

Abstract
The case of a 55-year-old man who attempted suicide by ingesting <100 mL of 28% sodium chlorite solution is presented. On arrival in the intensive care unit, the patient appeared cyanotic with lowered consciousness and displayed anuria and chocolate brown serum.
Initial laboratory tests revealed 40% of methemoglobin. The formation of methemoglobin was effectively treated with methylene blue (10% after 29 hours).
To remove the toxin, and because of the anuric acute renal failure, the patient received renal replacement therapy. Despite these therapeutic measures, the patient developed hemolytic anemia and disseminated intravascular coagulation, which were treated with red blood cell transfusion and intermittent hemodialysis. These interventions led to the improvement of his condition and the patient eventually fully recovered. Patient gave written informed consent.
This is the third known case of chlorite poisoning that has been reported. Based upon this case, we suggest the management of sodium chlorite poisoning to comprise the early administration of methylene blue, in addition to renal replacement therapy and transfusion of red blood cells.
INTRODUCTION
Ingestion of sodium chlorite leads to an intoxication characterized by methemoglobin (MetHb) formation, hemolysis, and acute renal failure.1 To our best knowledge, very little data is available regarding the pathophysiology and treatment of sodium chlorite intoxication as only two cases have been reported so far.1 Methylene blue (MB) is an effective antidote to methemoglobinemia as it activates nicotinamide adenine dinucleotide phosphate (NADPH)-MetHb reductase, following its conversion to leukomethylene blue by glucose-6-phosphate dehydrogenase (G6PD).3 Methemoglobinemia in chlorite poisoning, however, has been described as resistant to MB treatment.1 Here we report a case of a patient with sodium chlorite poisoning in which the associated severe methemoglobinemia was successfully treated with MB, resulting in a substantial decrease in MetHb formation. Nevertheless, the successful lowering of methemoglobinemia did not preclude the development of acute renal failure, which was effectively treated by renal replacement therapy (RRT).
CASE
Day 1
A 55-year-old man was admitted to our intensive care unit on May 23, 2011 after a suicide attempt. He had ingested about 100 mL of 28% sodium chlorite solution, 0.75 L of whiskey, and ibuprofen (12 tablets of 400 mg), after which he had attempted to hang himself.
His relatives found him lying on the floor, cyanotic, and with lowered consciousness, as judged by his inadequate response to verbal stimulus. Upon arrival of the ambulance, he vomited and was immediately transported to the hospital’s emergency department and subsequently admitted to our department.
On arrival, his Glasgow Coma Scale score was 13 (E4M5V4), body temperature 33.6°C, heart rate 110/min, blood pressure 150/80 mm Hg, respiratory rate 28/min, pulse oximetry (SpaO2) 65%. Physical examination revealed generalized skin cyanosis, neck markings after strangulation, a silent abdomen, and incontinence for urine and feces. Blood samples from the arterial line rendered chocolate-brown serum. Further physical examination showed no abnormalities.
The patient’s clinical history disclosed depression, a previous suicide attempt, and alcohol abuse. He did not take any regular medication. The patient was sedated, intubated, and mechanically ventilated (ventilator Savina, Dräger Medical GmbH, Lübeck, Germany; ventilation mode was bilevel positive airway pressure).
The initial laboratory evaluation showed the following: hemoglobin 179 g/L, erythrocytes 5.45 × 1012/L, hematocrit 0.54, leukocytes 28.5 × 109/L, platelet count 279 × 109/L, body urea nitrogen (BUN) 5.5 mg/dL (Figure 1), creatinine 142 μmol/L (Figure 2), potassium 7.5 mM, osmolality 351 mosm/L, arterial lactate 6.5 mM, and myoglobin 433 ng/mL. Bilirubin was elevated to 28 mM whereas liver enzymes and coagulation factors were normal.
FIGURE 1.
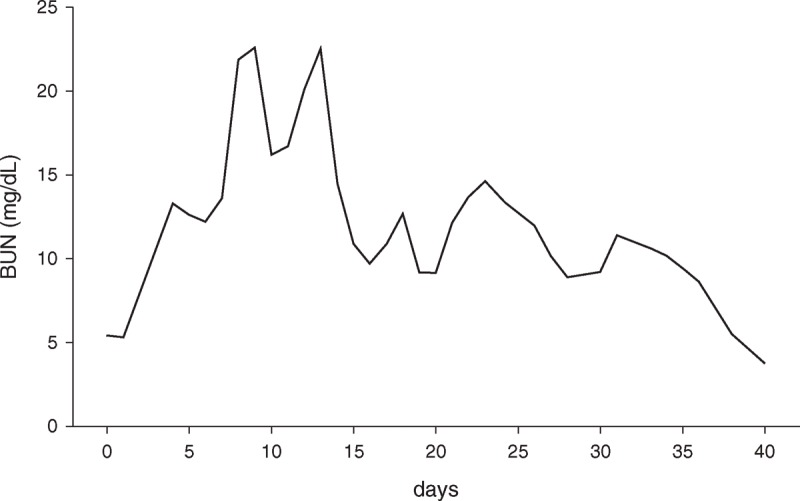
Plasma level of BUN over time of hospitalization. BUN = body urea nitrogen.
FIGURE 2.

Plasma level of creatinine over time of hospitalization.
Arterial blood gas analysis was performed while ventilated with 100% oxygen. The pH was 7.25, PaCO2 4.45 kPa, PaO2 52.45 kPa, BE −12.2 mmol/L, HCO3 14.1 mmol/L, SaO2 99%, and lactate 6, 48 mmol/L. The laboratory tests also revealed 40.1% of MetHb (Figure 3).
FIGURE 3.
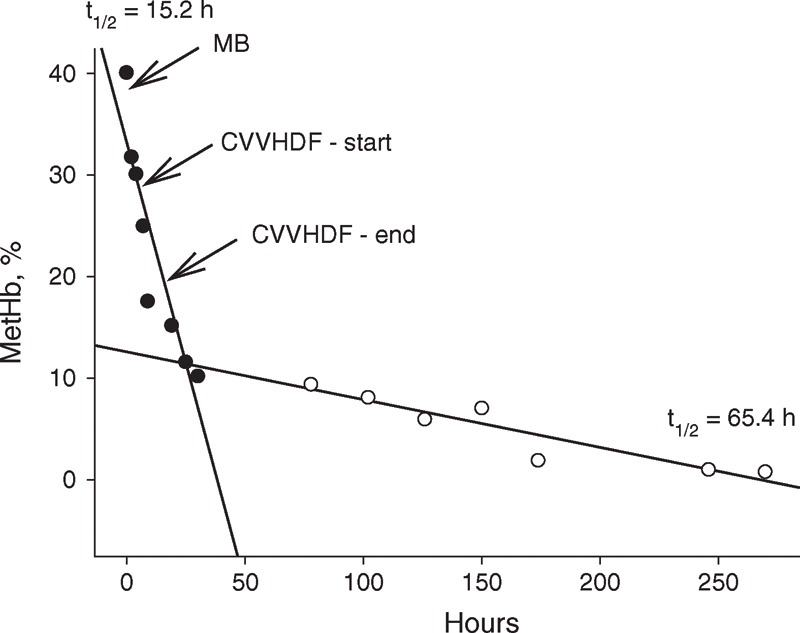
Plasma level of MetHb (%) over time of hospitalization. Note that there are clearly 2 kinetics of MetHb elimination: one that occurred within 24 hours with a half-life of about 15 hours (MB administration, RRT) and second with a half-life of about 65 hours (ordinary body elimination mechanisms). Abbreviations: CVVHDF = continuous venovenous hemodiafiltration, MetHb = methemoglobin.
Toxicology revealed 2 mg/g of alcohol in blood, ibuprofen was found both in gastric content and urine. Gastric fibroscopy was performed to check for burns in the gastrointestinal tract caused by sodium chlorite. Multiple superficial necrotic spots were found in the antrum, the body of the stomach, and the duodenum, whereas edema of the pylorus was observed. After 3 days, ulcerations were found in the upper part of esophagus, stomach, and duodenum.
On admission, the electrocardiogram showed peaked T waves. A chest x-ray and a CT scan of the brain were normal.
Antidote therapy with MB was instituted 2 hours after admission by an intravenous bolus of 34 mg MB (approximately 0.5 mg/kg, Bleu Patenté V sodique Guerbet 2.5%, Guerbet, Roissy CdG Cedex, France). Four and half hours after admission, because of acute renal failure (anuria, hyperkalemia, metabolic acidosis), continuous venovenous hemodiafiltration (CVVHDF) was initiated on the multiFiltrate (Fresenius Medical Care, Bad Homburg, Germany) with hemofilter (Ultraflux AV600S, Fresenius Polysulfone, Fresenius Medical Care AG & Co. KGaA, Bad Homburg, Germany), with a blood flow of 250 mL/min, a hemodialysis flow of 1800 mL/h, and a substitution hemofiltration solution flow of 35 mL/kg/h for a total duration of 10.5 hours. Later that day, another 4 hours of hemodialysis was performed because of hyperkalemia (Fresenius Medical Care 4008B; dialysis filter, FX80, Fresenius Helixone, High-flux [Fresenius Medical Care AG & Co. KGaA, Bad Homburg, Germany]).
After 14 hours, the lactate level dropped to 2.55 mmol/L whereas the level of MethHb decreased to 15.2%, further declining to 10.2% after 29 hours (Figure 3). Within 24 hours, the patient developed hemolytic anemia and disseminated intravascular coagulation (DIC, hemoglobin levels dropped from 179 to 116 g/L, thrombocytes from 279 to 164, D-dimer 29.1 mg/mL, international normalized ratio: 1.8, activated partial thromboplastin time ratio: 1.4). Over the course of the next 4 days, the hemolytic anemia deteriorated. Hemoglobin levels on the fifth day dropped to 59 g/L and platelet count to 77 × 109/L. In response, 4 units of packed red blood cells were transfused in 3 hours, amounting in total to 1074 mL).
Twenty hours after admission, a norepinephrine drip was instituted (0.16 μg/kg/min) because of hypotension (80/60 mm Hg) during the next 37 hours.
Clinical Course >24 Hours
On day 8, the patient developed bronchopneumonia and sepsis (body temperature 39°C, antibiotics were initiated).
On day 10, the gastric and esophageal ulceration appeared improved, and on day 13 the patient was weaned from mechanical ventilation. Day 17 onward, intermittent hemodialysis (IHD) was performed every second day. By day 22, the patient started to urinate (2500 mL of urine/24 h, with blood urea 12 mg/dL (Figure 1), creatinine 428 μmol/L (Figure 2), and an estimated GFR of 15.6 mL/min). He was subsequently transferred to a medium care facility without further need of hemodialysis. In total, hemodialysis had been performed 16 times during hospitalization.
By day 43, the patient was transferred to a psychiatric ward and discharged from hospital on day 64. Patient gave written informed consent.
DISCUSSION
Here we report a case of a patient poisoned by sodium chlorite in a suicide attempt, who then developed methemoglobinemia, hemolysis, and acute renal failure.
The patient ingested <100 mL of 28% sodium chlorite, which led to a 40% rise in MetHb level. He was treated with MB, which decreased MetHb levels to about 10% within 24 hours. The lowering of MetHb, however, proved insufficient to prevent development of subsequent hemolysis and acute renal failure.
To our knowledge, only two prior cases of acute sodium chlorite intoxication have been reported in medical literature previously. In the first case (Lin and Lim1), a patient was treated with continuous arteriovenous hemofiltration and administration of multiple doses of MB. MB in this case had no effect on MetHb formation (MetHb about 50%). In the second case (Romanovsky et al,2 MetHb about 10%), the patient was treated with IHD, continuous venovenous hemofiltration (CVVH), and red blood cell exchange. In both cases, the patients developed hemolysis, DIC, and acute renal failure that required IHD for renal recovery, and both patients survived. Our patient (MetHb about 40%) was treated with early administration of MB CVVHDF, followed by daily IHD and red blood cell transfusions. Reduction of methemoglobinemia by MB did not prevent the development of acute renal failure.
Mechanism of MetHb Formation
MetHb formation after chlorite poisoning is due to oxidation,5 causing the change of the iron ion of hemoglobin from the ferrous state (Fe2+) to the ferric state (Fe3+) leading to the loss of the ability of hemoglobin to bind oxygen.4 In the physiological condition, the low level of MetHb (1%–3%) is sustained by 2 important mechanisms. The first is the hexose-monophosphate shunt pathway in the erythrocyte in which the oxidizing agents are reduced by glutathione. The second, and more important, mechanism preventing MetHb formation are 2 enzyme systems, diaphorase I and diaphorase II. These 2 enzyme systems require nicotinamide adenine dinucleotide (NADH) and NADPH and reduce the ferric iron ion into its original ferrous state, thus, turning MetHb into hemoglobin.
Chlorite-Induced Methemoglobinemia
Oxidative agents are capable of heme iron oxidation, and thus cause MetHb formation. In 1964, chlorite ingestion was associated with MetHb formation for the first time.6
When MetHb concentration is increased, tissue hypoxia may occur, giving rise to symptoms including cyanosis, shortness of breath, exercise intolerance, fatigue, dizziness, and loss of consciousness. Severe methemoglobinemia (>50%) causes dysrhythmias, seizures, and coma, whereas death is imminent when MetHb levels exceed >70%. Arterial blood with elevated MetHb appears with a characteristic chocolate-brown color.
Based on our and the previous 2 reported cases, it seems that MetHb formation is dependent on the amount of chlorite ingested. In the case of Lin and Lim,1 the patient who was presented with >50% MetHb had ingested a teacup of 10% chlorite, whereas in the second case (Romanovsky et al2), MetHb was around 6% in a patient who had ingested a mouthful of 28% chlorite diluted with water in a cup. Our patient had ingested <100 mL of 28% chlorite, rendering a MetHb level of 40%. This suggests that oxidation of hemoglobin by chlorite is dose-dependent, and even a relatively small amount of chlorite, as in the case of Romanovsky et al,2 can cause MetHb formation.
The Effectiveness of MB
To date, no standard treatment procedures for chlorite poisoning have been established, due to the lack of reported cases. Because of the chemical similarity, chlorite () poisoning resembles chlorate () poisoning. However, chlorate is far more common in households than chlorite, hence poisoning with chlorate is much better documented. Chlorate poisoning displays similar symptoms, that is, oxidative damage to red blood cells and kidney, and also causes methemoglobinemia followed by hemolysis and anuric acute renal failure.7
MB counteracts methemoglobinemia in 2 ways. First, MB is reduced to leukomethylene blue by the erythrocyte’s MetHb reductase in the presence of NADPH, which subsequently reduces MetHb to hemoglobin. Additionally, MB activates diaphorase II up to 5 times its normal level.3
MB is commonly used in the treatment of methemoglobinemia.3 Nevertheless, the effectiveness of MB in the treatment of methemoglobinemia caused by chlorite (or chlorate) poisoning is debated. Some authors found MB to be effective while others not. MB is reported to be ineffective in many cases of chlorate poisoning. Steffen and Seitz7 reported a case of unsuccessful use of MB in chlorate poisoning and Singelmann et al8 further showed by experiments on erythrocytes that chlorate denatures the G6PD, possibly explaining the ineffectiveness of MB. However, Lee et al9 successfully used MB in their case of chlorate poisoning. The lack of effectiveness of MB in chlorite/chlorate poisoning is attributed to the denaturation of the erythrocyte G6PD by sodium chlorite (or chlorate).10 Consequently, MB-induced conversion of MetHb back to hemoglobin fails, because its action is dependent on nicotinamide-adenine dinucleotide phosphate (NADP+) produced by G6PD, as outlined above. Importantly, MB is ineffective in patients with an inherited deficiency of G6DH, as it was assumed in the case reported by Romanovsky et al.2 Therefore, special caution needs to be taken with these patients. Exchange transfusion may play a role in the management of severe hemolysis or in G6DH deficiency associated with chlorite-induced methemoglobinemia in which MB is rather contraindicated.
A failure of MB to reverse MetHb may perhaps originate from a concentration of chlorite, the longer exposure needed for G6PD denaturation, and even from an individual variability in the sensitivity of G6PD dehydrogenase to oxidants. Lee et al,9 who successfully used MB in chlorate poisoning, postulates that a “window of opportunity” exists when MB is used within 6 hours after ingestion of chlorate, prior to hemolysis. Indeed, in our case, MB was used within this period (2 hours after admission—before institution of hemolysis). Therefore, the early use of MB seems to be essential in its therapeutic effect.
Unfortunately, we did not assess denaturation of G6PD in the erythrocytes, so we cannot exclude its presence at the time of MB administration.
Furthermore, MB is more effective in normal erythrocytes as opposed to ruptured erythrocytes. It is likely that both of the patients reported by Lee et al9 and by us received MB prior to hemolysis. In the presence of hemolysis, high-dose MB (20–30 mg/kg) can even induce MetHb formation.9 Ascorbic acid, N-acetylcysteine and tocopherol may be used as an alternative to MB, but these treatments currently lack proof of efficacy.
While no experimental studies in man have been conducted, experience from the clinics show that MB increases the rate of MetHb conversion back to hemoglobin up to 6-fold.3 Improvement usually occurs within 30 minutes after MB injection,9,11 which is in line with our observation of MetHb level decreasing from 40.1% to 31.8% within 2 hours after MB administration. Despite this lowering, it is however difficult to provide a conclusive statement on whether MB lowered MetHb in the presented case. The decline in MetHb we observed appears very modest compared with the one reported in a case of chlorate poisoning by Lee at al,9 in which MetHb level dropped from 66.8% to <1% within 4 hours after MB. Except for the different poisoning agents, that is, chlorite versus chlorate, the difference in MB effect on MetHb may also represent a dosing issue, as we employed 0.5 mg/kg MB, whereas Lee et al9 used 2 mg/kg. Alternatively, the poisoning with chlorite in our case might have (already) caused partial inactivation of G6PD, leading to a slower rate of MetHb reduction by MB. The considerable time lag between ingestion of chlorite and hospitalization in our patient would then emphasize the importance of early MB administration.
Remarkably, the decrease in MetHb seemed to follow a 2-phase kinetic profile. The rapid decline, lasting about 30 hours, seems related to the administration of MB. Such notion is supported by MB having a half-life of about 6 hours, which would render the drug ineffective after about a day. The second phase, showing a 4-fold lower elimination rate, is caused by spontaneous decrease.
Importantly, MB infusion does not seem to cause negative effects in patient with chlorite poisoning when administered before the occurrence of hemolysis. Therefore, pending finite data, our recommendation would be to institute MB treatment prior to hemolysis, up to 6 hours past ingestion.
Acute Kidney Injury
In this case and other 2 cases, the reduction of methemoglobinemia did not prevent the development of acute kidney injury (AKI). Because of the different levels of methemoglobinemia in these cases, the development of AKI seems unrelated to methemoglobinemia. Indeed, AKI is not a common consequence of methemoglobinemia. More likely, AKI develops independently of methemoglobinemia in response to the direct toxic effect of chlorite on renal tissue. Lin and Lim1 reported renal tubulointerstitial changes with interstitial infiltration, edema, and moderate damage of tubules, but no changes at the glomerular level in biopsy of the chlorite poisoned patient. Furthermore, chlorite ingestion in rats was reported by Heffernan et al5 to cause increased kidney/body weight ratio. Jackson et al12 reported that sodium chlorate acutely constricts renal vessels that might lead to a decrease in renal blood flow causing anuric acute renal failure. The chemical similarity between chlorate and chlorite suggests that in our case the cause of AKI might have been mediated via the same mechanism, that is, an acute toxic effect on renal tubules or arterioles. To reduce chlorite toxicity, it is therefore advisable to remove chlorite from circulation by early institution of RRT.
Renal Replacement Therapy
RRT can be applied intermittently, such as IHD or continuous renal replacement therapy (CRRT), such as in CVVHDF, which was used in our case. IHD achieves removal of fluid, solutes, and toxins. Rapid fluid removal during IHD has, however, been suggested to lead to hypotension, with the potential for further renal injury and prolongation of AKI.13 The kinetics of chlorite is largely unknown. Thus, it is unknown, whether chlorite is rapidly removed or inactivated, or is released back from the tissues into the blood stream, thereby prolonging exposure of tissues. IHD would be more effective in chlorite removal if the rate of chlorite removal in plasma was linear, that is, not influenced by its back-releasing from tissues. If otherwise, then CRRT may be the better choice because of its continuous and prolonged nature. Thus, at present, it is unclear whether continuous RRT is superior to IHD in preventing hemolysis and removing chlorite from plasma. An additional benefit of continuous RRT is the facilitation of the management of fluid balance in hemodynamically unstable critically ill patients, as it was in our case.13
Effect of Chlorite on Hemolysis, DIC, and AKI
Our patient developed within 24 hours hemolysis and DIC, which deteriorated in the course of the next 4 days. It is of note that in the currently presented case as well as in 2 previously reported cases hemolysis, DIC, and AKI occurred. As studies varied considerably with respect to the level of MetHb, this observation suggests that the MetHb is not causally related to the hemolysis in chlorite poisoning. Rather, the hemolysis seems caused by a toxic action of the chlorite per se, possibly related to its oxidative properties. Moreover, to our knowledge, there is no data available in the current literature demonstrating that MetHb causes or precedes hemolysis.
However, Singelmann et al8 reported that in vitro chlorate induces hemolysis due to membrane protein polymerization, thus increasing erythrocyte membrane rigidity, denaturation of globin (the essential protein subunit of hemoglobin), crosslinking of membrane proteins, and inactivation of membrane enzymes causing the disruption of erythrocytes membranes.
Such proposition seems in accord with the chain of events typical for hemolytic anemia caused by oxidative compounds in vivo, featuring denaturation of hemoglobin and Heinz body hemolytic anemia.14
Similar to hemolysis, DIC may be directly related to the oxidative properties of chlorite, as it is likely a consequence of hemolysis because of the coagulating properties of the hemolyzed erythrocyte “ghost cells”.15
Thus, most likely, the oxidative effect of chlorite, rather than its induction of MetHb, induces the majority of symptoms, including hemolysis, DIC, and damage of renal tissue.
Finally, in the absence of severe methemoglobinemia, we believe that the intermittent lactic acidosis resulted from a combination of decreased oxygen-carrying capacity and reduced tissue perfusion.
Chlorate salts are common household chemicals used for cleaning or as bleach because of their oxidative capabilities. Chlorite salts are however less frequent in households, but an aqueous solution of 28% sodium chlorite (NaClO2) in distilled water is branded by the product name mineral miraculous solution (http://jimhumble.org), which is used orally as diluted solution. Our case report shows that chlorite ingestion is life threatening, even in small amounts (Romanovsky et al2). Therefore, leaflets should warn consumers to take special caution and measures, and thus avoid accidental poisoning.
Taken together, our report fuels the notion that early administration of MB, that is, before development of hemolysis, exerts a beneficial effect in chlorite poisoning because of a rapid decrease in MetHb levels, leading to an improved oxygenation of tissues. In addition, the management of sodium chlorite intoxication appears to benefit from early institution of RRT to manage AKI and to speed up clearance of chlorite. Finally, transfusion of red blood cells is warranted to recover red blood cell count.
Footnotes
Abbreviations: AKI = acute kidney injury, BUN = body urea nitrogen, CRRT = continuous renal replacement therapy, CVVH = continuous venovenous hemofiltration, CVVHDF = continuous venovenous hemodiafiltration, DIC = disseminated intravascular coagulation, G6PD = glucose-6-phosphate dehydrogenase, IHD = intermittent hemodialysis, MB = methylene blue, MetHb = methemoglobin, NADPH = nicotinamide adenine dinucleotide phosphate, RRT = renal replacement therapy.
The authors have no funding and conflicts of interest to disclose.
References
- 1.Lin JL, Lim PS. Acute sodium chlorite poisoning associated with renal failure. Ren Fail. 1993;15:645–648. [DOI] [PubMed] [Google Scholar]
- 2.Romanovsky A, Djogovic D, Chin D. A case of sodium chlorite toxicity managed with concurrent renal replacement therapy and red cell exchange. J Med Toxicol. 2013;9:67–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clifton J, Leikin JB. Methylene blue. Am J Ther. 2003;10:289–291. [DOI] [PubMed] [Google Scholar]
- 4.Saeui C, Charlton N, Brady WJ. Biochemical issues in emergency medicine: diagnostic and therapeutic considerations of selected toxic presentations. Am J Emerg Med. 2012;30:231–235. [DOI] [PubMed] [Google Scholar]
- 5.Heffernan WP, Guion C, Bull RJ. Oxidative damage to the erythrocyte induced by sodium chlorite, in vitro. J Environ Pathol Toxicol. 1979;2:1501–1510. [PubMed] [Google Scholar]
- 6.Symon K, Musil J, Knotek Z, et al. [Risk of use of the chlorine dioxide in waterworks’ treatment of water. Hygienic evaluation]. Cesk Hyg. 1964;9:475–481. [PubMed] [Google Scholar]
- 7.Steffen C, Seitz R. Severe chlorate poisoning: report of a case. Arch Toxicol. 1981;48:281–288. [DOI] [PubMed] [Google Scholar]
- 8.Singelmann E, Wetzel E, Adler G, et al. Erythrocyte membrane alterations as the basis of chlorate toxicity. Toxicology. 1984;30:135–147. [DOI] [PubMed] [Google Scholar]
- 9.Lee E, Phua DH, Lim BL, et al. Severe chlorate poisoning successfully treated with methylene blue. J Emerg Med. 2013;44:381–384. [DOI] [PubMed] [Google Scholar]
- 10.Singelmann E, Steffen C. Increased erythrocyte rigidity in chlorate poisoning. J Clin Pathol. 1983;36:719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradberry SM. Occupational methaemoglobinaemia. Mechanisms of production, features, diagnosis and management including the use of methylene blue. Toxicol Rev. 2003;22:13–27. [DOI] [PubMed] [Google Scholar]
- 12.Jackson RC, Elder WJ, Mcdonnell H. Sodium-chlorate poisoning complicated by acute renal failure. Lancet. 1961;2:1381–1383. [DOI] [PubMed] [Google Scholar]
- 13.Sigler MH, Manns M. Continuous renal replacement therapy in the critically ill patient with acute renal failure. Pros and cons. ASAIO J. 1994;40:928–930. [DOI] [PubMed] [Google Scholar]
- 14.Hopkins J, Tudhope GR. The effects of drugs on erythrocytes in vitro: Heinz body formation, glutathione peroxidase inhibition and changes in mechanical fragility. Br J Clin Pharmacol. 1974;1:191–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ranghino A, Costantini L, Deprado A, et al. A case of acute sodium chlorate self-poisoning successfully treated without conventional therapy. Nephrol Dial Transplant. 2006;21:2971–2974. [DOI] [PubMed] [Google Scholar]