

Abstract
Deciphering of genetic variants plays a critical role in research and clinic of genetic disorders, such as the well known neurodegenerative disease Parkinson disease (PD). To combine pool of targeted genes and next-generation sequencing (NGS), investigators could obtain high efficient but low-cost sequencing data of interested genes. Aim to discover genetic variants that might contribute to PD, we selected 48 candidate genes involved in different pathways and conducted a pilot study to screen nonsynonymous SNPs (nsSNPs) in 4 pooled samples from 237 sporadic Chinese PD patients. Using our custom-designed NimbleGen array and Illumina HiSeq2000, a total of 4 novel nsSNPs (c. 352G>T in STK39, c. 823G>T in DGKQ, c. 36T>A in DLA-DRB5, and c. 1981G>T in GRN) were discovered but not validated by Sanger sequencing. Additionally, we also selected 6 annotated nsSNPs without report in previous PD studies and validated by Sanger sequencing. However, genotyping analysis of 6 validated nsSNPs in 50 PD patients and 50 controls showed no significant differences in cases compared with controls. These data represent the first documentation and validation of these mutations in PD using target gene capture sequencing. Additional replication studies in other populations and functional research are merited to better evaluate precapture multiplex protocol and validate the role of the 6 nsSNPs in PD risk.
INTRODUCTION
Genetic variation, including single-nucleotide polymorphisms (SNPs), plays a significant role in both Mendelian and non-Mendelian diseases. Recently, the understanding of the genetics of Parkinson disease (PD) has seen remarkable developments 5 known genes are proved be causative for monogenic forms,1–5 while more than 11 loci were identified as risk factors for the development of common forms of PD.6,7 Mutations in SNCA and LRRK2 were identified as a cause for dominant forms of PD, while mutant PARK2, PINK1, and DJ-1 were shown to motivate recessive forms of PD. Above 5 genes were all recognized by genetic linkage approaches, which required large pedigrees with affected and unaffected people. Given that a great number of genes and people sequenced by Sanger sequencing, this method must be time consuming and costly. Since LRRK2 was identified in family patients by Sanger in 2004, then no other genes have been found. However, a large sample size of sporadic PD cases still remain unexplained genetically.
The recent emergence of genome technologies provided wide-spectrum vision for the etiology of many diseases and was gradually making its way into clinical laboratories. From 2005, some next-generation sequencing (NGS) platforms have been launched, such as Illumina HiSeq2000, Roche 454, and ABI SOLiD, all of which will generate a great amount of sequence codes. Although NGS technology with high-throughput screening can be applied to decipher gene variants, its high cost still impede wide application in resequencing projects of whole human genome. Therefore, target sequence of interested genomic regions is an important approach for promoting its applications. The combination of target gene capture with NGS has been used in research and diagnosis on several complicated disorders and common diseases.8–10 Target-capture platforms, like Agilent SureSelect target-enrichment system and Nimblegen solid-phase hybrid array, have been developed to either capture the entire human exome or regions of interest. Recently, Nimblegen announced that the new precapture multiplex protocol enables multiple DNA samples to be barcoded and captured in a single SeqCap EZ Library for exome or custom capture experiments.
Previous studies proved that target gene capture-sequencing technology applied into detection of rare variants with high fidelity, throughput, and speed but low cost.10 In this study, we designed a customed, solid-phase array (Roche NimbleGen, Inc., Madison, WI) to capture 48 genes involved in PD pathogenic pathways (Table 1). After target regions enrichment, sequence by the HiSeq2000 Analyzer (Illumina, San Diego, CA) was conducted to analyze the variants of these genes in 4 pooled blood genomic DNA samples mixed from 237 Chinese sporadic PD patients. Furthermore, we performed the Sanger sequencing followed by PCR to validate the data obtained from NGS platform in 50 PD patients and 50 controls.
TABLE 1.
The Information of 48 Genes for Capture Sequence

MATERIALS AND METHODS
Study Subjects
Two hundred and thirty-seven unrelated patients with sporadic PD (94 female and 143 male) were recruited from the Neurology Department of Xiamen First Hospital. The average age and the average onset age of patients were 60.3 ± 11.3 (25–83) years and 56.4 ± 10.8 (23–80) years, respectively. Patients were examined by 2 neurologists based on diagnostic criteria of United Kingdom PD Brain Bank.11 All patients were divided into the early onset PD (onset before 50 years; n = 51) and the late onset PD (onset at or after 50 years; n = 186). Written informed consent form was obtained from all of the patients and the protocols of the study was approved by the Xiamen First Hospital Ethics Committee.
DNA Extraction
Genomic DNA was extracted from whole peripheral blood of the subjects using MagCore® Genomic DNA Whole Blood Kit (Cat. No. MGB400–04, RBC Bioscience Taiwan) on an automatic extraction machine MagCore HF16 (RBC Bioscience Corp., Taiwan). DNA quantification was determined by the NanoDrop ND2000 (Nanodrop Technologies, Wilmington, DE, USA) spectrophotometer, and the integrity of extracted genomic DNA was examined by 1% agarose electrophoresis. A total of 237 extracted genomic DNA samples were mixed into 4 pooled samples used for target capture and NGS.
Target Gene Capture and NGS
A custom capture array (NimbleGen, Roche) was designed to enrich 48 candidate genes implicated in the pathology of PD according to GeneReviews (NCBI). These genes included all exons, splice sites, and the immediately adjacent intron sequences. Genomic DNA was captured on a NimbleGen’ array following the manufacturer's protocols. Briefly, approximately 1 mg genomic DNA of participants was fragmented to a size range of 20 to 300 bp by ultrasonoscope. The fragmented DNA was purified and treated with T4 DNA polymerase, T4 phosphonucleotide kinase, and the Klenow fragment of Escherichia coli DNA polymerase. Adapters from Illumina were ligated onto the polished ends and confirmed by PCR. PCR primer has a barcode sequence which was used as the sample index signature. The resulting library was hybridized to a custom NimbleGen’ array. After hybridization, the DNA fragments bound to the array were washed and eluted by 300 mL of elution buffer (Qiagen, Hilden, Germany). Eluted library was sent to Illumina HiSeq2000 Analyzers for sequencing according to the manufacture's protocol. The following data filtering and analysis were totally followed the previous analysis pipeline.10
Genotyping
In order to confirm the DNA sequence variants screened by NGS, the target sites and the flanking sequences were amplified in recruited 100 samples (50 PD patients and 50 controls) individually with specific primers designed using Primer 5.0. PCR products were purified and sequenced by Sanger sequencing through ABI3730 (Applied Biosystems, Foster City, CA, USA), and sequence data were analyzed with Lasergene 8 software (DNAStar, WI, USA).
Statistical Analysis
All statistical analyses were performed using SPSS18.0 software. The frequencies of allele and genotype were compared between cases and controls by Fisher exact test. Statistically significant difference was accepted at a two-sided P < 0.05.
RESULTS
Total Sequence Enrichment and Target Region Mapping
To discover DNA variants of PD associated genes, we designed a unique capture array to enrich the 48 genes involved in PD. The enriched regions were sequenced by HiSeq2000 Analyzer (Illumina). Using target gene capture sequencing, we obtained a total of 87,234,014 to 103,783,232 high-quality reads across the 4 pooled samples from 237 sporadic PD patients. Although target region capture average ratio was 56.35%, 94.07% of the yielded clean reads could be mapping to the reference human genome (HG19). The majority of coverage ratio was over 94%, and average depth of target genes was more than 100-fold (Figure 1). Therefore, sequencing coverage was fully adequate to detect gene variants within the majority of the targeted regions.
FIGURE 1.
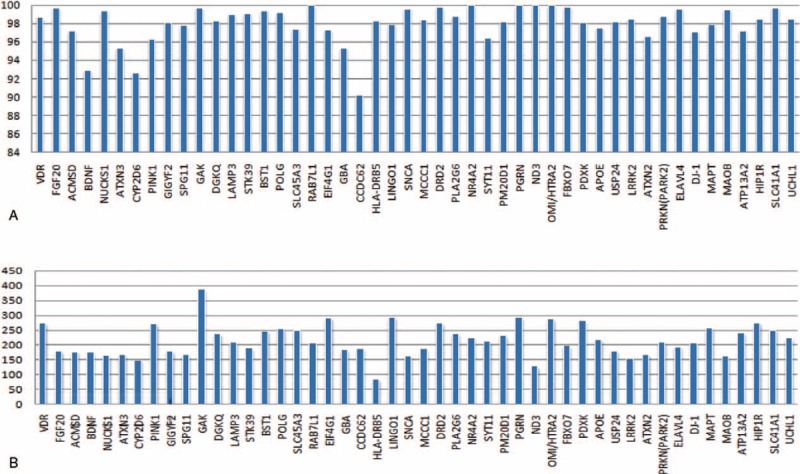
The coverage ratio and average depth distribution in target genes. (A) The coverage ratio distribution of target genes. The coverage ratio of each gene was more than 94%, except for CCDC62 (90.23%), CYP2D6 (92.61%), and BDNF (92.96%). (B) The average depth distribution of target genes. The average depth of each target gene was over 100-fold, except for HLA-DRB5 (83.09×).
Discovery of Novel Nonsynonymous SNPs (nsSNPs)
A total of 6455 common genetic variants across the 4 pooled samples were found by NGS, among which 109 variants were located in exons including 59 nsSNPs. In our study, we focused on both novel nsSNPs and annotated nsSNPs without report in previous PD studies, but they may implement strong influence on the structure and/or functions of protein leading to the rare phenotype of PD. As the result, we discovered 4 nsSNPs as novel after mapping our sequence data to The Single Nucleotide Polymorphism Database (dbSNP) 136 built containing data from 1000 genomes. Additionally, we identified 6 annotated nsSNPs without report in PD studies by PubMed search.
Validation of 4 Novel nsSNPs
To confirm the 4 novel nsSNPs (c. 352G>T in STK39, c. 823G>T in DGKQ, c. 36T>A in DLA-DRB5, and c. 1981G>T in GRN) discovered in 48 candidate genes of 4 pooled samples, Sanger sequencing was used to validate the results in 50 PD patients and 50 controls (Figure 2). However, 4 novel nsSNPs were not validated as they are likely subject to sequencing errors.
FIGURE 2.
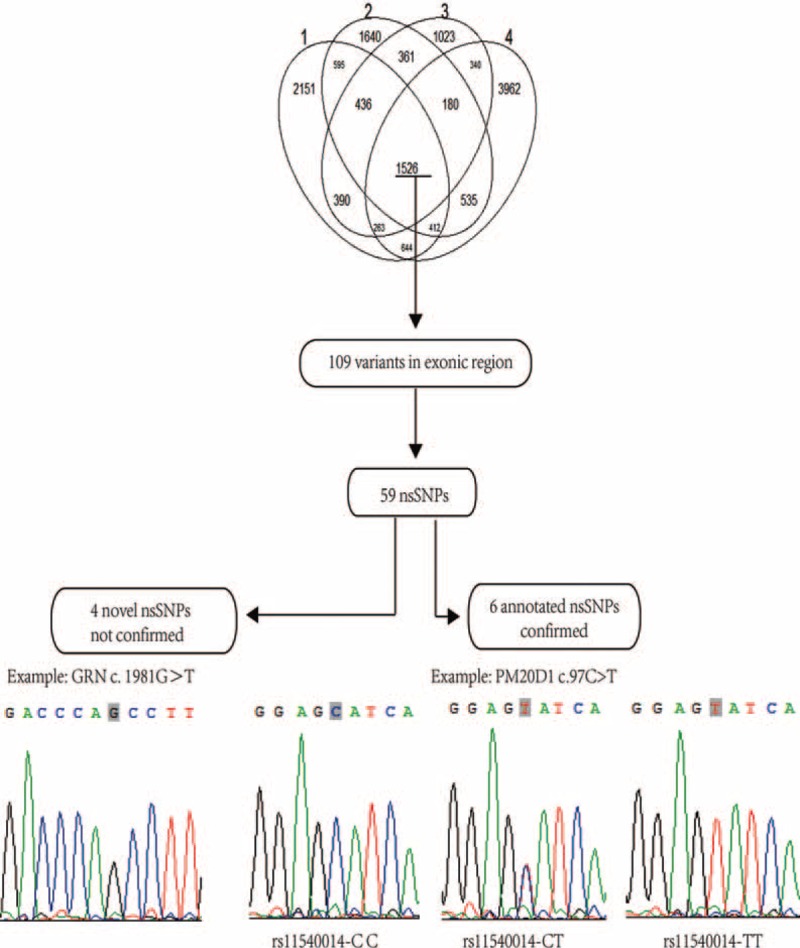
Schematic overview of results from discovery to validation. The number of common mutations was 6455 among 4 pooled samples from 237 sporadic PD patients. Validation and genotyping analysis of 4 novel nsSNPs and 6 annotated nsSNPs by Sanger sequencing. Genotyping was performed for 50 PD patients and 50 controls. Total numbers of validated and not validated nsSNPs are indicated. c.1981G>T in GRN and c.97C>T (rs11540014) in PM20D1 represent specific examples of not confirmed and confirmed calls, respectively.
Genotyping and Association Analysis of 6 Annotated nsSNPs
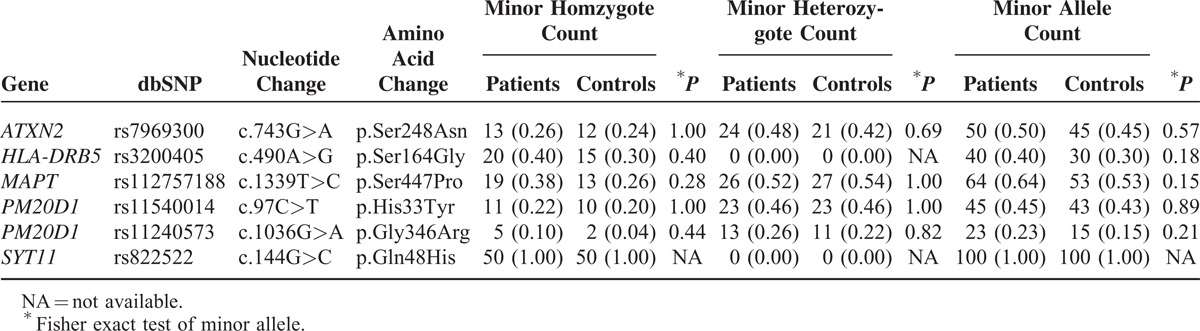
Using the Sanger sequencing, we performed validation and genotyping analysis of 6 annotated nsSNPs in 50 PD patients and 50 controls. Genotyping results are shown in Table 2. We detected 6 annotated nsSNPs in both patients and controls. Minor allele frequency (MAF) of these nsSNPs was not significant according to nominal P-values by Fisher exact test. We found rs822522 in SYT11 that was only allele “C” in 100 Chinese people, which was consistent to dbSNP report. Except for rs11240573 in PM20D1, MAFs of the most nsSNPs reached up to 5%, but none of them showed significant differences in frequencies between controls and cases. Genotypic analysis of minor homozygote or minor heterozygote also shown no significant differences in frequencies between controls and patients.
TABLE 2.
Genotype and Allele Analysis of 6 Annotated nsSNPs in 50 PD Patients and 50 Controls
DISCUSSION
This is a pilot study to explore the hypothesis that sporadic PD patients have individually rare but collectively more frequent functional variants in 48 PD associated genes in Chinese population by precapture multiplex protocol. We capitalized on advances in NGS to identify novel, potentially casual variants in these genes involved in major pathways of PD. Thus far, there was no such kind of a study has been reported in genetics studies of PD. In our study, we focused on 4 novel nsSNPs and 6 annotated nsSNPs without report in previous PD studies, which were discovered from 4 pooled genomic DNA samples mixed from 237 sporadic PD patients. Among them, 4 novel nsSNPs were not confirmed to be real variants given that there are billions of nucleotides in a human genome, even low experimental error rates yield many errors in variant calls. Nevertheless, 6 annotated nsSNPs were validated to be true positive. Our genotyping analysis of 6 annotated nsSNPs in 50 PD patients and 50 controls indicated that none of them was significant. Our data represent the first documentation of using target gene capture sequencing to identify rare variants from pooled samples in Chinese population. Additional replication studies in different populations and functional studies are warranted to better evaluate precapture multiplex protocol and validate the role of the 6 nsSNPs in PD risk.
The NGS platforms have dramatically developed throughout the laboratories. Although various aspects of DNA sequencing have been improved but a higher accuracy than the traditional Sanger sequencing does not appear to be one of them. Different kinds of sequencing errors signals might result from each process, from initially sample preparation to final data analysis. Generally, the error rate of NGS was between 0.1% and 1.0%.12 Thus, it is important to mitigate sequencing error through replicate and increase samples in NGS.13 In many investigations, individual samples and pooled samples are generally 2 kinds of prepared samples for sequencing. From other perspective, sequencing every diploid sample individually is a kind of pooled-line method because 2 haploid genomes are sequenced together. To call variants accurately for both haploids, the diploid needs to be sequenced to a sufficient depth (eg, 20×).14 However, individual samples are commonly not reached to such a depth (eg, the 1000 Human Genome Project15), in fact the majority of technology-detected aggregate properties of these individual sequences. For polymorphisms estimation issues, little information would be lost using sequencing pooled samples and also the efficiency would be largely improved when the sample number is large.16 It has been shown that the dual sequencing application could minimize the confounding effects of sequencing errors. It is the fact that dual platform is a standard pattern for NGS sequencing, on whichever platform, required to be backed up by other genotyping method, commonly Sanger sequencing.
As other multifactorial or complex disease, the interaction between environmental factors (such as a negative association with smoking) and genetic factors is response for pathogenesis of PD. Recent advances in high-throughput genetic analysis have brought new insights into the disorders, and PD was considered to be more than one disease entity. Table 1 lists loci with mutations have a causing or additive effect on PD. These loci include those traditionally noted as “Parkinson loci” (such as the new autosomal dominant gene EIF4G117) as well as others that are not, which are emerging as important in the pathogenesis of PD. However, our study has some limitations. Ligation of barcodes to fragmented PCR amplicons may result in uneven sequence coverage of different samples. The use of error-correcting barcodes would allow a greater proportion of pooled sequences to be deconvoluted.18 Because of the difference in the probe: sample ratio for the array and solution capture methods, we may perform target enrichment by solution hybridization for pooled samples. Future large studies are warranted to confirm our findings.
CONCLUSION
In summary, target gene capture sequence combines NGS with the capture of nucleotide sequencing from the subset of “high interest” genes. This method was evaluated by capturing specific sequences from 48 PD related genes in Chinese population. As expected, we found both positive and negative results. Nevertheless, we conclude that target gene capture sequencing is an effective way to identify rare variants of target disease genes. It is expected that following on the success of genome-wide association studies, in the next few years, target sequencing will reveal more novel genes involved in genetic diseases.
Footnotes
Abbreviations: MAF = minor allele frequency, NGS = next-generation sequencing, nsSNP = nonsynonymous SNP, PD = Parkinson disease, SNP = single-nucleotide polymorphism.
This work was supported by the Science and Technology Project of Fujian Province (No. 2012D062), China.
ZL and QL contributed equally to this work.
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304:1158–1160. [DOI] [PubMed] [Google Scholar]
- 2.Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003; 299:256–259. [DOI] [PubMed] [Google Scholar]
- 3.Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392:605–608. [DOI] [PubMed] [Google Scholar]
- 4.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997; 276:2045–2047. [DOI] [PubMed] [Google Scholar]
- 5.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004; 44:601–607. [DOI] [PubMed] [Google Scholar]
- 6.Nalls MA, Plagnol V, Hernandez DG, et al. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet 2011; 377:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.A two-stage meta-analysis identifies several new loci for Parkinson's disease. PLoS Genet 2011; 7:e1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonnefond A, Durand E, Sand O, et al. Molecular diagnosis of neonatal diabetes mellitus using next-generation sequencing of the whole exome. PLoS One 2010; 5:e13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levin JZ, Berger MF, Adiconis X, et al. Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts. Genome Biol 2009; 10:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei X, Ju X, Yi X, et al. Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. PLoS One 2011; 6:e29500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma Q, An X, Li Z, et al. P268S in NOD2 associates with susceptibility to Parkinson's disease in Chinese population. Behav Brain Funct 2013; 9:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol 2008; 26:1135–1145. [DOI] [PubMed] [Google Scholar]
- 13.Robasky K, Lewis NE, Church GM. The role of replicates for error mitigation in next-generation sequencing. Nat Rev Genet 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harismendy O, Ng PC, Strausberg RL, et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol 2009; 10:R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abecasis GR, Altshuler D, Auton A, et al. A map of human genome variation from population-scale sequencing. Nature 2010; 467:1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Z, Li X, Ling S, et al. Estimating DNA polymorphism from next generation sequencing data with high error rate by dual sequencing applications. BMC Genomics 2013; 14:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chartier-Harlin MC, Dachsel JC, Vilarino-Guell C, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet 2011; 89:398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mamanova L, Coffey AJ, Scott CE, et al. Target-enrichment strategies for next-generation sequencing. Nat Methods 2010; 7:111–118. [DOI] [PubMed] [Google Scholar]