of ligand–receptor mismatch owing to species-specific variation. A well-recognized example is the fact that human MET (the hepatocyte growth factor (HGF) receptor) does not bind mouse HGF84. How such differences might influence therapy responses in preclinical studies remains to be determined.
Finally, inherent genomic instability characteristics of human cancers may modulate therapeutic response and resistance; therefore, nGEMMs with similar genomic instability would probably be more predictive preclinical therapeutic models. The HIM tumours can circumvent this particular issue by using telomerase-deficient cells. Treating human tumours originating in the mouse, in a non-xenograft cell line, will give rise to new insights that are difficult to obtain using mouse tumours only. Wu and colleagues32 have demonstrated that using the breast HIM model, treatment with trastuzumab is efficacious. This will enable better characterization and understanding of trastuzumab responses in primary human tumours, which has not been possible so far in GEMMs owing to the inability of trastuzumab to bind to mouse ERBB2. Although more work is needed, the successful use of these models in preclinical studies could dramatically reduce the time and cost of drug development and so warrant further investigation. This will become even more important as we embark on the exploration of combination strategies.
Archived tumours in translational research
A major limitation of xenograft transplantation models using established human tumour cell lines is the inability to model the heterogeneity of a patient population. The replication of such heterogeneity is a key advantage of GEMMs and nGEMMs, as such models are typically engineered with one or more initiating genetic events that are not sufficient for full transformation. As reflected by the latency and incomplete penetrance of these models, and consistent with recent hallmark reviews of cancer biology85,86 that postulate the requirement for mutations in 8–12 key pathways, tumours that emerge de novo from these GEMMs and nGEMMs have additional spontaneously acquired mutations, which lead to molecular and biological variations that mirror the heterogeneity of a patient population. Such heterogeneity can be exploited, as exemplified by drug studies in germline non-small-cell lung cancer (NSCLC) and pancreatic ductal adenocarcinoma (PDAC) KRAS models using EGFR (erlotinib) and VEGF (anti-mouse VEGFA antibody mimicking bevacizumab) inhibitors30. However, a challenge introduced by this genetic diversity in tumours that emerge from GEMMs and nGEMMs is the difficulty of obtaining matched pairs of pre-treatment and post-treatment tumour tissues for comparative analyses. Therefore, tumour archives have become popular as a way of preserving this feature of heterogeneity in a tumour population while enabling access to pre-treatment tumour materials. Watters et al.40 exploited this concept by establishing a well-characterized archive of 107 propagated tumours from a breast ERBB2 chimeric model. Testing the archive with a γ-secretase inhibitor responder–non-responder analysis revealed a set of biomarkers that predicted response to the compound. Additionally, archives from chimeric tumours retain the characteristics of the primary tumours, as has been demonstrated for a lung NSCLC KRAS archive (Y. Zhou, A. Bressel, T. Zi, D. Potz, Z. Cai, I. Chiu, M. Robinson and J. H., unpublished observations). Archives from HIM transplantation models can also be established (M. Wu, K. Clark, Z. Cai, N. Deng, I. Chiu, M. Robinson and J. H., unpublished observations). Similarly, Jimeno et al.8 have developed an archive of primary patient explants for pancreatic tumours. These tumour archives are another platform for studies of drug response and resistance that is complementary to GEMMs and nGEMMs.
Conclusion and perspective
One of the most exciting aspects of nGEMMs is their applicability to preclinical drug development. The essential interaction of normal stroma with the tumour cells allows for testing drugs that not only interfere with tumour cell growth but that also target the tumour–stroma interaction. Moreover, the ability to establish cohorts of de novo tumours enables population-based empirical testing of drug responses. Finally, the use of HIM models using cells from human donors now enables the generation of tumours arising from primary human cells at orthotopic sites, thus avoiding many of the problems that are associated with prolonged cell culture and xenografts. However, it is worth remembering that each nGEMM has its limitations. For example, humanized nGEMMs are limited by the availability of donors, the species incompatibility of ligands and the absence of a memory-based immune system. Mouse nGEMMs still require improvement in the reproducibility of tumour phenotype and in the more widespread use of immunocompetent hosts. Nonetheless, nGEMMs of cancer provide valuable systems for designing and testing new therapeutics, including rapid cohort generation, reproducibility of phenotypes and improvements in modelling the tumour microenvironment.
In summary, no single model is likely to recapitulate all aspects of the complex genetics and biology of human cancers; therefore, understanding the strength and limitation of each model is necessary to maximally leverage these complimentary engineered model systems to facilitate the development of drugs and drug combinations in the future.
At a glance.
Genetically engineered mouse models (GEMMs) have been invaluable in advancing our knowledge of tumour biology. However, accelerated cancer gene discovery through large-scale cancer genomics and an increasing desire to use GEMMs for preclinical therapeutic studies have strained the capacity of germline GEMMs.
Non-germline genetic engineering approaches allow for accelerated and flexible genetic manipulation of models.
Chimeric models develop tumours in the context of normal stroma, with reduced timelines and mouse housing cost.
Transplantation models allow flexible and speedy manipulation of tissue stem and/or progenitor cells with multiple genetic tools (such as knock out, transgenes and RNA interference).
Human donor tissue models (or human in mouse models) allow the de novo development of primary human tumours in mouse stroma by manipulating primary human cells.
Therapeutic studies in vivo benefit from the wealth of complex GEMM and non-GEMM models to guide drug discovery.
Glossary
- Germline model
Mouse model that carries genetic modifications in its germline and which is maintained through breeding
- Inducible model
Mouse model that activates the expression of a transgene through a transactivator transgene that is, tTA or rtTA
- Non-germline GEMM (nGEMM)
Mouse model that carries genetic modifications in some of its somatic cells but not in the germline cells. Each model has to be individually generated through, for example, transplantation and injection
- Mosaic model
Germline model that acquires modifications of the germline genetic modification in some of its somatic cells
- Conditional GEMM
Model that acquires an activation or inactivation of the original genetic modification in somatic cells through the temporal or spatial expression of a modifier such as Cre
- Chimeric model
Mouse model that has been generated by ESC manipulation followed by the injection of these cells into a pre-implantation embryo. The resulting chimeric animal is the model animal
- Transplantation model
Mouse model in which part of a tissue is modified by transplanting tissue stem cells that carry genetic modifications
- Human in mouse (HIM) model
Transplantation model in which the transplanted cells are human tissue stem cells
Footnotes
Competing interests statement
The authors declare competing financial interests; see Web version for details.
References
- 1.Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nature Rev Drug Discov. 2006;5:741–754. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- 2.Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nature Genet. 2000;24:57–60. doi: 10.1038/71691. [DOI] [PubMed] [Google Scholar]
- 3.Roberts RB, et al. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci USA. 2002;99:1521–1526. doi: 10.1073/pnas.032678499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jonkers J, Berns A. Conditional mouse models of sporadic cancer. Nature Rev Cancer. 2002;2:251–265. doi: 10.1038/nrc777. [DOI] [PubMed] [Google Scholar]
- 5.Chin L, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. doi: 10.1038/22788. The first conditional transgenic mouse model to demonstrate solid tumour regression on extinction of the initiating oncogene. [DOI] [PubMed] [Google Scholar]
- 6.Su LK, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 7.Corpet DE, Pierre F. How good are rodent models of carcinogenesis in predicting efficacy in humans? A systematic review and meta-analysis of colon chemoprevention in rats, mice and men. Eur J Cancer. 2005;41:1911–1922. doi: 10.1016/j.ejca.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Jimeno A, et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol Cancer Ther. 2009;8:310–314. doi: 10.1158/1535-7163.MCT-08-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edelmann W, et al. Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell. 1997;91:467–477. doi: 10.1016/s0092-8674(00)80433-x. [DOI] [PubMed] [Google Scholar]
- 10.Meuwissen R, Linn SC, van der Valk M, Mooi WJ, Berns A. Mouse model for lung tumorigenesis through Cre/lox controlled sporadic activation of the K-Ras oncogene. Oncogene. 2001;20:6551–6558. doi: 10.1038/sj.onc.1204837. [DOI] [PubMed] [Google Scholar]
- 11.Tuveson DA, et al. Endogenous oncogenic K-rasG12D stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 12.Dinulescu DM, et al. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nature Med. 2005;11:63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- 13.Flesken-Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Res. 2003;63:3459–3463. [PubMed] [Google Scholar]
- 14.Engelman JA, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nature Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Indra AK, et al. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999;27:4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bosenberg M, et al. Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis. 2006;44:262–267. doi: 10.1002/dvg.20205. [DOI] [PubMed] [Google Scholar]
- 17.Dankort D, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nature Genet. 2009;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhomen N, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt-Supprian M, Wunderlich FT, Rajewsky K. Excision of the Frt-flanked neoR cassette from the CD19cre knock-in transgene reduces Cre-mediated recombination. Transgenic Res. 2007;16:657–660. doi: 10.1007/s11248-007-9100-4. [DOI] [PubMed] [Google Scholar]
- 20.Gossen M, et al. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- 21.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 22.Jain M, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297:102–104. doi: 10.1126/science.1071489. [DOI] [PubMed] [Google Scholar]
- 23.D’Cruz CM, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nature Med. 2001;7:235–239. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- 24.Rao G, et al. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene. 2004;23:6156–6162. doi: 10.1038/sj.onc.1207818. [DOI] [PubMed] [Google Scholar]
- 25.Shih AH, et al. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer Res. 2004;64:4783–4789. doi: 10.1158/0008-5472.CAN-03-3831. [DOI] [PubMed] [Google Scholar]
- 26.Momota H, Nerio E, Holland EC. Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res. 2005;65:7429–7435. doi: 10.1158/0008-5472.CAN-05-1042. [DOI] [PubMed] [Google Scholar]
- 27.Pao W, Klimstra DS, Fisher GH, Varmus HE. Use of avian retroviral vectors to introduce transcriptional regulators into mammalian cells for analyses of tumor maintenance. Proc Natl Acad Sci USA. 2003;100:8764–8769. doi: 10.1073/pnas.1133333100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Regales L, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest. 2009;119:3000–3010. doi: 10.1172/JCI38746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marumoto T, et al. Development of a novel mouse glioma model using lentiviral vectors. Nature Med. 2009;15:110–116. doi: 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh M, et al. Modelling therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nature Biotechnol. 2010 May 23; doi: 10.103a8/nbt.1640. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Y, et al. Chimeric mouse tumor models reveal differences in pathway activation between ERBB family- and KRAS-dependent lung adenocarcinomas. Nature Biotechnol. 2010;28:71–78. doi: 10.1038/nbt.1595. EGFR- but not KRAS-driven chimeric lung adenocarcinomas generated from engineered ESCs show near-complete tumour regression on treatment with an EGFR inhibitor. [DOI] [PubMed] [Google Scholar]
- 32.Wu M, et al. Dissecting genetic requirements of human breast tumorigenesis in a tissue transgenic model of human breast cancer in mice. Proc Natl Acad Sci USA. 2009;106:7022–7027. doi: 10.1073/pnas.0811785106. Normal breast cancer cells grown as organoids produce orthotopic tumours in mice with histopathological features that more closely resemble the primary tumour than the features of tumours generated from cells cultured on plastic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev. 1999;13:2670–2677. doi: 10.1101/gad.13.20.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zender L, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–1267. doi: 10.1016/j.cell.2006.05.030. Liver progenitor cells harvested from embryonic mice and engineered with various oncogenes produce carcinomas orthotopically in syngeneic hosts and can be used to identify new oncogenes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pear WS, et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- 36.Le LQ, Shipman T, Burns DK, Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell. 2009;4:453–463. doi: 10.1016/j.stem.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trimboli AJ, et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461:1084–1091. doi: 10.1038/nature08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paik JH, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jongsma J, et al. A conditional mouse model for malignant mesothelioma. Cancer Cell. 2008;13:261–271. doi: 10.1016/j.ccr.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 40.Watters JW, et al. De novodiscovery of a γ-secretase inhibitor response signature using a novel in vivo breast tumor model. Cancer Res. 2009;69:8949–8957. doi: 10.1158/0008-5472.CAN-09-1544. [DOI] [PubMed] [Google Scholar]
- 41.Hemann MT, et al. An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nature Genet. 2003;33:396–400. doi: 10.1038/ng1091. [DOI] [PubMed] [Google Scholar]
- 42.Takada A, Takada Y. Proliferation of donor hematopoietic cells in lethally irradiated host mice. Transplantation. 1972;13:276–280. doi: 10.1097/00007890-197203000-00013. [DOI] [PubMed] [Google Scholar]
- 43.Schmitt CA, et al. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell. 2002;1:289–298. doi: 10.1016/s1535-6108(02)00047-8. [DOI] [PubMed] [Google Scholar]
- 44.Schwaller J, et al. Transformation of hematopoietic cell lines to growth-factor independence and induction of a fatal myelo- and lymphoproliferative disease in mice by retrovirally transduced TEL/JAK2 fusion genes. EMBO J. 1998;17:5321–5333. doi: 10.1093/emboj/17.18.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fridman JS, et al. Tumor promotion by Mdm2 splice variants unable to bind p53. Cancer Res. 2003;63:5703–5706. [PubMed] [Google Scholar]
- 46.Wendel HG, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21:3232–3237. doi: 10.1101/gad.1604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Phan VT, et al. Cooperation of cytokine signaling with chimeric transcription factors in leukemogenesis: PML-retinoic acid receptor alpha blocks growth factor-mediated differentiation. Mol Cell Biol. 2003;23:4573–4585. doi: 10.1128/MCB.23.13.4573-4585.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hemann MT, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–811. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmitt CA, Rosenthal CT, Lowe SW. Genetic analysis of chemoresistance in primary murine lymphomas. Nature Med. 2000;6:1029–1035. doi: 10.1038/79542. [DOI] [PubMed] [Google Scholar]
- 50.Zuber J, et al. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009;23:877–889. doi: 10.1101/gad.1771409. nGEMM leukaemia models driven by an MLLT1 gene fusion, but not ones driven by a RUNX1 gene fusion, are refractory to chemotherapy, mirroring the prognostic course of therapy in patients harbouring either of the respective fusions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wendel HG, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–337. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 52.Mullighan CG, Williams RT, Downing JR, Sherr CJ. Failure of CDKN2A/B (INK4A/B-ARF)-mediated tumor suppression and resistance to targeted therapy in acute lymphoblastic leukemia induced by BCR-ABL. Genes Dev. 2008;22:1411–1415. doi: 10.1101/gad.1673908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lauchle JO, et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature. 2009;461:411–414. doi: 10.1038/nature08279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Varticovski L, et al. Accelerated preclinical testing using transplanted tumors from genetically engineered mouse breast cancer models. Clin Cancer Res. 2007;13:2168–2177. doi: 10.1158/1078-0432.CCR-06-0918. [DOI] [PubMed] [Google Scholar]
- 55.Rottenberg S, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. 2008;105:17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rottenberg S, et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci USA. 2007;104:12117–12122. doi: 10.1073/pnas.0702955104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Evers B, et al. A tissue reconstitution model to study cancer cell-intrinsic and -extrinsic factors in mammary tumourigenesis. J Pathol. 2010;220:34–44. doi: 10.1002/path.2655. [DOI] [PubMed] [Google Scholar]
- 58.Bachoo RM, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 59.Zindy F, et al. Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 2007;67:2676–2684. doi: 10.1158/0008-5472.CAN-06-3418. [DOI] [PubMed] [Google Scholar]
- 60.Xing D, Orsulic S. A mouse model for the molecular characterization of brca1-associated ovarian carcinoma. Cancer Res. 2006;66:8949–8953. doi: 10.1158/0008-5472.CAN-06-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alcantara Llaguno SR, Chen J, Parada LF. Signaling in malignant astrocytomas: role of neural stem cells and its therapeutic implications. Clin Cancer Res. 2009;15:7124–7129. doi: 10.1158/1078-0432.CCR-09-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uren AG, et al. Large-scale mutagenesis in p19ARF-and p53-deficient mice identifies cancer genes and their collaborative networks. Cell. 2008;133:727–741. doi: 10.1016/j.cell.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Starr TK, et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science. 2009;323:1747–1750. doi: 10.1126/science.1163040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zender L, et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell. 2008;135:852–864. doi: 10.1016/j.cell.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Witt AE, et al. Functional proteomics approach to investigate the biological activities of cDNAs implicated in breast cancer. J Proteome Res. 2006;5:599–610. doi: 10.1021/pr050395r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bric A, et al. Functional identification of tumor-suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell. 2009;16:324–335. doi: 10.1016/j.ccr.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meacham CE, Ho EE, Dubrovsky E, Gertler FB, Hemann MT. In vivo RNAi screening identifies regulators of actin dynamics as key determinants of lymphoma progression. Nature Genet. 2009;41:1133–1137. doi: 10.1038/ng.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wattel E, et al. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 1994;84:3148–3157. [PubMed] [Google Scholar]
- 70.Jiang H, et al. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009;23:1895–1909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoffman RM. Orthotopic metastatic (MetaMouse) models for discovery and development of novel chemotherapy. Methods Mol Med. 2005;111:297–322. doi: 10.1385/1-59259-889-7:297. [DOI] [PubMed] [Google Scholar]
- 72.Maser RS, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447:966–971. doi: 10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2009;31:9–18. doi: 10.1093/carcin/bgp268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rudolph KL. Telomeres and telomerase influence the course of human diseases, aging and carcinogenesis. Curr Mol Med. 2005;5:133–134. doi: 10.2174/1566524053586617. [DOI] [PubMed] [Google Scholar]
- 75.Farazi PA, Glickman J, Horner J, Depinho RA. Cooperative interactions of p53 mutation, telomere dysfunction, and chronic liver damage in hepatocellular carcinoma progression. Cancer Res. 2006;66:4766–4773. doi: 10.1158/0008-5472.CAN-05-4608. [DOI] [PubMed] [Google Scholar]
- 76.Olive KP, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 77.Fan H, Oro AE, Scott MP, Khavari PA. Induction of basal cell carcinoma features in transgenic human skin expressing Sonic Hedgehog. Nature Med. 1997;3:788–792. doi: 10.1038/nm0797-788. The first in a series of papers using normal human skin cells to orthotopically generate carcinomas and melanomas in mice by grafting with stromal elements. [DOI] [PubMed] [Google Scholar]
- 78.Chudnovsky Y, Adams AE, Robbins PB, Lin Q, Khavari PA. Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nature Genet. 2005;37:745–749. doi: 10.1038/ng1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Khavari PA. Modelling cancer in human skin tissue. Nature Rev Cancer. 2006;6:270–280. doi: 10.1038/nrc1838. [DOI] [PubMed] [Google Scholar]
- 80.Lazarov M, et al. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nature Med. 2002;8:1105–1114. doi: 10.1038/nm779. [DOI] [PubMed] [Google Scholar]
- 81.Atillasoy ES, et al. UVB induces atypical melanocytic lesions and melanoma in human skin. Am J Pathol. 1998;152:1179–1186. [PMC free article] [PubMed] [Google Scholar]
- 82.Barabe F, Kennedy JA, Hope KJ, Dick JE. Modeling the initiation and progression of human acute leukemia in mice. Science. 2007;316:600–604. doi: 10.1126/science.1139851. A HIM leukaemia model using human cord blood transduced with an MLLT1 gene fusion can be used to identify LICs. [DOI] [PubMed] [Google Scholar]
- 83.Kuperwasser C, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci USA. 2004;101:4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rong S, et al. Tumorigenicity of the met proto-oncogene and the gene for hepatocyte growth factor. Mol Cell Biol. 1992;12:5152–5158. doi: 10.1128/mcb.12.11.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 86.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nature Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]